
 |
 |
 |
 |
 |
Artigo
|
|
| Avaliação da eficiência fototérmica de óxidos de grafeno reduzidos com diferentes dimensões Evaluation of the photothermal efficiency of reduced graphene oxides with different sizes. |
|
Isabela A. A. BessaI; Mikaelly O. B. SousaI; Fernanda D. MarquesII; Elisa Q. C. da CostaI; Ana Beatriz C. SouzaI; Dayenny L. D'AmatoI; Camille C. de MelloI; Diego O. da CostaI I Departamento de Química Inorgânica, Universidade Federal Fluminense, 24020-141 Niterói - RJ, Brasil Recebido: 30/04/2025; *e-mail: cmronconi@id.uff.br Editor Convidado responsável pelo artigo: Suzana B. Peripolli Reduced graphene oxide (rGO) has emerged as a promising material for photothermal applications due to its ability to absorb near-infrared (NIR) light and efficiently convert it into heat. In this study, we investigated the influence of sheet size on the photothermal properties of rGO by employing two exfoliation methods - ultrasonic bath and probe sonication - at varying durations. The resulting materials were characterized by scanning electron microscopy (SEM), infrared and Raman spectroscopy, and X-ray diffraction (XRD). Subsequent chemical reduction with ascorbic acid was confirmed through spectroscopic analyses. SEM images and particle size distribution analyses revealed that longer sonication times, particularly with ultrasonic probes, produced smaller and more uniform flakes. Despite significant differences in lateral dimensions, photothermal experiments under NIR laser irradiation (830 nm) revealed similar temperature increases for all samples, indicating that the size reduction does not play a major role in enhancing the photothermal response under the tested conditions. These findings suggest that the photothermal performance of rGO is primarily governed by the restoration of conjugated sp2 domains during reduction, rather than by the sheet dimensions. The results provide valuable insights into the design of rGO-based photothermal agents, especially for biomedical applications where internalization depends on particle size. INTRODUÇÃO O grafeno é uma camada única de átomos de carbono com hibridização sp2 dispostos em uma estrutura hexagonal, semelhante a um favo de mel.1 Desde a sua descoberta em 2004,2 o grafeno tem se destacado como um dos principais representantes dos nanomateriais bidimensionais. No entanto, a produção de monocamadas de grafeno puras, com baixa densidade de defeitos e em larga escala, ainda representa um grande desafio técnico. Métodos como a deposição química de vapor, embora eficazes, são economicamente inviáveis para aplicações em larga escala.3 Como alternativa, a rota química envolvendo a oxidação do grafite e sua posterior redução tem se mostrado uma abordagem viável para obtenção do óxido de grafeno reduzido (rGO).4 O rGO tem sido amplamente explorado em substituição ao grafeno devido às suas propriedades ópticas,5,6 térmicas,7 adsortivas,8,9 eletrônicas10-12 catalíticas13,14 e fototérmicas.15,16 Especialmente em nanomedicina, a sua extensa rede de ligações π conjugadas, aliada à sua elevada área superficial, possibilita um alto carregamento de fármacos e interações biomoleculares eficientes.17 Além disso, essas ligações formam uma rede eletronicamente conjugada, capaz de absorver radiação na região do infravermelho próximo (NIR) de maneira eficiente, convertê-la em energia térmica e dissipar o calor por meio de vibrações estruturais.18,19 Esse efeito, chamado de efeito fototérmico, intimamente relacionado à estrutura do material, posiciona o rGO como um candidato promissor para aplicações biomédicas20,21 e fotocatalíticas.22,23 Estudos recentes demonstraram o uso de rGO em diferentes contextos biomédicos,24-26 incluindo formulações para o tratamento de glioblastomas,27 infecções bacterianas28 e câncer de pulmão,29 sempre explorando sua capacidade de gerar calor sob irradiação NIR. Em geral, observa-se que a eficiência fototérmica está fortemente relacionada à estrutura eletrônica e à extensão dos domínios sp2, restaurados durante a etapa de redução.20 Trabalhos anteriores do nosso grupo16,30 demonstraram a eficácia de folhas de rGO funcionalizadas com polímeros como agentes quimiofototérmicos no tratamento da Leishmaniose. Nesse estudo, as folhas de rGO, com dimensões na escala de micrômetros, foram eficientemente internalizadas por macrófagos, que são células do sistema imunológico capazes de fagocitar partículas grandes de até 30 µm.31-33 O calor gerado pelo efeito fototérmico potencializou a morte do parasita, validando o potencial terapêutico do rGO. No entanto, para aplicações em células cancerosas, que não possuem capacidade de fagocitose e dependem de mecanismos como difusão passiva e endocitose, partículas menores são necessárias.34,35 Neste contexto, em um trabalho mais recente,17 foi observado que as folhas de rGO da ordem de mícrons podem induzir morte celular por sufocamento, mostrando a importância do controle das dimensões do material para otimizar sua eficiência terapêutica. A redução do tamanho das folhas de rGO para 400 nm aumentou a internalização celular. Diante disso, o presente trabalho tem como objetivo investigar como diferentes métodos e tempos de esfoliação afetam o tamanho das folhas de rGO e, consequentemente, sua eficiência fototérmica (Esquema 1). Considerando que a redução do tamanho das folhas pode impactar a capacidade de absorção e dissipação de energia, combinamos técnicas de esfoliação (banho ultrassônico e sonda ultrassônica) com caracterizações estruturais, análises por microscopia eletrônica de varredura e ensaios térmicos sob irradiação NIR. Dessa forma, buscamos compreender de forma sistemática a influência do tamanho lateral das folhas na resposta fototérmica do material.
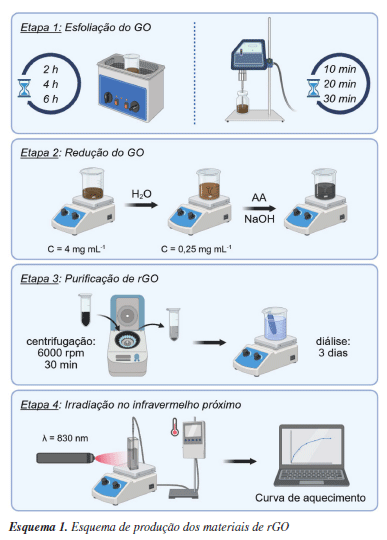
PARTE EXPERIMENTAL Materiais Os seguintes reagentes foram utilizados sem tratamento prévio: ácido ascórbico (AA, P.A., Reagen), ácido clorídrico (HCl, 35-37%, Isofar), ácido sulfúrico (H2SO4, 98%, Vetec), grafite em flocos (≥ 99%, Sigma-Aldrich), hidróxido de sódio (NaOH, P.A., Sigma-Aldrich), nitrato de sódio (NaNO3, ≥ 99%, Vetec), pentóxido de fósforo (P4O10, ≥ 99%, Sigma-Aldrich), permanganato de potássio (KMnO4, 97%, Sigma-Aldrich), peróxido de hidrogénio (H2O2, ≥ 30%, Sigma-Aldrich), persulfato de potássio (K2S2O8, P.A., Vetec). Os solventes tetracloroetileno (TCE), acetona e isopropanol (P.A., Synth) foram empregados na preparação dos substratos revestidos com as amostras de rGO para microscopia eletrônica de varredura (MEV). Métodos Síntese do óxido de grafeno O óxido de grafeno foi sintetizado a partir da oxidação do grafite pelo método de Hummers modificado,36 seguindo a metodologia previamente reportada pelo nosso grupo.8,30,37,38 Para a preparação da dispersão, 600 mg de grafite oxidado foram dispersos em 150 mL de água deionizada e macerados com um bastão de vidro formando uma dispersão de concentração de 4 mg mL-1. A dispersão foi então fracionada em três béqueres de 50 mL e três frascos de 40 mL, cada um contendo 25 mL da suspensão. As amostras nos béqueres foram submetidas à esfoliação em banho de ultrassom (Ecosonics, 160 W, 40 kHz, 3,8 L) por períodos de 2, 4, e 6 h. Já as amostras dos frascos foram processadas por esfoliação com sonda de ultrassom (Sonics Vibracell, 125 W, 20 kHz, sonda de titânio de 6 mm de diâmetro) a 40% de amplitude, por 10, 20, e 30 min, utilizando um ciclo de pulsos de 5 s ligado / 1 s desligado. Síntese do óxido de grafeno reduzido Em um béquer de 600 mL, 20 mL das dispersões foram diluídas em 300 mL de água destilada até atingir uma concentração de 0,25 mg mL-1, seguida da adição de 400 mg de AA (7,1 mM).39 O pH do meio foi então ajustado para 11 com NaOH e as dispersões foram mantidas sob agitação à temperatura ambiente por 48 h. Após esse período, as amostras foram submetidas à centrifugação (3622 rcf, 30 min), redispersas em água e submetidas à diálise empregando membranas de celulose (14 kDa, Sigma-Aldrich) por 72 h. Como resultado, foram obtidas dispersões finais com concentração de 1,0 mg mL-1. As amostras foram nomeadas de acordo com o método e tempo de esfoliação. Caracterizações As análises de espectroscopia vibracional na região do infravermelho próximo foram obtidas no espectrofotômetro Thermo Scientific Nicolet iS50 FTIR, equipado com acessório de reflectância total atenuada (ATR), obtidos a 298 K, com resolução de 4 cm-1 e 32 varreduras por espectro. Os espectros Raman foram medidos em um microscópio Raman confocal Witec Alpha300 AR com comprimento de onda de excitação de 532 nm (rede de difração 600 L mm-1, potência 0.5 mW). As bandas D e G foram modeladas usando a função gaussiana no software OriginPro, versão 9.0 (OriginLab Corporation, USA, 2019). As análises por difração de raios-X de policristal (PDRX) foram realizadas usando um difratômetro Empyrean 3rd Generation PANalytical/Malvern no modo spinner com radiação Kα (λ = 1,5418 Å) em uma faixa de 2θ de 5-50°. Microscopia eletrônica de varredura (MEV) A preparação das amostras para análise em MEV foi realizada por meio da deposição dos materiais, previamente diluídos em água, sobre substratos semicondutores. Para isso, discos de silício (Si) com óxido natural (lote No. 5/0716; diâmetro: 100 ± 0,5 mm; tipo N, dopado com fósforo; orientação (100) ± 0,5°; resistividade: 3-7 Ω cm; espessura: 400 ± 25 μm) foram cortados em placas de aproximadamente 0,5 cm de aresta. Em seguida, os substratos foram submetidos a uma pré-limpeza sequencial com TCE, acetona e isopropanol, seguida de tratamento em Plasma Cleaner (modelo 1020, Fischione Instruments) por 5 min, sob vácuo, utilizando atmosfera composta por 75,0 ± 0,20% de argônio e 25,0 ± 0,20% de oxigênio (mol/mol).40 Para a deposição, as dispersões de rGO (0,25 mg mL-1) foram diluídas para 0,01 mg mL-1, com sonicação em banho por 10 min para homogeneização e dispersão. A caracterização morfológica e dimensional do material depositado foi realizada no microscópio Helios Nanolab 650, FEI, com feixe acelerado a 5 kV e desacelerado a 500 V, utilizando detecção por through-the-lens detector (TLD). As imagens foram processadas no ImageJ, versão 1.54p (National Institutes of Health, USA, 2017), e os dados analisados no Microsoft Excel, versão 365 (Microsoft, USA, 2011) e OriginPro, versão 9.0. As condições específicas para as medidas granulométricas estão detalhadas na Tabela 1.
