
 |
 |
 |
 |
 |
Artigo
|
|
| Avaliação da combinação da nebulização discreta e processos de microextração aplicados à determinação de molibdênio por espectrometria de absorção atômica com chama (FAAS) Combined discrete nebulization and microextraction process for molybdenum determination by flame atomic absorption spectrometry (FAAS) |
|
Jenny A. Oviedo; Amanda M. D. de Jesus; Lucimar L. Fialho; Edenir R. Pereira-Filho*
Departamento de Química, Universidade Federal de São Carlos, CP 676, 13560-970 São Carlos - SP, Brasil Recebido em 15/05/2013 *e-mail: erpf@ufscar.br Simple and sensitive procedures for the extraction/preconcentration of molybdenum based on vortex-assisted solidified floating organic drop microextraction (VA-SFODME) and cloud point combined with flame absorption atomic spectrometry (FAAS) and discrete nebulization were developed. The influence of the discrete nebulization on the sensitivity of the molybdenum preconcentration processes was studied. An injection volume of 200 µL resulted in a lower relative standard deviation with both preconcentration procedures. Enrichment factors of 31 and 67 and limits of detection of 25 and 5 µg L-1 were obtained for cloud point and VA-SFODME, respectively. The developed procedures were applied to the determination of Mo in mineral water and multivitamin samples. INTRODUÇAO Molibdênio é um elemento encontrado em níveis traço em solos e apresenta um papel importante no metabolismo do nitrogênio nas plantas, sendo essencial para o crescimento de organismos biológicos, incluindo plantas e animais.1 Do ponto de vista fisiológico, a concentraçao de Mo no sangue humano é de 5 µg L-1. A deficiência desse elemento na dieta humana inibe o crescimento celular e aumenta a susceptibilidade à cárie, enquanto que um aumento da concentraçao no sangue está associado com o risco de gota e esclerose.2 Dentre os métodos mais comuns para determinaçoes de baixas concentraçoes de Mo tem-se a espectrofotometria na regiao do UV-VIS,3-6 espectrometria de absorçao atômica com chama (FAAS, Flame atomic absorption spectrometry),7,8 espectrometria de absorçao atômica com forno de grafite (GFAAS, Graphite furnace AAS),9-11 espectrometria de emissao óptica com plasma indutivamente acoplado (ICP OES, Inductively couple optical emission spectrometry),12 espectrometria de massa com plasma indutivamente acoplado (ICP-MS, ICP mass spectrometry)13 e métodos eletroquímicos.14-16 A espectrometria de absorçao atômica (AAS) é uma das técnicas mais utilizadas para a determinaçao de metais e alguns semi-metais. Dentre os diferentes atomizadores empregados, a utilizaçao da chama é frequente devido à sua simplicidade e baixo custo. Contudo, a determinaçao direta de Mo em amostras ambientais é dificultada devido à sua baixa concentraçao.17 Consequentemente, procedimentos de separaçao e pré-concentraçao geralmente sao necessários antes das medidas analíticas por FAAS para melhorar a sensibilidade do procedimento analítico. O método mais amplamente utilizado para separaçao e pré-concentraçao de quantidades traço de íons metálicos é a extraçao líquido-líquido (LLE, Liquid liquid extraction). As principais desvantagens no uso da LLE sao baixa frequência analítica e necessidade de grandes quantidades de solventes orgânicos, os quais muitas vezes apresentam custo elevado e/ou sao tóxicos. Visando superar essa limitaçao, têm sido desenvolvidos novos métodos de microextraçao e pré-concentraçao,18-20 