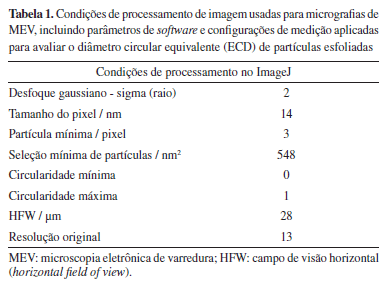
A determinação do tamanho lateral das folhas de rGO foi realizada com base no diâmetro circular equivalente (ECD), um parâmetro geométrico definido como apropriados para materiais bidimensionais, conforme estabelecido em normas técnicas para caracterização por MEV e em protocolos atualizados de procedimentos analíticos tal como descrito no Guia europeu de medidas de tamanho lateral para nanomateriais.41-44 Estudo da propriedade fototérmica Para avaliar o efeito fototérmico dos materiais produzidos, 1 mL das dispersões aquosas de rGO nas concentrações de 1,0, 0,5 e 0,1 mg mL-1 foram irradiadas perpendicularmente em uma cubeta de quartzo, utilizando um laser contínuo de GaAs (comprimento de onda do laser (λ) = 830 nm; irradiância (E) = 1,6 W cm-2; potência (P) = 0,2 W; área do feixe (A) = 0,126 cm2) (Fluence MAXX, HTM). A variação de temperatura foi monitorada com um termopar acoplado à câmera FLIR TG267 (Teledyne FLIR). Os experimentos foram realizados em triplicata e conduzidos à temperatura ambiente. Cálculo da eficiência fototérmica As curvas de aumento de temperatura em função do tempo foram ajustadas de acordo com o modelo descrito por Roper e Hoepfner.45 As curvas foram ajustadas no software OriginPro 9.0 a uma função exponencial derivada da Lei de resfriamento de Newton, que descreve a variação da temperatura T de um corpo ao longo do tempo t em um ambiente com temperatura constante (Equação 1): 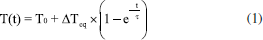 onde T é temperatura no tempo t, Teq é a temperatura de equilíbrio (ou temperatura máxima atingida), T0 é a temperatura ambiente (ou temperatura inicial) e é uma constante arbitrária de proporcionalidade. Combinando essa abordagem ao balanço de energia do sistema, é possível estimar a eficiência fototérmica (η) dos materiais analisados em cada concentração (Equação 2): 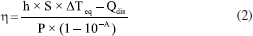 onde h é o coeficiente de transferência de calor, S é a área de troca de calor, Qdis é o calor dissipado pelo controle (água), P é a potência do laser (0,2 W) e A é a absorvância no comprimento de onda de incidência do laser (830 nm). O produto desconhecido h × S é obtido a partir da relação da Equação 3, onde mi é a massa dos componentes, em gramas, e Cpi é a capacidade calorífica dos componentes. Para dispersões diluídas (até 1 mg mL-1), a contribuição da capacidade calorífica do material é muito baixa e, portanto, considera-se apenas a capacidade calorífica da água (4,18 J °C g-1).  Os valores obtidos dos ajustes estão dispostos nas Tabelas 3S-5S (Material Suplementar).
RESULTADOS E DISCUSSÃO Síntese dos materiais óxido de grafite, óxido de grafeno (GO) e óxido de grafeno reduzido (rGO) A produção de monocamadas de grafeno sem defeitos em larga escala ainda representa um desafio técnico e financeiro para a comunidade científica. Como alternativa para explorar as propriedades desse material, uma abordagem viável envolve a oxidação do grafite para obtenção de óxido de grafite, seguida de sua esfoliação para gerar óxido de grafeno (GO) e posterior redução a óxido de grafeno reduzido (rGO). O óxido de grafite é composto por domínios de carbonos sp2 e sp3, e grupos oxigenados, como epóxidos e hidroxilas no plano basal, e carbonilas e carboxilas nas extremidades.46 A inserção desses grupos aumenta a distância interplanar e enfraquece a interação entre as folhas, facilitando a esfoliação.47 Na literatura, existem três metodologias principais para a oxidação do grafite a óxido de grafite: Brodie,48 Staudenmaier49 e Hummers.50 A extensão da oxidação do material varia conforme o método selecionado. Dentre essas abordagens, a utilizada neste trabalho foi a metodologia de Hummers modificada, que incorpora uma etapa preliminar de pré-oxidação para aumentar o grau de oxidação, facilitando, assim, a esfoliação do material.51 Durante essa etapa, o P4O10 atua como agente desidratante, removendo moléculas de água residuais do grafite. Esse processo é fundamental para que o K2S2O8, em meio fortemente ácido, intercale com as folhas do grafite, formando a espécie de grafite intercalado, viabilizando a etapa de oxidação propriamente dita.52,53 A reação se iniciou entre NaNO3 e H2SO4 concentrado sob aquecimento, formando NO2, O2 e H2O (reação 4).  Em seguida, a adição de KMnO4 em meio fortemente ácido leva à formação da espécie MnO3+ (reação 5) que, posteriormente, reage com excesso de permanganato para formar Mn2O7 (reação 6).  De acordo com trabalhos da literatura, a espécie Mn2O7 se intercala com as folhas de grafite e, sob aquecimento, o Mn2O7 se reduz a MnO2, liberando O2 e O3 (reação 7).54,55 Essa intercalação é favorecida pela etapa de pré-oxidação que aumenta a distância interplanar entre as folhas do grafite e permite a inserção do Mn2O7. 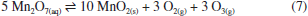 O MnO2 formado é posteriormente reduzido a Mn(II) (reação 8), enquanto o excesso de Mn2O7 pode reagir com água, formando o ácido permangânico (reação 9). 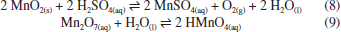 Para completar o processo, o meio reacional foi tratado com H2O2, reduzindo todo o Mn(VII) a Mn(II) (reação 10), que foi removido por sucessivas lavagens com água.54 Embora o método de Hummers modificado promova uma oxidação quase completa das folhas de grafite, ele também induz mudanças estruturais, como enrugamento e formação de defeitos (buracos), além de gerar resíduos de metais pesados. Apesar dessas limitações, continua sendo a abordagem mais utilizada.54 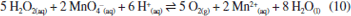 A geração de oxigênio molecular nas reações 1 e 4, assim como de ozônio na reação 4, contribui diretamente para a oxidação do grafite. Em particular, o ozônio desempenha um papel fundamental nesse processo, pois é um agente oxidante significativamente mais forte (E0 = 2,08 V) em comparação com o oxigênio molecular (E0 = 1,23 V).56 Além disso, o permanganato também participa ativamente da oxidação ao reagir com as ligações C=C, promovendo a formação de epóxidos, o que influencia diretamente a estrutura e reatividade do material.55 A obtenção do óxido de grafeno (GO) a partir do óxido de grafite é realizada por esfoliação usando métodos térmicos,57 eletroquímicos58 ou sonoquímicos.59 Esse último é realizado por meio de sonicação na presença de um solvente polar, a qual utiliza a energia ultrassônica para romper as interações intermoleculares do óxido de grafite, enquanto o solvente estabiliza as folhas por ligações de hidrogênio e interações dipolo-dipolo.60 A vantagem desse método reside na eliminação do uso de alta temperatura, pressão e longo tempo de reação, bem como etapas de reação trabalhosas.61 As ondas ultrassônicas promovem colisões interparticulares e colisões entre as partículas e a parede do béquer, podendo gerar um acúmulo de tensão de tração.62-64 A energia gerada é suficiente para exercer tanto uma força normal, capaz de separar mecanicamente as camadas, quanto uma força lateral, que promove o deslizamento relativo entre elas.65 Além disso, a força gerada por essa técnica de esfoliação pode fragmentar partículas maiores ou camadas espessas de GO em estruturas menores. Esse efeito de fragmentação pode reduzir o tamanho lateral do GO que facilita a esfoliação, uma vez que folhas menores apresentam forças coletivas de interação de van der Waals mais fracas entre suas camadas, tornando o processo mais eficiente.66 No caso do uso de um banho ultrassônico, as ondas se propagam no meio líquido sem incidir diretamente sobre a amostra. Ainda assim, a energia gerada é suficiente para romper as interações entre as folhas de óxido de grafite, favorecendo a formação do GO. Por outro lado, quando se utiliza uma sonda ultrassônica, o contato direto com a amostra concentra a energia mecânica em uma região reduzida, promovendo um acúmulo de carga de tração nas folhas, o que leva à diminuição do tamanho das partículas, além de induzir defeitos na estrutura.62 Mesmo assim, a literatura66 reporta que, devido à cavitação acústica não uniforme, é difícil controlar as tensões de cisalhamento e compressão que atuam sobre o GO, o que resulta em folhas com tamanho médio variando de centenas de nanômetros a alguns micrômetros. Para avaliar o efeito da sonicação nas propriedades fototérmicas, utilizamos dois métodos - a sonda e o banho ultrassônico - aplicados por diferentes períodos, e os materiais resultantes foram caracterizados por meio de técnicas físico-químicas. Os espectros vibracionais na região do infravermelho dos materiais de GO esfoliados por banho e sonda de ultrassom exibiram perfis semelhantes, evidenciando as bandas características da oxigenação desses materiais (Figura 1a). As bandas atribuídas à deformação axial das ligações hidroxila (ν(O-H)), carbonila (ν(C=O)) e epóxido (ν(C-O-C)) são observadas nas regiões de 3377, 1719 e 1215 cm-1, respectivamente, e as bandas de deformação angular de hidroxilas (δ(O-H)) e ligação de carbono e oxigênio (δ(C-O)) são observadas em 1380 e 1046 cm-1 (Tabela 1S, Material Suplementar). Adicionalmente, em 1618 cm-1, destaca-se a banda correspondente à deformação axial da ligação C=C, associada aos domínios sp2 remanescentes.
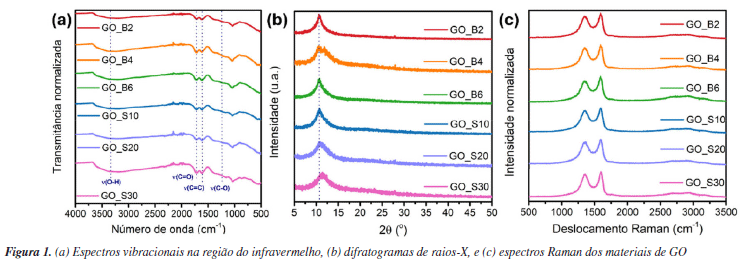
Para investigar se os processos de esfoliação causaram mudanças a longo alcance nos materiais de GO, foram realizadas análises de difração de raios-X de policristal (PDRX). O difratograma do grafite apresentou um pico característico e bem definido em 2θ = 26,6° referente ao plano cristalino (002), que corresponde à distância d(002) entre as folhas de grafeno (Figura 1S, Material Suplementar). A partir da Lei de Bragg (Equação 11), a distância interplanar foi calculada em 3,34 Å, valor em concordância com a literatura.67  Os difratogramas dos materiais de GO apresentaram um pico largo e deslocado para a região de baixo ângulo, entre 2θ = 10,6-11,3° (Figura 1b), conforme apresentado na Tabela 2. Esse deslocamento é típico do processo de oxidação do grafite para GO, no qual a introdução de grupos oxigenados resulta no aumento da distância interplanar entre as folhas. Consequentemente, os valores de d(002), calculados pela Lei de Bragg, encontram-se elevados em relação ao grafite precursor, variando entre 7,8 e 8,4 Å.

Ao comparar os diferentes métodos de esfoliação para o GO, observa-se, a partir dos dados da Tabela 2, que o tempo de exposição ao banho de ultrassom não causou alterações significativas nos valores de d(002) entre as amostras. No entanto, destaca-se a amostra GO_S30, que apresentou o menor valor de d(002), sugerindo uma leve diminuição na distância interplanar. Esse comportamento pode estar relacionado a um início de redução do GO promovido in situ pela ação da sonda ultrassônica.17,68,69 A espectroscopia Raman aplicada a materiais derivados de grafeno fornece informações valiosas sobre a estrutura da rede cristalina. Os espectros desses materiais apresentam duas bandas características: a banda G, em torno de ca. 1580 cm-1, atribuída ao estiramento simétrico do modo E2g das ligações C-C; e a banda D, próxima de ca. 1350 cm-1, associada ao modo de relaxação A1g dos anéis aromáticos. Essa última é ativada pela presença de defeitos estruturais que quebram a simetria local.70 Assim, a intensidade da banda D está diretamente relacionada ao grau de desordem do material, sendo influenciada por defeitos como extremidades, vacâncias, anéis com diferentes números de átomos de carbono e variações na hibridização.71-73 Além da intensidade, a largura total à meia altura (FWHM, do inglês full width at half maximum) da banda D também fornece informações sobre a qualidade cristalina, impurezas e o tamanho dos cristalitos.74 Variações na FWHM da banda D refletem mudanças na razão entre hibridizações sp2/sp3, afetando os modos vibracionais e a largura dos picos Raman.75 Para quantificar o nível de desordem frequentemente se analisa a razão entre as intensidades das bandas D e G (ID/IG), sendo que valores elevados desta razão indicam maior número de defeitos ou maior desordem estrutural. O espectro Raman do grafite apresenta as bandas D e G, sendo a banda D consideravelmente menos intensa que a banda G (Figura 2S, Material Suplementar), resultando em uma razão ID/IG igual a 0,09, conforme apresentado na Tabela 2. Essa razão baixa é compatível com o grau elevado de ordenamento da estrutura do grafite. Com a inserção de grupos oxigenados durante o processo de oxidação do grafite para o GO, a banda D se torna mais alargada e intensa e, consequentemente, a razão ID/IG tende a aumentar, refletindo a transição parcial da hibridização dos átomos de carbono de sp2 para sp3 (Figura 1c, Tabela 2). Essa mudança promove a introdução de defeitos e distorções na rede, intensificando a banda D e, consequentemente, elevando a razão ID/IG.76 Isso é observado para todos os materiais de GO sintetizados nesse trabalho. Contudo, assim como observado nos difratogramas, as diferentes técnicas e tempos de esfoliação não parecem influenciar significativamente o nível de organização estrutural do material, sendo observadas apenas variações incrementais na razão ID/IG.