como por exemplo, a microextraçao com gota orgânica solidificada (SFODME, Solidified floating organic drop microextraction)21-24 e o ponto nuvem.25 Esses métodos utilizam um volume pequeno de solvente orgânico, possuem alta eficiência de extraçao, elevados fatores de pré-concentraçao e baixos limites de detecçao, além de serem seguros, simples e rápidos, potencializando a determinaçao de analitos em baixas concentraçoes. A maioria das publicaçoes identificadas na literatura relacionadas à microextraçao tem como principal objetivo a determinaçao de analitos orgânicos, contudo, tem sido publicados vários trabalhos que mostram o uso de procedimentos de pré-concentraçao, na extraçao e separaçao de espécies inorgânicas. Além disso, percebe-se na literatura consultada que a maioria das aplicaçoes sao referentes a matrizes de baixa complexidade, tais como amostras de água.26-27 Uma alternativa que tem como objetivo melhorar a sensibilidade nas medidas por FAAS é a introduçao discreta de amostras.28-30 A aquisiçao de sinal analítico com uso de aspiraçao contínua é realizada em altura de pico, uma vez que a amostra é aspirada continuamente em direçao ao atomizador. Alternativamente, a introduçao discreta de amostras gera sinais transientes, o que requer a aquisiçao do sinal analítico através da área do pico. As principais vantagens da nebulizaçao discreta como uma alternativa de introduçao de amostra sao a maior sensibilidade analítica, que pode ser observada com a avaliaçao dos coeficientes angulares obtidos nas curvas analíticas de calibraçao, o menor volume de amostra necessário, diminuindo o tempo de limpeza do sistema de introduçao de amostra, evitando possíveis entupimentos do capilar do nebulizador do FAAS e reduçao dos depósitos de carbono no queimador.31 A importância de combinar os procedimentos de microextraçao com medidas analíticas por FAAS utilizando nebulizaçao discreta, é a melhora na eficiência de atomizaçao quando comparado com a introduçao contínua de amostra. A alta viscosidade dos produtos de microextraçao dificulta a aspiraçao e formaçao do aerossol na etapa da nebulizaçao contínua diminuindo a sensibilidade. O presente estudo tem como objetivo investigar as características de um procedimento que combina a introduçao discreta de amostras e a SFODME assistida por vórtex e a CPE (Cloud Point Extraction) para a determinaçao de Mo em água e em complexo multivitamínico por FAAS.
PARTE EXPERIMENTAL Instrumentaçao As medidas foram feitas em um espectrômetro de absorçao atômica com chama (Varian AA240FS, Mulgrave, Austrália) equipado com uma lâmpada de arco de deutério para a correçao da radiaçao de fundo. Como fonte de radiaçao, foi usada uma lâmpada de catodo oco de Mo. Os parâmetros instrumentais empregados foram: comprimento de onda de 313,3 nm, 7,0 mA de corrente elétrica aplicada na lâmpada de catodo oco e 0,5 nm de resoluçao espectral. A chama óxido nitroso-acetileno foi utilizada para a determinaçao de Mo com vazoes de 11,0 e 7,6 L min-1, respectivamente. Altura e área de pico foram utilizadas para monitorar os sinais analíticos para a nebulizaçao contínua ou com injeçao de volume discreto da amostra, respectivamente. Para a aspiraçao discreta por FAAS, uma ponteira de micropipeta com capacidade de 1000 µL foi diretamente conectada à tubulaçao do nebulizador pneumático através de um capilar de politetrafluoroetileno (PTFE) e fixada em um suporte universal. As microinjeçoes manuais de 200 µL foram realizadas com o auxílio de uma micropipeta de volume variável de 100 a 1000 µL (Eppendorf, Alemanha). Um pHmetro (PHS-3B, Phtek, China) foi utilizado para medir valores de pH. Na microextraçao com gota utilizou-se um agitador de tipo vórtex (Thermolyne type 37600 mixer, Dubuque, IA, EUA) e no procedimento de ponto nuvem foi utilizado um banho termostatizado (AquaWave 9374, Barnstead|Lab-Line, Alemanha). Em ambos os procedimentos uma centrifuga (Hermle/Labnet Z200A, Alemanha) foi necessária para os processos de extraçao e separaçao. O preparo das amostras de complexo multivitamínico foi feito utilizando um bloco digestor (Q-327M242, Quimis, Sao Paulo, SP, Brasil). Reagentes e materiais Todos os reagentes utilizados foram de grau analítico. As soluçoes foram preparadas com água ultrapura obtida de um sistema de purificaçao Milli-Qr (Millipak-40 Filter Unit 0,22 µm NPT, Bedford, MA, EUA) com resistividade de 18,2 MΩ cm. Os materiais de vidro e polipropileno foram previamente lavados e mantidos em banho de ácido nítrico 10% v v-1 por 24 h e lavados posteriormente com água ultrapura. As soluçoes de Mo foram preparadas por diluiçoes sucessivas com água ultrapura a partir de uma soluçao estoque de 1000 mg L-1 (Qhemis High Purity, Sao Paulo, Brasil). Para os testes de microextraçao com gota e ponto nuvem, usaram-se soluçoes de 8-hidroxiquinolina (8-HQ) (Vetec, Rio de Janeiro, RJ, Brasil) 0,5% m v-1 para complexaçao do Mo, soluçao tampao em pH 4,75 e 4,50 preparada a partir do acetato de sódio (Labsynth, Sao Paulo, SP, Brasil) e ácido acético glacial (Qhemis, Sao Paulo, Brasil) em concentraçoes e proporçoes apropriadas para ajuste do pH e etanol 99,5% (Tec Lab, Sao Paulo, SP, Brasil) para diluiçao da fase rica que continha o analito. Na microextraçao em gota foi utilizado ainda 1-undecanol (Sigma-Aldrich, Saint Louis, MO, EUA) como solvente orgânico e HCl 0,15 mol L-1 para dissoluçao da 8-HQ. No método do ponto nuvem foi utilizado como agente surfactante α-[p-(1,1,3,3-tetrametilbutil)fenil]-w- hidroxipoli(oxietileno) (Triton X-114) (Sigma-Aldrich) 5% m v-1. Procedimentos As condiçoes utilizadas para microextraçao com gota foram aqueles otimizados no trabalho de Oviedo et al.32 Em um tubo de polipropileno de 15 mL foi adicionado 120 µL de soluçao estoque de Mo contendo 100 mg L-1, 1 mL de tampao acetato pH 4,75 e 2,5 mL de 8-HQ 0,5% m/v foram misturados e o volume foi ajustado para 8 mL. Para a formaçao do complexo foram necessários 10 min. Em seguida, 60 µL de 1-undecanol foram adicionados. O tubo foi agitado com o uso do vórtex por 2 min para assegurar a completa extraçao. A mistura foi centrifugada por 2 min a 2000 rpm. Após esse processo o solvente orgânico ficou na superfície da soluçao. O tubo foi transferido para um banho de gelo e o solvente orgânico solidificou na parede do mesmo após 10 min, formando um anel na superfície. A soluçao aquosa (fase pobre) foi descartada e o solvente orgânico foi liquefeito no mesmo tubo quando mantido à temperatura ambiente. Finalmente, a fase que continha o analito foi diluída com 500 µL de etanol para posterior determinaçao por FAAS com aspiraçao contínua e com aspiraçao discreta injetando manualmente um volume de 200 µL. Para o procedimento de extraçao em ponto nuvem, foi realizado inicialmente um planejamento fatorial 24 com o intuito de otimizar as seguintes variáveis: tipo de agente complexante (PAN,1 - (2-piridilazo) - 2-naftol e 8-HQ, 8-Hidroquinolina), volume (0,5 e 1 mL) das soluçoes dos agentes complexantes, concentraçao (0,1 e 0,5% m v-1), e o pH (4,5 e 3,75). Posteriormente, foi realizado outro planejamento fatorial 24, no qual foram analisadas as variáveis: volume (0,5 e 1 mL), tempo de complexaçao (10 e 20 min), concentraçao (5 e 10% v v-1), e tipo do agente surfactante (Triton X-100 e Triton X-114). Após a identificaçao das melhores condiçoes, foi realizado um estudo sobre o tipo (etanol, metanol e uma mistura 1:1 de etanol HNO3) e o volume (200 e 400 µL) de solvente utilizado para diluiçao da fase rica. Posterior à otimizaçao, seguiu-se com o procedimento adicionando em tubos de polipropileno 10 mL da soluçao padrao de Mo 5 mg L-1 com 1 mL da soluçao de tampao acetato pH 4,50 e 0,5 mL da soluçao do complexante 8-HQ, após 10 min adicionou-se 0,5 mL do surfactante Triton X-114 com concentraçao de 5% m v-1 e os tubos foram levados ao banho termostatizado por 20 min sob uma temperatura de 45 ºC. Posteriormente, os tubos foram centrifugados a 3500 rpm por 10 min. Após a separaçao, a fase contendo predominantemente água (fase pobre) foi retirada com o auxílio de uma micropipeta e a fase rica em surfactante que continha o analito foi diluída com 200 µL de etanol. Preparo de amostra Para a aplicaçao dos procedimentos de microextraçao combinados com nebulizaçao discreta foi realizada uma digestao total de uma amostra de complexo multivitamínico adquirido no comércio local de Sao Carlos - SP. Antes da digestao, a amostra foi moída manualmente utilizando almofariz e pistilo de ágata até homogeneizaçao. Foi pesado 100 mg de amostra e transferidos para tubos do bloco digestor. Posteriormente, foram adicionados 3 mL de HNO3 concentrado e aquecido sob temperatura de 100 ºC por 1 h. Após o resfriamento dos tubos, foi adicionado 1 mL de H2O2 e a temperatura do bloco foi aumentada vagarosamente até atingir 180 ºC. Os digeridos foram retirados do bloco digestor quando o volume dos mesmos foi de aproximadamente 0,5 mL. Os digeridos foram transferidos para tubos de polipropileno com capacidade de 15 mL e o volume final ajustado com água deionizada até 10 mL para posterior extraçao e determinaçao de Mo. As amostras de água mineral nao passaram por nenhum pré-tratamento.
RESULTADOS E DISCUSSAO Extraçao por ponto nuvem Em todos os estudos para otimizaçao das condiçoes do procedimento de extraçao por ponto nuvem, a resposta monitorada foi o fator de enriquecimento (F.E.), o qual foi calculado pela relaçao das absorbâncias médias da soluçao padrao antes e após a extraçao. As Tabelas 1 e 2 mostram as respostas para os dois planejamentos fatoriais 24 realizados.
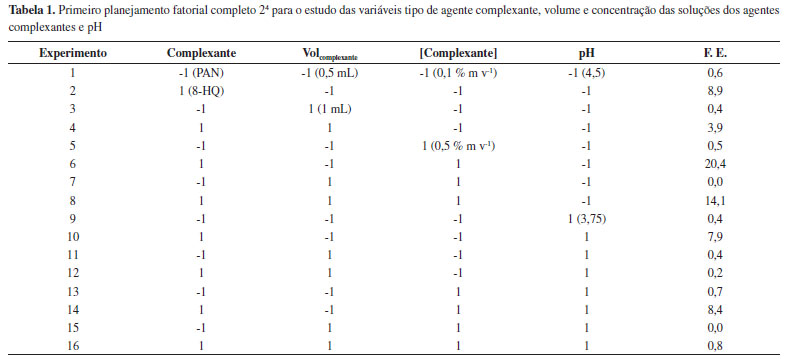
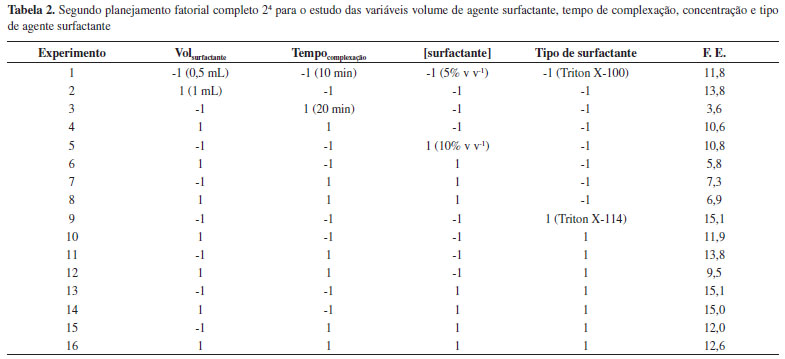
Avaliando os resultados obtidos no primeiro planejamento, podemos perceber que os maiores efeitos observados foram para o tipo de complexante (efeito = 8) e o pH (efeito = -4). Desta forma, a situaçao ideal seria utilizar a 8-HQ (nível 1) e o pH 4,5 (nível -1). No segundo planejamento as variáreis mais importantes foram o tipo de surfactante (efeito = 4) e tempo de complexaçao (efeito = -3). Novamente a melhor condiçao de trabalho é obtida ao utilizar o Triton X-114 (nível 1) e o tempo de 10 min (nível -1). As demais variáveis de ambos planejamentos nao apresentaram efeitos significativos nas faixas estudadas. A Tabela 3 apresenta as respostas para o estudo sobre o tipo e volume de solvente utilizado na etapa de diluiçao.