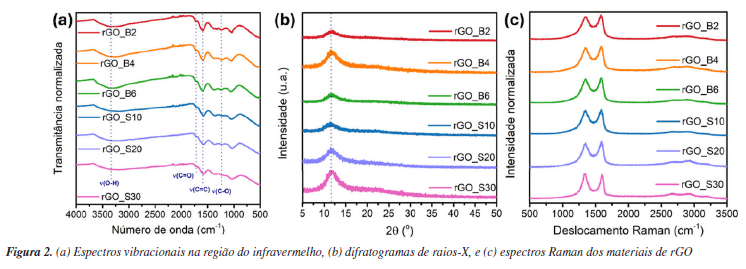
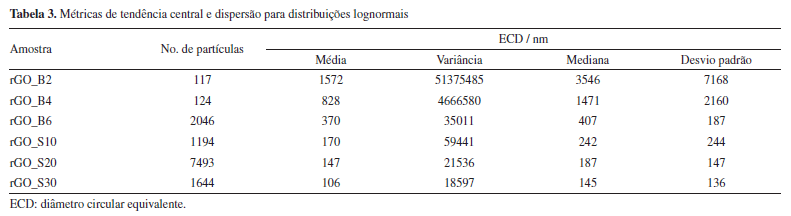
A partir dos valores de ID/IG, é possível estimar o tamanho lateral dos cristalitos (La) e a distância média entre defeitos (LD) por meio das Equações 12 e 13,77-79 nas quais λL representa o comprimento de onda do laser utilizado. O parâmetro La está associado à extensão média dos domínios grafíticos ordenados, compostos por átomos de carbono sp2; quanto maior esse valor, maior a continuidade e organização da rede cristalina.77 Por sua vez, LD corresponde ao espaçamento médio entre defeitos estruturais - como vacâncias, grupos funcionais oxigenados ou distorções - e serve como uma medida direta da densidade de imperfeições que interrompem a conjugação eletrônica do material.78 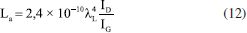 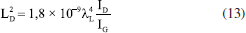 Em geral, valores elevados de La e LD são indicativos de alta cristalinidade e baixa desordem, enquanto valores mais baixos revelam uma estrutura mais fragmentada e rica em defeitos. Para LD > 10 nm, o material ainda preserva regiões ordenadas e contínuas de estrutura sp2, e a interação entre defeitos vizinhos é mínima. Essa condição é importante porque garante que a Equação 13 seja válida, já que ela foi desenvolvida para amostras com baixa densidade de defeitos (ou seja, defeitos dispersos).78 Quando LD é menor que 10 nm, os defeitos estão muito próximos e começam a interagir, o que invalida os pressupostos do modelo e exige outras abordagens para caracterizar o grau de desordem. Portanto, LD > 10 nm é um indicativo de baixa a moderada desordem e permite o uso confiável das relações empíricas derivadas da espectroscopia Raman para quantificação de defeitos. O grafite apresentou valores de La e LD aproximadamente iguais a 213 e 40 nm, respectivamente, valores típicos de uma rede altamente ordenada e com extensa conjugação sp2. Já as amostras de GO apresentaram valores muito menores tanto de La quanto de LD, indicando uma rede significativamente mais defeituosa (Tabela 2), não sendo observadas mudanças significativas entre os métodos de preparo. Após o tratamento sonoquímico, as dispersões foram submetidas à redução química com ácido ascórbico (AA E0 = −0,9 V),80 Em trabalhos anteriores,16,17,30 a hidrazina (E0 = −1,16 V)81 foi utilizada como agente redutor por ser conhecida por reduzir principalmente os grupos epóxidos e, portanto, restaurar a sistema π.82,83 Embora não tenhamos observado efeitos adversos advindos de seu uso nos estudos in vitro, a hidrazina é um composto tóxico e explosivo.84 Por isso, neste estudo, optamos por um agente redutor mais verde e tão eficiente quanto a hidrazina.85 De acordo com a literatura,39,85,86 a redução do GO pelo AA ocorre principalmente por meio da conversão de epóxidos e hidroxilas. Por meio de espectroscopia de fotoelétrons excitados por raios-X (XPS), Yoshimura e colaboradores86 demonstraram que, em meio alcalino, essa redução leva à diminuição predominante da concentração de epóxidos, hidroxilas e carbonilas. Mais recentemente, Ortiz-Anaya e Nishina39 propuseram que, em meio ácido, a redução do GO ocorre via ataque nucleofílico do AA ao grupo epóxido, gerando hidroxilas, um carbocátion intermediário e, por fim, restaurando a ligação C=C do rGO e com a formação do subproduto ácido 2,3-dicetogulônico. No espectro de infravermelho dos materiais de rGO (Figura 2a), observa-se uma diminuição na intensidade das bandas associadas aos grupos oxigenados, cujas atribuições estão detalhadas na Tabela 1S. Destaca-se, em particular, a banda correspondente ao estiramento da ligação de carbonila (ν(C=O)), sugerindo que a redução ocorreu predominantemente nas extremidades do material. Embora a literatura relacione a redução do GO por AA principalmente devido à eliminação de epóxidos e hidroxilas, como discutido anteriormente, estudos mais recentes sugerem que, sob condições brandas, como temperatura ambiente, a redução pode ocorrer majoritariamente nos grupos carbonilados. Essa seletividade é atribuída à maior reatividade desses grupos frente ao AA em ambientes menos agressivos, o que permite a modulação das propriedades do rGO sem comprometer sua estabilidade coloidal.75,86 Um exemplo dessa abordagem seletiva foi relatado por Chen e colaboradores,87 em que usaram componentes extracelulares bacterianos Lysinibacillus sp., em contato com o GO. Os autores demonstraram que essas bactérias foram capazes de reduzir o GO por meio da secreção de AA sob condições brandas (30 °C), obtendo espectros de infravermelho semelhantes aos aqui apresentados, o que corrobora a hipótese de que a redução ocorreu majoritariamente nas bordas do material. Zanoni e colaboradores75 investigaram o mecanismo de redução do GO pelo AA e pela N-acetilcisteína (NAC) por meio de cálculos teóricos baseados em teoria do funcional da densidade (DFT, do inglês density functional theory), aliados às análises eletroquímicas e espectroscópicas. Eles observaram que o processo envolve principalmente a conversão dos grupos epóxi e carbonila em grupos hidroxila, promovendo a restauração parcial da rede grafítica. A redução dos grupos carbonila mostrou-se termodinamicamente mais favorável, ocorrendo por meio de uma única transferência de hidrogênio do AA, com formação de um grupo hidroxila e liberação do radical HA•, que se afasta espontaneamente da superfície sem barreira energética significativa. Por outro lado, a redução dos grupos epóxi exige duas transferências de hidrogênio sequenciais: a primeira forma um grupo hidroxila e o radical HA• e a segunda resulta na geração de água e ácido desidroascórbico (DHA), que se desliga da superfície. Apesar de ser altamente exergônica, essa via apresenta uma barreira cinética mais alta. Esses resultados mostram que a redução das carbonilas é energeticamente favorável, bem como acessível do ponto de vista cinético, contribuindo para a regeneração seletiva de regiões da estrutura conjugada do grafeno. É importante destacar que diversos fatores como concentração de GO, concentração do agente redutor e tempo de redução influenciam diretamente no grau de redução dos materiais, sendo desafiador comparar diretamente com os trabalhos da literatura. Os difratogramas de raios-X dos materiais de rGO apresentaram um pico característico do plano cristalino (002) na região de 2θ = 11,4-12°, correspondente a uma distância interplanar de aproximadamente 7,6 Å (Figura 2b, Tabela 1). Em comparação ao GO, não foi observado um deslocamento significativo desse pico para ângulos maiores, o que indica uma baixa taxa de reempilhamento das folhas de rGO. Esse comportamento pode estar associado ao método de redução utilizado. Como discutido anteriormente, a redução sob condições brandas com AA tende a ocorrer preferencialmente nas extremidades das folhas, enquanto o plano basal permanece em grande parte inalterado. Consequentemente, a baixa extensão de redução nas regiões centrais não contribui significativamente para a diminuição da distância interplanar. Além disso, o AA pode atuar como um agente estabilizante eficaz, promovendo a dispersão coloidal do material e inibindo o reempilhamento das folhas durante e após o processo de redução.84 Por fim, os espectros Raman revelaram um aumento discreto na razão ID/IG, sendo esse aumento mais evidente nos materiais esfoliados por sonda ultrassônica (Figura 2c). Em teoria, a redução do GO a rGO deveria diminuir a intensidade da banda D, pois o processo tende a restaurar a estrutura grafítica. No entanto, observa-se o efeito oposto: a remoção dos grupos oxigenados acaba introduzindo mais defeitos estruturais, o que justifica o aumento da razão ID/IG. Ainda assim, o parâmetro FWHM da banda D, que está diretamente relacionado à eficiência do processo de redução, mostra-se reduzido após a conversão de GO em rGO, conforme indicado na Tabela 2. Esse estreitamento da banda D, juntamente com o aumento da razão ID/IG, sugere uma reestruturação parcial dos domínios sp2, especialmente nos materiais obtidos por esfoliação com sonda ultrassônica. Dessa forma, a utilização da sonda parece ter influenciado não apenas na dispersibilidade dos materiais, mas também no seu tamanho, favorecendo uma maior área de contato e, consequentemente, uma redução mais eficaz. Em relação aos parâmetros La e LD, a comparação entre as amostras de GO e rGO apresenta a mesma tendência observada na largura da banda D e na razão ID/IG, com as amostras produzidas por sonda exibindo valores menores de La e LD, indicando uma rede mais defeituosa. Esses resultados mostram que, embora a redução do GO para rGO possa restabelecer parcialmente a estrutura grafítica, ela não é capaz de eliminar completamente os defeitos gerados durante a oxidação, mantendo uma densidade de defeitos muito superior à do grafite puro. Microscopia eletrônica de varredura (MEV) Com o objetivo de avaliar se os diferentes tratamentos sonoquímicos tiveram impactos significativos no tamanho dos materiais produzidos, as amostras foram analisadas por microscopia eletrônica de varredura (MEV) e análises estatísticas. A Figura 3 apresenta as imagens de MEV obtidas para todas as amostras analisadas, juntamente com as distribuições lognormais correspondentes do diâmetro circular equivalente (ECD). A análise ECD das partículas mostrou tendências relacionadas ao tempo de exposição em sistemas de banho e sonda ultrassônica. As distribuições foram ajustadas a um modelo lognormal para representar com mais precisão a dispersão dimensional das partículas, considerando a natureza assimétrica dos dados.
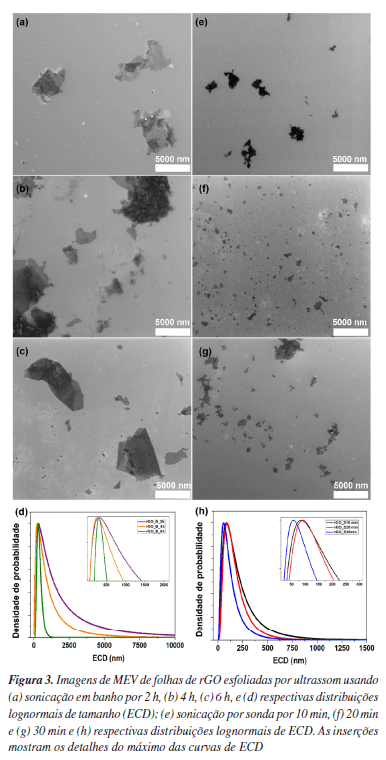
Para as amostras tratadas com banho ultrassônico foi observado um estreitamento perceptível da distribuição lognormal após 6 h de tratamento. Em contraste, tempos de tratamento mais curtos (2 e 4 h) resultaram em distribuições mais amplas. A maior variabilidade no tamanho das partículas é devido à atuação irregular das tensões mecânicas ao longo do material. Essa distribuição não homogênea de energia pode levar tanto à esfoliação incompleta de algumas regiões quanto à fragmentação excessiva em outras, comprometendo a uniformidade morfológica das folhas de GO obtidas. Essa tendência sugere que o aumento do tempo de esfoliação leva a uma redução na dispersão do tamanho, resultando em um conjunto mais homogêneo de partículas em termos de ECD. Esse comportamento é consistente com a natureza da distribuição lognormal, na qual a largura da curva está relacionada ao desvio padrão logarítmico (σ), representando a dispersão em uma escala logarítmica. Um σ menor implica em uma distribuição mais estreita e, portanto, maior uniformidade dimensional. Para as partículas esfoliadas por sonda ultrassônica, também foi observado um leve estreitamento da distribuição no maior tempo de tratamento avaliado (30 min). Embora a diferença na largura da curva entre os tratamentos de 10, 20 e 30 min tenha sido pequena, a amostra tratada por 30 min apresentou uma mediana de ECD ligeiramente menor, indicando uma fragmentação mais efetiva das partículas naquele momento. A redução da mediana, juntamente com o leve estreitamento da curva, sugere que a ação mecânica da ultrassonografia por 30 min favoreceu uma distribuição de tamanho mais centrada, com partículas menores e redução da assimetria positiva. Isso porque a densidade de energia transmitida ao sistema foi mais elevada e/ou concentrada, promovendo uma esfoliação mais eficiente e maior incidência de fragmentação, o que favorece a obtenção de folhas de GO com distribuição de tamanho mais uniforme. As medidas de dispersão referentes às amostras analisadas estão apresentadas na Tabela 3.
Estudo da propriedade fototérmica A propriedade fototérmica de um material está relacionada à sua capacidade de absorver luz e converter energia luminosa em calor.88 No caso dos nanomateriais, essa conversão ocorre por três mecanismos principais: aquecimento plasmônico localizado, relaxamento não radiativo em semicondutores, e vibrações térmicas de moléculas. Materiais à base de carbono, como o grafeno e seus derivados, absorvem luz e geram calor por meio de vibrações da rede cristalina.88 As ligações C=C possuem elétrons que podem ser facilmente excitados dos orbitais π para os orbitais π* sob irradiação de baixa energia. Quando a energia dos fótons incidentes é suficiente para induzir uma transição eletrônica no material, os elétrons π são promovidos do estado fundamental (HOMO, orbital molecular de maior energia ocupado) para um nível de energia superior (LUMO, orbital molecular de menor energia desocupado). O subsequente relaxamento desses elétrons para o estado fundamental ocorre via acoplamento vibrônico, resultando na liberação do excesso de energia na forma de calor. Além disso, a conjugação ou hiperconjugação dos orbitais π pode influenciar significativamente as transições eletrônicas entre HOMO e LUMO, reduzindo a energia da banda proibida à medida que o número de ligações π aumenta.88 Portanto, é importante avaliar se o efeito fototérmico de materiais a base de grafeno é dependente do tamanho de suas folhas. Como discutido anteriormente, o preparo do GO a partir do grafite envolve a oxidação das folhas, introduzindo grupos hidroxila e epóxido em ambos os lados do plano basal, além de grupos carboxila nas extremidades.89 Essas modificações aumentam a separação entre as nanofolhas, favorecendo sua esfoliação e levando à formação de regiões alifáticas (sp3) intercaladas com domínios aromáticos (sp2). A distribuição aleatória desses domínios depende do grau de oxidação do material.51,90,91 A posterior redução do GO elimina parcialmente os grupos oxigenados, restaurando os domínios sp2 e ampliando a absorção na região do infravermelho próximo (NIR).91 Como resultado, o rGO se apresenta como um excelente agente fototérmico a partir de relaxamentos de rede.88,92,93 Além disso, esse efeito é particularmente relevante para aplicações biológicas, como a fototerapia, pois dentro da janela de transparência biológica (700-950 nm), a absorção e o espalhamento da luz pelos tecidos são minimizados.94 Isso permite o aquecimento local sem causar danos significativos às células adjacentes, tornando o rGO um candidato promissor para aplicações médicas baseadas em aquecimento localizado. Considerando futuras aplicações biológicas, as dispersões foram preparadas em água Milli-Q. Os espectros de absorção na região do ultravioleta visível (UV-Vis) estão apresentados na Figura 3S e as atribuições na Tabela 2S (Material Suplementar). As amostras de GO apresentam uma banda em ca. 230 nm atribuída à transição p-p* das ligações C=C em regiões de domínio sp2, e um ombro em torno de 300 nm, relacionado à transição n-p* das ligações C=O nas regiões sp3.95 Após o processo de redução, a absorção em ca. 300 nm desaparece, e a banda principal sofre um deslocamento batocrômico para ca. 246 nm, indicando a restauração da conjugação π. Esse deslocamento, associado à menor energia requerida para a transição p-p*, pode ser utilizado como indicativo da eficiência do processo de redução.96 Na Figura 4 são apresentadas as curvas de aquecimento dos seis materiais produzidos em diferentes concentrações. A curva correspondente à água Milli-Q, utilizada como controle, não mostrou variação expressiva de temperatura ao longo do tempo de irradiação, confirmando que o efeito fototérmico observado é exclusivamente atribuído aos materiais analisados. As curvas de variação de temperatura (DT) ao longo do tempo, para ambos os métodos de esfoliação, revelam uma clara dependência com a concentração: quanto menor a concentração, menor o DT. Na Figura 5 são apresentados os dados de DT em função da concentração.