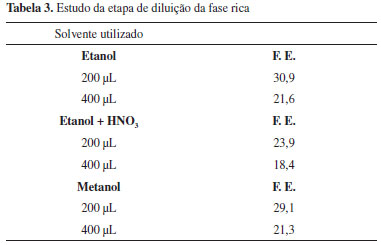
A melhor condiçao da diluiçao da fase rica foi quando se utilizou 200 µL de etanol. O estudo do efeito do volume sobre os sinais analíticos das soluçoes introduzidas por nebulizaçao discreta foi necessário devido à dificuldade de introduzir pequenos volumes no FAAS, pois a formaçao de microbolhas de ar na amostra pode alterar o perfil dos sinais transientes. A Tabela 4 apresenta as condiçoes finais otimizadas empregadas na pré-concentraçao por ponto nuvem para determinaçao de Mo.

O procedimento CPE foi aplicado em soluçoes contendo 5 mg L-1 de Mo e utilizou-se uma ponteira de micropipeta conectada diretamente ao tubo de aspiraçao do nebulizador. Volumes variáveis entre 50 a 300 µL dessa soluçao foram inseridos. Quando a introduçao da amostra foi feita de forma discreta, os sinais transientes gerados foram medidos em área de pico e quanto maior o volume de injeçao, maior o valor de absorbância integrada. A Figura 1 mostra os perfis dos sinais analíticos obtidos para Mo empregando diferentes volumes de injeçao.
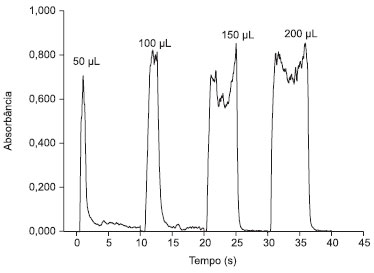 Figura 1. Perfis dos sinais analíticos obtidos para Mo (5 mg L-1) para diferentes volumes de injeçao com a extraçao por ponto nuvem
Uma desvantagem quando a introduçao de amostra foi feita de forma discreta é o maior desvio nas medidas devido principalmente às injeçoes manuais. Neste trabalho, a introduçao de 200 µL foi mais adequada considerando-se a boa repetibilidade e o baixo desvio padrao relativo. Os desvios padrao relativos variaram de 7,5 a 18% (n = 3). A melhora na sensibilidade do procedimento CPE combinado à nebulizaçao discreta foi comprovada estudando-se as curvas analíticas de calibraçao obtidas com soluçoes padrao inorgânicas (soluçoes aquosas convencionais) e orgânicas (fase rica diluída em etanol) para o Mo e introduzindo as soluçoes no FAAS de forma discreta e contínua. Na Tabela 5 estao apresentados os parâmetros de desempenho obtidos das curvas analíticas de calibraçao para concentraçoes de 0,3 a 4,0 mg L-1.
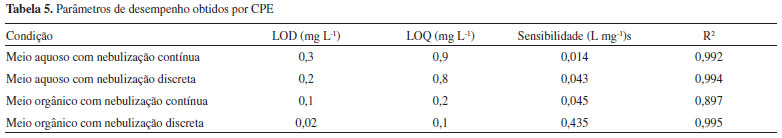
Os parâmetros de desempenho do procedimento desenvolvido foram avaliados por meio do cálculo dos limites de detecçao (LOD) e de quantificaçao (LOQ) definidos como: LOD = 3σ/s e LOQ = 10σ/s, onde s é o coeficiente angular (sensibilidade) das curvas analíticas, e σ é o desvio padrao de 10 medidas consecutivas do branco. Os menores limites de detecçao e quantificaçao, 0,02 e 0,1 mg/L respectivamente, foram obtidos com o procedimento CPE utilizando nebulizaçao discreta (injeçao de 200 µL da fase rica). A razao dos coeficientes angulares da curva analítica de calibraçao com soluçoes em meio orgânico com nebulizaçao discreta e da curva analítica de calibraçao em meio aquoso com nebulizaçao contínua forneceu o fator de enriquecimento de 31 vezes. Microextraçao com gota orgânica solidificada As microextraçoes com gota foram realizadas em soluçoes com concentraçao de 1,5 mg L-1 de Mo. Da mesma forma que para o procedimento de CPE, avaliou-se o efeito do volume de injeçao para a microextraçao com gota. Foram testados volumes entre 50 e 300 µL. Na Figura 2 observa-se um aumento gradual na absorbância integrada para volumes maiores injetados. Nesse caso, os desvios padrao relativos variaram de 1,6 a 20% (n = 3). Os volumes de 150 e 200 µL apresentaram melhor repetibilidade entre as medidas. O volume de amostra escolhido para a microinjeçao manual foi de 200 µL, que apresentou um sinal mensurável com boa repetibilidade (RSD = 1,6%) entre injeçoes e se mostrou adequado para a obtençao das curvas analíticas de calibraçao.
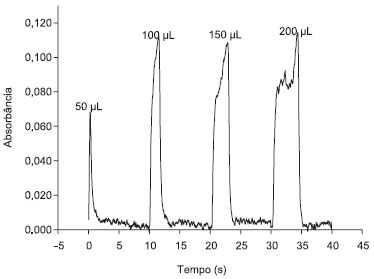 Figura 2. Perfis dos sinais analíticos obtidos para Mo para diferentes volumes de injeçao com a microextraçao com gota
Para estudar o aumento na sensibilidade após o método de microextraçao com gota na determinaçao de Mo por FAAS devida à nebulizaçao discreta, compararam-se os coeficientes angulares obtidos para as curvas analíticas de calibraçao para Mo em meio ácido (curva em meio aquoso) e após a microextraçao com gota, introduzindo a soluçao no nebulizador de forma discreta e contínua. Na Tabela 6 estao apresentadas os parâmetros de desempenho obtidos para as curvas analíticas de calibraçao em meio aquoso e em meio orgânico para soluçoes contendo de 0,3 a 4,0 mg L-1. Pode-se observar, quando comparados os coeficientes angulares, que há um aumento na sensibilidade quando a introduçao da soluçao é feita de forma discreta, sendo que esse aumento é mais significativo quando se faz a determinaçao após a microextraçao do Mo.