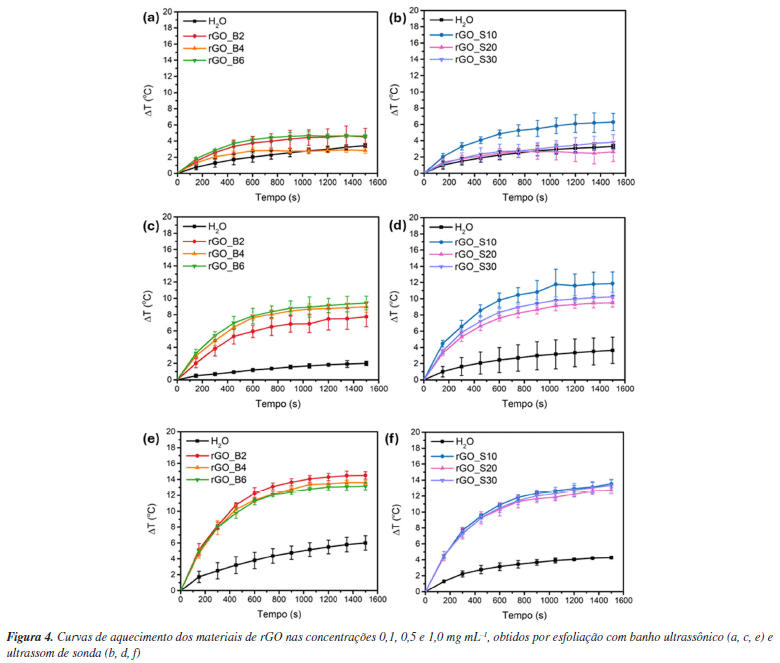
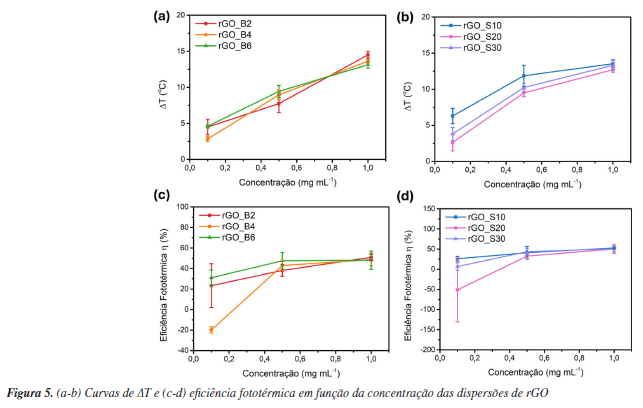
Em 0,1 mg mL-1, o DT registrado foi muito próximo ao da água, indicando que, nas condições experimentais adotadas, o efeito fototérmico dos materiais não é detectável nessa concentração. Na concentração 1 mg mL-1, ambos os métodos de esfoliação resultaram em ΔT médio semelhante, em torno de 13 °C. Já na concentração intermediária de 0,5 mg mL-1, os materiais obtidos via ultrassom de sonda apresentaram um ΔT levemente superior (ca. 10 °C) em relação aos esfoliados por banho ultrassônico (ca. 9 °C). Esses resultados sugerem que a eficiência na conversão de luz em calor está mais fortemente associada à restauração dos domínios sp2 promovida durante o processo de redução do que ao tamanho médio das folhas resultantes da esfoliação. A excitação dos plasmons de superfície e a subsequente dissipação de energia via decaimento não radiativo ocorrem dentro dos domínios sp2 do rGO. Dessa forma, mesmo que as folhas sejam menores na esfoliação por sonda, o efeito fototérmico se mantém desde que haja uma quantidade suficiente desses domínios para absorver e converter energia térmica de maneira eficiente. Além disso, como o efeito fototérmico ocorre localmente em cada folha, a redução no tamanho das partículas não compromete a capacidade total do material de gerar calor sob irradiação. A eficiência da conversão fototérmica, calculada a partir da Equação 2, fornece a fração de luz convertida em calor.97 Como observado nas Figuras 5c e 5d, na concentração de 0,1 mg mL-1, a eficiência é muito baixa e, em alguns casos, até negativa, indicando que nesta concentração não há diferença significativa. A conversão de energia luminosa em calor não depende apenas da composição química do material conversor, mas também de sua concentração, do volume da solução e de parâmetros do laser utilizado, como o comprimento de onda, a área do feixe e a potência.98 Diante da grande variabilidade nos métodos de preparo, nos tipos de materiais produzidos e nas condições empregadas nos experimentos fototérmicos, torna-se difícil estabelecer uma comparação direta com os dados reportados na literatura.
CONCLUSÕES Neste trabalho foi investigado como diferentes métodos e tempos de esfoliação influenciam o tamanho das folhas de óxido de grafeno reduzido (rGO) e sua eficiência fototérmica. A esfoliação por sonda ultrassônica mostrou-se mais eficaz na fragmentação das folhas, resultando em partículas com distribuição de tamanho mais homogênea e medianas significativamente menores. A redução química com ácido ascórbico, conduzida sob condições brandas, preservou a estabilidade coloidal dos materiais e restaurou, de forma seletiva, os domínios conjugados sp2, fator essencial para a absorção de luz na região do infravermelho próximo (NIR). Apesar das diferenças nas dimensões das folhas, os experimentos de aquecimento sob irradiação NIR mostraram que todas as amostras apresentaram desempenho fototérmico semelhante, com variações de temperatura diretamente proporcionais à concentração, mas praticamente independentes do tamanho das folhas. Esses resultados indicam que, nas condições estudadas, a eficiência fototérmica do rGO está mais relacionada à extensão da rede eletronicamente conjugada do que às dimensões laterais do material. Essa observação é particularmente relevante para aplicações biomédicas, pois permite a otimização do tamanho das folhas para favorecer a internalização celular, sem comprometer a eficiência na conversão de luz em calor. Assim, este estudo contribui para o entendimento do papel do tamanho de partículas em sistemas fototérmicos e fornece subsídios importantes para o desenvolvimento de nanomateriais funcionalizados com aplicações terapêuticas.
MATERIAL SUPLEMENTAR O material suplementar desse trabalho (atribuições das bandas nos espectros de infravermelho dos materiais GO e rGO, espectro vibracional Raman, difratograma de raios-X do grafite comercial, espectros eletrônicos dos materiais GO e rGO, com suas respectivas atribuições, e tabelas contendo os valores dos ajustes das curvas fototérmicas) está disponível em http://quimicanova.sbq.org.br/, na forma de arquivo PDF, com acesso livre.
DECLARAÇÃO DE DISPONIBILIDADE DE DADOS Os dados que suportam os resultados deste estudo serão disponibilizados pelo autor correspondente mediante solicitação.
AGRADECIMENTOS Os autores agradecem às agências brasileiras de fomento pelo apoio financeiro: Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, 409082/2022-8 e 312949/2020-0), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, bolsa de I. A. A. B.) e Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ, CNE E-26/200.418/2023 e Rede Nano E-26/010.000981/2019). Agradecemos ao Laboratório de Espectroscopia Molecular (http://www.uff.br/lame/), aos Laboratórios Multiusuários de Caracterização de Materiais (http://www.uff.br/lamate/), Laboratório Multiusuário de raios-X (Lab-X) do Centro Brasileiro de Pesquisas Físicas (CBPF) pelas análises de DRX (https://www.gov.br/cbpf/pt-br/laboratorios/lab-x-laboratorio-multiusuario-de-raios-x-do-cbpf) e ao Laboratório de Difração e Espectroscopia pelas análises de espectroscopia Raman (http://www.inmetro.gov.br/metcientifica/materiais/lades.asp).
CONTRIBUIÇÕES DO AUTOR Isabela A. A. Bessa foi responsável por conceituação, investigação, curadoria de dados, análise formal, redação (rascunho original, revisão e edição); Mikaelly O. B. Sousa por investigação, curadoria de dados, análise formal, redação de rascunho original; Fernanda D. Marques por investigação, curadoria de dados, redação de rascunho original; Elisa Q. C. da Costa, Ana Beatriz C. Souza, Dayenny L. D'Amato, Camille C. de Mello, Diego O. da Costa, Wilson S. Nascimento, Johnnys S. Hortêncio por investigação, curadoria de dados, análise formal; Bráulio S. Archanjo por curadoria de dados, aquisição de financiamento, administração de projetos, recursos, redação (revisão e edição); Célia M. Ronconi por conceituação, curadoria de dados, aquisição de financiamento, administração de projetos, recursos e redação (revisão e edição).