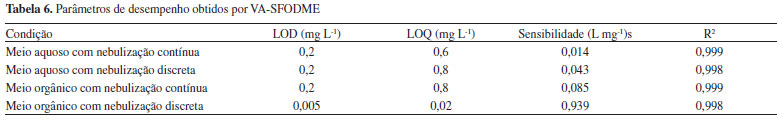
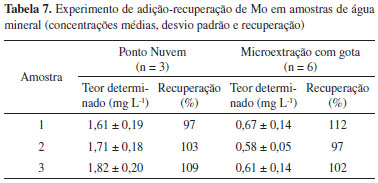
O LOD para a curva analítica obtida com soluçoes em meio aquoso com aspiraçao discreta é maior quando comparado com a aspiraçao contínua; a sensibilidade aumentou, porém o desvio do branco é maior devido às injeçoes manuais. Para as curvas de calibraçao com soluçoes em meio orgânico observou-se que quando se combinou a VA-SFODME com a introduçao discreta de amostra, obteve-se um ganho de sensibilidade de 11 vezes comparado com o obtido por nebulizaçao contínua, o que leva à obtençao de baixos LOD que viabilizariam a determinaçao de Mo em níveis traço. O fator de enriquecimento do procedimento desenvolvido combinando microextraçao e nebulizaçao discreta foi de 67. Neste ponto é importante mencionar que para a nebulizaçao contínua e discreta a forma de aquisiçao dos sinais é diferente. No primeiro caso as medidas sao efetuadas com o monitoramento da altura dos sinais analíticos e as medidas para nebulizaçao discreta foram realizadas levando em consideraçao a área dos sinais e, provavelmente a precisao com nebulizaçao continua deveria ser maior pelo fato de nao necessitar de injeçao manual. Entretanto, os parâmetros de desempenho medidos (Tabelas 5 e 6) tanto no procedimento de SFODME como no CPE foram bem melhores com a nebulizaçao discreta, mostrando que a variabilidade das medidas nessa forma de aquisiçao de sinais nao comprometeu a qualidade dos resultados. Determinaçao de Mo em amostras de água mineral e complexo multivitamínico Após aperfeiçoar o processo de injeçao discreta e adotando as melhores condiçoes de extraçao de Mo, foram escolhidas três amostras de água mineral e efetuados testes de adiçao-recuperaçao (Tabela 7). Uma concentraçao de 1,66 mg L-1 foi adicionada previamente à extraçao CPE e uma concentraçao de 0,60 mg L-1 de Mo previamente à VA-SFODME. As recuperaçoes obtidas variaram de 97 a 112 % evidenciando a exatidao dos procedimentos. As concentraçoes basais de Mo ficaram abaixo dos LOD para ambos os métodos propostos.
Molibdênio também foi determinado em amostra de complexo multivitamínico que, de acordo com o valor rotulado, continha 15 µg g-1 de Mo (Tabela 8). Para VA-SFODME e CPE, as porcentagens de recuperaçao ficaram em 94 e 98 %, respectivamente, mostrando a aplicabilidade dos procedimentos desenvolvidos para diferentes tipos de amostras e matrizes complexas.

CONCLUSAO A nebulizaçao discreta como uma alternativa de introduçao de amostra na chama mostrou-se eficiente e sensível para a determinaçao de Mo em diferentes tipos de amostras. Os parâmetros analíticos calculados nas Tabelas 5 e 6 mostraram uma diferença para os dois procedimentos de extraçao utilizados. Essa diferença pode ser devido ao fato de que a afinidade do complexo formado com o solvente orgânico (SFODME) é maior do que quando se faz o uso de surfactantes (CPE) ocorrendo assim à extraçao de maneira mais eficiente nos procedimentos de microextraçao (SFODME). Esses fatores sao o que levaram às diferentes características analíticas (entre elas o fator de enriquecimento) entre os procedimentos de CPE e SFODME. As vantagens mais importantes que podem ser observadas na combinaçao da nebulizaçao discreta com processos de microextraçao e pré-concentraçao sao a obtençao de menores LOD e a necessidade de menores quantidades de amostra para a determinaçao de Mo. Além disso, sao procedimentos simples com potencial para aplicaçoes em análises de rotina.
AGRADECIMENTOS A Coordenaçao de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) e ao Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) pelas bolsas concedidas e à Fundaçao de Amparo à Pesquisa do Estado de Sao Paulo (FAPESP) pelo apoio financeiro.