REFERÊNCIAS 1. Lin, H.; Buerki-Thurnherr, T.; Kaur, J.; Wick, P.; Pelin, M.; Tubaro, A.; Carniel, F. C.; Tretiach, M.; Flahaut, E.; Iglesias, D.; Vázquez, E.; Cellot, G.; Ballerini, L.; Castagnola, V.; Benfenati, F.; Armirotti, A.; Sallustrau, A.; Taran, F.; Keck, M.; Bussy, C.; Vranic, S.; Kostarelos, K.; Connolly, M.; Navas, J. M.; Mouchet, F.; Gauthier, L.; Baker, J.; Suarez-Merino, B.; Kanerva, T.; Prato, M.; Fadeel, B.; Bianco, A.; ACS Nano 2024, 18, 6038. [Crossref] 2. Geim, A. K.; Novoselov, K. S.; Nat. Mater. 2007, 6, 183. [Crossref] 3. Saha, J. K.; Dutta, A.; Waste Biomass Valorization 2022, 13, 1385. [Crossref] 4. De Silva, K. K. H.; Huang, H. H.; Joshi, R. K.; Yoshimura, M.; Carbon 2017, 119, 190. [Crossref] 5. Lundie, M.; Sljivančanin, Z.; Tomić, S.; J. Mater. Chem. C 2015, 3, 7632. [Crossref] 6. Ferreira, D. R. C.; Serodre, T.; Gastelois, P. L.; Macedo, W. A. A.; Martins, E. M. N.; Furtado, C. A.; J. Braz. Chem. Soc. 2024, 35, e-20240164. [Crossref] 7. Kim, C.; Bin; Lee, J.; Cho, J.; Goh, M.; Carbon 2018, 139, 386. [Crossref] 8. Ribeiro, M.; Bessa, I.; da Silva, A.; Ligiero, C.; Osta, L.; Silva, L.; Araujo, J.; Archanjo, B.; Ronconi, C.; dos Santos, T.; J. Braz. Chem. Soc. 2024, 35, e-20240093. [Crossref] 9. da Silva, N. V. M.; Ladeira, A. C. Q.; Furtado, C. A.; J. Mol. Liq. 2024, 413, 126001. [Crossref] 10. Navarro-Rodriguez, M.; Camús, V.; Cros, A.; Garro, N.; Somoza, A. M.; Palacios-Lidon, E.; Appl. Surf. Sci. 2024, 642, 158611. [Crossref] 11. da Silva, F.; Rocha, D.; Silva, M.; Nossol, E.; Muñoz, R.; Semaan, F.; Dornellas, R.; J. Braz. Chem. Soc. 2020, 31, 429. [Crossref] 12. Zamboni, R. L.; Kalinke, C.; Ferreira, L. M. C.; Papi, M. A. P.; Orth, E. S.; Banks, C. E.; Marcolino-Júnior, L. H.; Bergamini, M. F.; Anal. Methods 2025, 17, 2214. [Crossref] 13. Fonsaca, J. E. S.; Hostert, L.; Zarbin, A. J. G.; Orth, E. S.; J. Mater. Chem. A 2024, 12, 8124. [Crossref] 14. Santos, Y.; Hostert, L.; Almeida, T.; Zarbin, A.; Souza, V.; Orth, E.; J. Braz. Chem. Soc. 2024, 35, e-20240063. [Crossref] 15. Amarnath, M.; Basu, H.; Basu, R.; Singh, S.; Chandwadkar, P.; Acharya, C.; Kailasa, S. K.; Patra, C. N.; Materials Today Sustainability 2024, 26, 100725. [Crossref] 16. Vitorino, L. S.; dos Santos, T. C.; Bessa, I. A. A.; Santos, E. C. S.; Verçoza, B. R. F.; de Oliveira, L. A. S.; Rodrigues, J. C. F.; Ronconi, C. M.; Colloids Surf. B Biointerfaces 2022, 209, 112169. [Crossref] 17. Bessa, I. A. A.; Cruz, J. V. R.; de Sousa, M. O. B.; Marques, F. D.; Archanjo, B. S.; Rocco, M. L. M.; Nascimento, V.; Ribeiro Pinto, L. F.; dos Santos, T. C.; Costa, N. M.; Ronconi, C. M.; ACS Appl. Nano Mater. 2025, 8, 4047. [Crossref] 18. Tarcan, R.; Todor-Boer, O.; Petrovai, I.; Leordean, C.; Astilean, S.; Botiz, I.; J. Mater. Chem. C 2020, 8, 1198. [Crossref] 19. Balandin, A. A.; Nat. Mater. 2011, 10, 569. [Crossref] 20. Wu, J.; Li, Z.; Li, Y.; Pettitt, A.; Zhou, F.; Technol. Cancer Res. Treat. 2018, 17. [Crossref] 21. Hashemi, M.; Omidi, M.; Muralidharan, B.; Smyth, H.; Mohagheghi, M. A.; Mohammadi, J.; Milner, T. E.; ACS Appl. Mater. Interfaces 2017, 9, 32607. [Crossref] 22. Mondal, A.; Prabhakaran, A.; Gupta, S.; Subramanian, V. R.; ACS Omega 2021, 6, 8734. [Crossref] 23. Van Tuan, P.; Hung, N. D.; Ha, L. T.; J. Mol. Struct. 2025, 1330, 141515. [Crossref] 24. de Melo-Diogo, D.; Lima-Sousa, R.; Alves, C. G.; Correia, I. J.; Biomater. Sci. 2019, 7, 3534. [Crossref]. 25. Liu, X.; Wu, X.; Xing, Y.; Zhang, Y.; Zhang, X.; Pu, Q.; Wu, M.; Zhao, J. X.; ACS Appl. Bio Mater. 2020, 3, 2577. [Crossref] 26. Hu, Y.; He, L.; Ma, W.; Chen, L.; Polym. Int. 2018, 67, 1648. [Crossref] 27. Dash, B. S.; Lu, Y. J.; Chen, J. P.; ACS Appl. Mater. Interfaces 2024, 16, 13543. [Crossref] 28. Yan, S.; Xu, S.; Wang, Y.; You, J.; Guo, C.; Wu, X.; Adv. Healthcare Mater. 2024, 13, 2400884. [Crossref] 29. Zhang, D. Y.; Zheng, Y.; Tan, C. P.; Sun, J. H.; Zhang, W.; Ji, L. N.; Mao, Z. W.; ACS Appl. Mater. Interfaces 2017, 9, 6761. [Crossref] 30. Vitorino, L. S.; dos Santos, T. C.; Bessa, I. A. A.; Santos, E. C. S.; Verçoza, B. R. F.; de Oliveira, L. A. S.; Rodrigues, J. C. F.; Ronconi, C. M.; Data Brief 2022, 41, 107841. [Crossref] 31. Lendeckel, U.; Venz, S.; Wolke, C.; ChemTexts 2022, 8, 12. [Crossref] 32. Bessa, I. A. A.; D'Amato, D. L.; Souza, A. B. C.; Levita, D. P.; Mello, C. C.; da Silva, A. F. M.; dos Santos, T. C.; Ronconi, C. M.; ACS Infect. Dis. 2024, 10, 2485. [Crossref] 33. Zent, C. S.; Elliott, M. R.; FEBS J. 2017, 284, 1021. [Crossref] 34. Wu, C.; Wu, Y.; Jin, Y.; Zhu, P.; Shi, W.; Li, J.; Wu, Q.; Zhang, Q.; Han, Y.; Zhao, X.; RSC Adv. 2019, 9, 13855. [Crossref] 35. da Silva, A. F. M.; da Costa, N. M.; Fernandes, T. S.; Bessa, I. A. A.; D'Amato, D. L.; Senna, C. A.; Lohan-Codeço, M.; Nascimento, V.; Palumbo, A.; Archanjo, B. S.; Pinto, L. F. R.; dos Santos, T. C.; Ronconi, C. M.; ACS Appl. Nano Mater. 2022, 5, 13805. [Crossref] 36. Pereira, A. F.; Schmidt, A.; Neves, B. R. A.; de Oliveira, C. K. B. Q. M.; Zarbin, A. J. G.; Nanoscale 2025, 17, 9974. [Crossref] 37. dos Santos, T. C.; Ronconi, C. M.; J. CO2 Util. 2017, 20, 292. [Crossref] 38. dos Santos, T. C.; Mancera, R. C.; Rocha, M. V. J.; da Silva, A. F. M.; Furtado, I. O.; Barreto, J.; Stavale, F.; Archanjo, B. S.; Carneiro, J. W. M.; Costa, L. T.; Ronconi, C. M.; J. CO2 Util. 2021, 48, 101517. [Crossref] 39. Ortiz‐Anaya, I.; Nishina, Y.; ChemPlusChem 2023, 88, e202300328. [Crossref] 40. Reinhardt, K. A.; Kern, W.; Handbook of Silicon Wafer Cleaning Technology, 3rd ed.; Elsevier: New York, 2018. 41. ISO 13322-1:2014 (en): Particle Size Analysis - Image Analysis Methods, Part 1: Static Image Analysis Methods, 2014. [Link] acessado em outubro 2025 42. ISO 19749:2021 (en): Nanotechnologies - Measurements of Particle Size and Shape Distributions by Scanning Electron Microscopy, ISO: Geneva, 2021. [Link] acessado em outubro 2025 43. ISO 17200:2020 (en): Nanotechnology - Nanoparticles in Powder Form - Characteristics and Measurements, ISO: Geneva, 2020. [Link] acessado em outubro 2025 44. Dapkunas, S. J.; Jillavenkatesa, A.; Lum, L. S. H.; NIST Recommended Practice Guide: Particle Size Characterization; National Institute of Standards and Technology (NIST): Gaithersburg, 2001. [Crossref] 45. Roper, D. K.; Ahn, W.; Hoepfner, M.; J. Phys. Chem. C 2007, 111, 3636. [Crossref] 46. Wu, J.; Lin, H.; Moss, D. J.; Loh, K. P.; Jia, B.; Nature Reviews Chemistry 2023, 7, 162. [Crossref] 47. Yang, Z.; Sun, Y.; Ma, F.; Appl. Surf. Sci. 2020, 529, 147075. [Crossref] 48. Brodie, B. C.; Philos. Trans. R. Soc. London 1858, 149, 249. [Crossref] 49. Staudenmaier, L.; Ber. Dtsch. Chem. Ges. 1898, 31, 1481. [Crossref] 50. Hummers, W. S.; Offeman, R. E.; J. Am. Chem. Soc. 1958, 80, 1339. [Crossref] 51. Kovtyukhova, N. I.; Ollivier, P. J.; Martin, B. R.; Mallouk, T. E.; Buzaneva, E. V.; Gorchinskiy, A. D.; Chem. Mater. 1999, 11, 771. [Crossref] 52. Dimiev, A. M.; Ceriotti, G.; Behabtu, N.; Zakhidov1, D.; Pasquali, M.; Saito, R.; Tour, J. M.; ACS Nano 2013, 7, 2773. [Crossref] 53. Dimiev, A. M.; Bachilo, S. M.; Saito, R.; Tour, J. M.; ACS Nano 2012, 6, 7842. [Crossref] 54. Chen, X.; Qu, Z.; Liu, Z.; Ren, G.; ACS Omega 2022, 7, 23503. [Crossref] 55. Yuan, H.; Ye, J.; Ye, C.; Yin, S.; Li, J.; Su, K.; Fang, G.; Wu, Y.; Zheng, Y.; Ge, M.; Tang, R.; Feng, G.; Qu, Y.; Zhu, Y.; Chem. Mater. 2021, 33, 1731. [Crossref] 56. Shriver, D.; Atkins, P.; Inorganic Chemistry, 5th ed.; W. H. Freeman: New York, 2009. 57. Shi, Y.; Wei, W.; Shen, L.; Bao, N.; Ind. Eng. Chem. Res. 2021, 60, 9787. [Crossref] 58. Gutiérrez‐Pineda, E.; Subrati, A.; Rodríguez‐Presa, M. J.; Gervasi, C. A.; Moya, S. E.; ChemEurJ 2023, 29, e202302450. [Crossref] 59. Li, C.; Lin, J.; Shen, L.; Bao, N.; Chem. Eng. Sci. 2020, 213, 115414. [Crossref] 60. Rossa, V.; Monteiro Ferreira, L. E.; Vasconcelos, S. C.; Shimabukuro, E. T.; Madriaga, V. G. C.; Carvalho, A. P.; Castellã Pergher, S. B.; da Silva, F. C.; Ferreira, V. F.; Conte Junior, C. A.; Lima, T. M.; RSC Adv. 2022, 12, 14084. [Crossref] 61. Muthoosamy, K.; Manickam, S.; Ultrason. Sonochem. 2017, 39, 478. [Crossref] 62. Mellado, C.; Figueroa, T.; Baez, R.; Meléndrez, M.; Fernández, K.; Materials Today Chemistry 2019, 13, 1. [Crossref] 63. Turner, P.; Hodnett, M.; Dorey, R.; Carey, J. D.; Sci. Rep. 2019, 9, 8710. [Crossref] 64. Tyurnina, A. V.; Tzanakis, I.; Morton, J.; Mi, J.; Porfyrakis, K.; Maciejewska, B. M.; Grobert, N.; Eskin, D. G.; Carbon 2020, 168, 737. [Crossref] 65. Yi, M.; Shen, Z.; J. Mater. Chem. A 2015, 3, 11700. [Crossref] 66. Weng, C.; Wu, J.; Shen, L.; Bao, N.; J. Mater. Sci. 2021, 56, 18946. [Crossref] 67. Jiao, X.; Qiu, Y.; Zhang, L.; Zhang, X.; RSC Adv. 2017, 7, 52337. [Crossref] 68. Abulizi, A.; Okitsu, K.; Zhu, J. J.; Ultrason. Sonochem. 2014, 21, 1174. [Crossref] 69. Liao, K. H.; Lin, Y. S.; Macosko, C. W.; Haynes, C. L.; ACS Appl. Mater. Interfaces 2011, 3, 2607. [Crossref] 70. Eckmann, A.; Felten, A.; Mishchenko, A.; Britnell, L.; Krupke, R.; Novoselov, K. S.; Casiraghi, C.; Nano Lett. 2012, 12, 3925. [Crossref] 71. Scardaci, V.; Compagnini, G.; C 2021, 7, 48. [Crossref] 72. Wu, J. B.; Lin, M. L.; Cong, X.; Liu, H. N.; Tan, P. H.; Chem. Soc. Rev. 2018, 47, 1822. [Crossref] 73. Thapliyal, V.; Alabdulkarim, M. E.; Whelan, D. R.; Mainali, B.; Maxwell, J. L.; Diamond Relat. Mater. 2022, 127, 109180. [Crossref] 74. Nandee, R.; Chowdhury, M. A.; Hossain, N.; Rana, M.; Mobarak, M. H.; Islam, A.; Aoyon, H.; Results Eng. 2024, 21, 101738. [Crossref] 75. Marrani, A. G.; Motta, A.; Palmieri, V.; Perini, G.; Papi, M.; Dalchiele, E. A.; Schrebler, R.; Zanoni, R.; Mater. Adv. 2020, 1, 2745. [Crossref] 76. Chadha, N.; Sharma, R.; Saini, P.; Carbon Lett. 2021, 31, 1125. [Crossref] 77. Cançado, L. G.; Takai, K.; Enoki, T.; Endo, M.; Kim, Y. A.; Mizusaki, H.; Jorio, A.; Coelho, L. N.; Magalhães-Paniago, R.; Pimenta, M. A.; Appl. Phys. Lett. 2006, 88, 163106. [Crossref] 78. Cançado, L. G.; Jorio, A.; Ferreira, E. H. M.; Stavale, F.; Achete, C. A.; Capaz, R. B.; Moutinho, M. V. O.; Lombardo, A.; Kulmala, T. S.; Ferrari, A. C.; Nano Lett. 2011, 11, 3190. [Crossref] 79. Cançado, L. G.; da Silva, M. G.; Ferreira, E. H. M.; Hof, F.; Kampioti, K.; Huang, K.; Pénicaud, A.; Achete, C. A.; Capaz, R. B.; Jorio, A.; 2D Mater. 2017, 4, 025039.[Crossref] 80. Hassan, O. B.; Chenavier, Y.; Maurel, V.; Pérard, J.; Bouzina, A.; Dubois, L.; Duclairoir, F.; Mater. Adv. 2025, 6, 143. [Crossref] 81. Rosca, V.; Koper, M. T. M.; Electrochim. Acta 2008, 53, 5199. [Crossref] 82. Park, S.; An, J.; Potts, J. R.; Velamakanni, A.; Murali, S.; Ruoff, R. S.; Carbon 2011, 49, 3019. [Crossref] 83. Gao, X.; Jang, J.; Nagase, S.; J. Phys. Chem. C 2010, 114, 832. [Crossref] 84. Zhang, J.; Yang, H.; Shen, G.; Cheng, P.; Zhang, J.; Guo, S.; Chem. Commun. 2010, 46, 1112. [Crossref] 85. Fernández-Merino, M. J.; Guardia, L.; Paredes, J. I.; Villar-Rodil, S.; Solís-Fernández, P.; Martínez-Alonso, A.; Tascón, J. M. D.; J. Phys. Chem. C 2010, 114, 6426. [Crossref] 86. de Silva, K. K. H.; Huang, H. H.; Yoshimura, M.; Appl. Surf. Sci. 2018, 447, 338. [Crossref] 87. Zhou, Y.; Lin, Z.; Han, Y.; Gan, L.; Cheng, Y.; Chen, Z.; Chemosphere 2024, 369, 143813. [Crossref] 88. Cui, X.; Ruan, Q.; Zhuo, X.; Xia, X.; Hu, J.; Fu, R.; Li, Y.; Wang, J.; Xu, H.; Chem. Rev. 2023, 123, 6891. [Crossref] 89. Yuan, R.; Yuan, J.; Wu, Y.; Chen, L.; Zhou, H.; Chen, J.; Appl. Surf. Sci. 2017, 416, 868. [Crossref] 90. Chen, Y. W.; Su, Y. L.; Hu, S. H.; Chen, S. Y.; Adv. Drug Delivery Rev. 2016, 105, 190. [Crossref] 91. Singh, R. K.; Kumar, R.; Singh, D. P.; RSC Adv. 2016, 6, 64993. [Crossref] 92. Yu, R.; Guo, Q.; Xia, F.; de Abajo, F. J. G.; Phys. Rev. Lett. 2018, 121, 057404. [Crossref] 93. Savchuk, Ol. A.; Carvajal, J. J.; Massons, J.; Aguiló, M.; Díaz, F.; Carbon 2016, 103, 134. [Crossref] 94. Li, C.; Chen, G.; Zhang, Y.; Wu, F.; Wang, Q.; J. Am. Chem. Soc. 2020, 142, 14789. [Crossref] 95. Kumar, N.; Setshedi, K.; Masukume, M.; Ray, S. S.; Carbon Lett. 2022, 32, 1031. [Crossref] 96. Go, S. H.; Kim, H.; Yu, J.; You, N. H.; Ku, B. C.; Kim, Y. K.; Solid State Sci. 2018, 84, 120. [Crossref] 97. Skinner, W. H.; Salimi, M.; Moran, L.; Blein-Dezayes, I.; Mehta, M.; Mosca, S.; Vaideanu, A. G.; Gardner, B.; Palombo, F.; Schätzlein, A. G.; Matousek, P.; Harries, T.; Stone, N.; J. Phys. Chem. C 2025, 129, 1864. [Crossref] 98. Freitas, S. C.; Belo, J. H.; Granja, A.; Canhota, M.; Silva, A. S.; Reis, S.; Crespo, H.; Araújo, J. P.; Sousa, C. T.; Adv. Mater. Interfaces 2023, 10, 2202214. [Crossref] |

On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access