REFERENCIAS 1. Kaiser, B. N.; Gridley, K. L.; Brady, J. N.; Phillips, T.; Tyerman, S. D.; Annals of Botany 2005, 96, 745. 2. Ivanov, V. M.; Kochelaeva, G. A.; Prokhorova, G. V.; J. Anal. Chem. 2002, 57, 758. 3. Tunçeli, A.; Rehbe, T. A.; Microchim. Acta 2004, 144, 69. 4. Comitre, A. L.; Reis, B. F.; Anal. Chim. Acta 2003, 479, 185. 5. Madrakian, T.; Afkhami, A.; Siri, R.; Mohammadnejad, M.; Food Chem. 2011, 127, 769. 6. Andrade, J. C.; Bruns R. E.; Eiras, S. P.; Analyst 1993, 118, 213. 7. Oliveira, S. R.; Gomes, Neto J. A.; Nóbrega, J. A.; Jones, B. T.; Spectrochim. Acta, Part B 2010, 65, 316. 8. Canfranc, E.; Abarca, A.; Sierra, I.; Marina, M. L.; J. Pharm. Biomed. Anal. 2001, 25, 103. 9. Filik, H.; Çengel, T.; Reşat, A.; J. Hazard. Mater. 2009, 169, 766. 10. Shijo, Y.; Suzuki, M.; Shimizu, T.; Aratake, S.; Uehara, N.; Anal. Sci. 1996, 12, 953. 11. Matsusaki, K.; Nomi, M.; Higa, M.; Sata, T.; Anal. Sci. 1999, 15, 145. 12. Liang, P.; Liu, Y.; Guo, Li.; J. Anal. At. Spectrom. 2004, 19, 1006. 13. Bettallo, A. C.; Gervasio, A. P.; Giné, M. F.; J. Anal. At. Spectrom. 2005, 20, 535. 14. Sun, Y-C.; Mierzwa, J.; Lan, C-R.; Talanta 2000, 52, 417. 15. Andrade, J. C.; Almeida, A. M.; Aleixo, L. M.; Coscione, A. R.; Abreu, M. F.; Anal. Chim. Acta 2003, 487, 243. 16. Andrade, J. C.; Almeida, A. M.; Coscione, A. R.; Aleixo, L. M.; Analyst 2001, 126, 892. 17. Şendil, O.; Türker, A. R.; Somer, G.; J. Anal. Chem. 2008, 63, 734. 18. Dadfarnia, S.; Shabani, A. M.; Anal. Chim. Acta 2010, 658, 107. 19. Pereira, M.; Arruda, M. A.; Microchim. Acta 2003, 141, 115. 20. Raynie, D. E.; Anal. Chem. 2010, 82, 4911. 21. Zanjani, M. R. K.; Yamini, Y.; Shariati, S.; Jönsson, J.; Anal. Chim. Acta 2007, 585, 286. 22. Ma, J.; Zhang, J.; Du, X.; Lei, X.; Li, J.; Microchim. Acta 2010, 168, 153. 23. Şahin, Ç. A.; Tokgӧz, I.; Anal. Chim. Acta 2010, 667, 83. 24. Rivas, R. E.; López, I.; Hernández, M.; Anal. Methods 2010, 2, 225. 25. Bezerra, M. A.; Conceiçao, A. L. B.; Ferreira, S. L. C.; Microchim. Acta 2006, 154, 149. 26. Al-Saidi, H. M.; Emara, A. A. A.; J. Saudi Chem. Soc. 2011 Aceito para publicaçao. DOI: 10.1016/j.jscs.2011.11.005 27. Gharehbaghi, M.; Shemiran, F.; Food Chem. Toxicol. 2011, 49, 423. 28. Guisi, M.; Ribeiro, A. S.; Vieira, M. A.; Curtius, A. J.; Revista Analytica 2007, 28, 58. 29. Sarica, D. Y.; Akim, D.; Ӧzden, T.; Turk. J. Chem. 2002, 26, 263. 30. Kmetov, V.; Stefanova, V.; Hristozov, D.; Georgieva, D.; Canals, A.; Talanta 2003, 59, 123. 31. Amais, R. S.; Garcia, E. E.; Monteiro, M. R.; Nóbrega, J. A.; Fuel 2012, 93, 167. 32. Oviedo J. A.; Fialho L. L.; Nóbrega J. A.; Spectrochim. Acta, Part B. Online publication complete: 15-Mar-2013, DOI information: 10.1016/j.sab.2013.02.005 |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access