
 |
 |
 |
 |
 |
Revisão
|
|
| A química de coordenação e a produção de combustíveis solares Coordination chemistry and solar fuel production |
|
Sinval F. Sousa; Antonio Otavio T. Patrocinio*
Instituto de Química, Universidade Federal de Uberlândia, Av. João Naves de Ávila, 2121, Santa Mônica, 38400-902 Uberlândia - MG, Brasil Recebido em 18/09/2013 *e-mail: otaviopatrocinio@iqufu.ufu.br Life on earth depends on the absorption and conversion of solar energy into chemical bonds, i.e. photosynthesis. In this process, sun light is employed to oxidize water into oxygen and reducing equivalents used to produce fuels. In artificial photosynthesis, the goal is to develop relatively simple systems able to mimic photosynthetic organisms and promote solar-to-chemical conversion. The aim of the present review was to describe recent advances in the application of coordination compounds as catalysts in some key reactions for artificial photosynthesis, such as water splitting and CO2 reduction. INTRODUÇÃO Atualmente, o tópico energia tem despertado o interesse de diversos setores da sociedade devido ao dilema de suprir a crescente demanda sem aumentar os danos ambientais já causados pela exploração dos combustíveis fósseis.1-5 O incentivo a novas tecnologias para energia renovável ganhou ainda mais força com estudos recentes que mostram o aumento da concentração de CO2 na atmosfera devido à queima de combustíveis fósseis, o que causa o chamado efeito estufa.6,7 Nesse contexto, o aproveitamento da energia solar torna-se extremamente atraente devido ao seu baixo impacto ambiental e à grande oferta de energia. Anualmente, o fluxo de radiação sobre a terra chega a 3,4×1024 J, o que supera em milhares de vezes o consumo de energia atual do mundo.7 De fato, o sol é nossa fonte primária de energia, responsável pela formação da biomassa, dos ventos, da maré, enfim, da vida no planeta. O aproveitamento da energia solar envolve sua conversão em outras formas de energia. Atualmente, já se encontram amplamente difundidos nas residências sistemas para aquecimento de água a partir da luz solar. Outra possibilidade envolve a conversão da energia luminosa em eletricidade por meio de sistemas fotovoltaicos. Tais sistemas são baseados em células solares tradicionais de silício ou, mais recentemente, em novas gerações de dispositivos como as DSCs (dye-sensitized solar cells) e as OPVs (organic photovoltaics), cuja eficiência e estabilidade têm avançado constantemente.8-15 A energia solar pode ainda ser convertida e armazenada na forma de ligações químicas, como ocorre nos organismos fotossintéticos, em que espécies químicas de alto conteúdo energético (carboidratos) são formadas a partir de CO2 e H2O, equação 1.
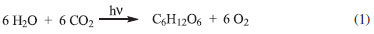
Biologicamente, a fotossíntese promove a quebra de água em oxigênio e prótons, enquanto a respiração combina essas espécies de forma controlada e eficiente para produzir metabólitos. Assim, a síntese desses metabólitos representa a fixação de hidrogênio e consequente armazenamento da energia solar na forma de ligações químicas. Estima-se que os organismos fotossintéticos produzem mais de 100 bilhões de toneladas de biomassa seca anualmente, o que equivale a 100 vezes o peso de toda a população humana na Terra e representa um estoque de energia de aproximadamente 100 TW.16 A produção de uma "folha artificial" capaz de gerar combustíveis a partir de substâncias abundantes e radiação solar, que possam ser armazenados e utilizados de acordo com a demanda em qualquer período do dia, vem de encontro com a busca da sociedade por fontes limpas de energia. A reprodução da fotossíntese fora do ambiente proteico dos organismos vivos constitui uma solução ideal para o problema energético e desafia químicos e cientistas de diversas áreas a desvendar "o segredo das plantas", como já colocado por Ciamician em seu artigo na Science em 1912.17 Dentre os diversos estudos sobre a conversão de energia solar, a investigação das propriedades (foto)químicas de complexos metálicos, em especial aqueles de configuração d6, tem atraído bastante interesse nas últimas décadas.18-33 As razões para o grande interesse nessa classe de compostos são várias, podendo-se citar:
Uma busca simples na base de dados Web of Science utilizando as expressões "Water Oxidation" ou "CO2 reduction" e "Metal Complex" retorna cerca de 410 entradas nos últimos 5 anos, o que mostra a grande atividade científica na área. Observa-se também um grande volume de investimentos na área com a formação de grandes redes de pesquisa ao redor do mundo.34-40 Nessa breve revisão, os avanços recentes e desafios na aplicação de complexos metálicos na conversão de energia solar em combustíveis serão discutidos e correlacionados com tópicos fundamentais da química de coordenação. Devido à vasta literatura na área e por limitações de espaço, esta revisão será focada em compostos de coordenação capazes de catalisar duas reações fundamentais para a produção de combustíveis solares: a oxidação da água e a redução do CO2. A fotossíntese natural como base para o desenvolvimento de catalisadores para oxidação da água Felizmente, diversos avanços recentes da ciência têm contribuído para desvendar o mecanismo fotossintético natural, seus componentes e principais etapas. Nas plantas e algas, a fotossíntese ocorre em dois sistemas conhecidos como fotossistemas I e II (PSI e PSII). No PS II, ocorre o chamado Ciclo de Kok,41 em que a energia solar é utilizada para oxidar a água a O2 e produzir prótons, que são transferidos por meio de cofatores ao PS I, responsável pela redução do CO2 (Ciclo de Calvin). A etapa crítica do processo é a foto-oxidação da água em oxigênio molecular, em que a energia dos fótons absorvidos pela clorofila e outros pigmentos é efetivamente convertida em equivalentes redox. A princípio, tal reação pode ser obtida a partir da luz na região do visível ou do infravermelho próximo, uma vez que a diferença entre os potenciais das semirreações de redução de H2 e oxidação da água é 1,23 V vs NHE , Esquema 1.42,43
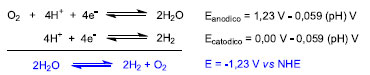 Esquema 1. Semirreações e respectivos potenciais eletroquímicos para a clivagem da água em H2 e O2
Termodinamicamente, a clivagem da água é um processo endergônico (ΔGº = 4,92 eV), envolvendo a transferência de quatro elétrons e quatro prótons, sendo que a reação deve ocorrer o mais próximo possível do potencial termodinâmico para maximizar a eficiência do processo. Dessa forma, a formação de intermediários radicalares, como o •OH, deve ser evitada, já que a oxidação subsequente exige agentes oxidantes fortes ou sobrepotenciais altos (Eº(•OH/H2O) = 2,31 V).44,45 Para evitar a formação de intermediários radicalares, é necessário um mecanismo no qual equivalentes redox sejam acumulados em um catalisador capaz de promover a formação da ligação O---O, liberar O2 e ser regenerado ao final do ciclo catalítico. Nos organismos fotossintéticos tal etapa ocorre no chamado Oxygen Evolving System (OEC), localizado no PS II. Um grande avanço no entendimento desse sistema foi dado com a determinação da estrutura cristalina do PS II da cianobactéria Thermosynechococus elongates por difratometria de raios-X de alta resolução.46,47 Tal estudo revelou a composição e a organização em nível atômico do OEC, bem como do ambiente proteico ao seu redor. Com base nesses dados, diversos grupos têm proposto mecanismos para a foto-oxidação da água e a formação da ligação O=O.46,48-59 Quimicamente, o OEC é um cluster contendo quatro íons manganês e um íon cálcio ligados por pontes oxo. Na estrutura proposta por Ferreira et al.,46 três íons manganês e o íon cálcio formam um "cubo" de fórmula molecular Mn3CaO4, sendo que o quarto íon manganês está ligado ao oxigênio adjacente ao íon Ca2+. O cluster metálico é rodeado por uma série de resíduos de aminoácidos que atuam como ligantes para os centros metálicos ou promovem ligações de hidrogênio, que atuam na desprotonação das moléculas de água. A ativação do ciclo catalítico se dá pela foto-oxidação de uma tirosina, Yz, pela clorofila P680•+, Figura 1. O cátion radical Yz•+ oxida o OEC (S0) gerando o primeiro de uma série de pelo menos quatro intermediários (S1 a S4) à medida que fótons são absorvidos. O estado S1 tem os íons manganês com estado de oxidação III, III, IV, IV60,61 e é termodinamicamente estável no escuro.
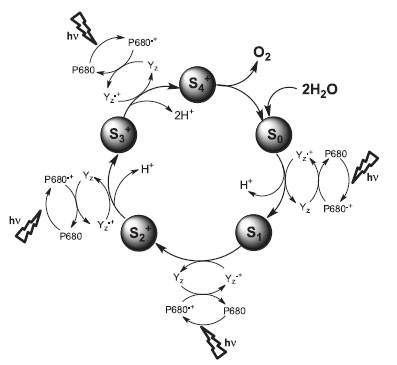 Figura 1. Ciclos de estados "S" do fotossistema II que resulta na oxidação da H2O em O2
O mecanismo de acumulação de equivalentes redox por meio da oxidação sucessiva do cluster Mn4Ca envolve etapas em que a transferência de elétrons está acoplada com a transferência de prótons. Esse processo evita a formação de intermediários instáveis e permite o acesso a espécies com estados de oxidação altos, que são efetivamente os catalisadores da oxidação da água, fixação de nitrogênio e outras reações biológicas importantes.62-64 O Esquema 2 apresenta os diferentes mecanismos para a transferência de elétron acoplada com prótons (PCET). No mecanismo ET-PT (electron transfer-proton transfer), a transferência de elétrons ocorre primeiro, o que provoca um aumento na constante de acidez do próton ligado ao centro doador (D) e consequente desprotonação. Já no mecanismo EPT (electron proton transfer), a transferência de prótons e elétrons ocorre de forma simultânea. Para tal, é necessária a associação entre doador e aceptor (A) por meio de ligações de hidrogênio. Há ainda um terceiro mecanismo em que elétrons são transferidos para o aceptor e, concomitantemente, prótons são transferidos para uma base de Brönsted (B) presente no meio, MS-EPT (multiple-site electron proton transfer). Maiores detalhes sobre os mecanismos de PCET, bem como exemplos envolvendo complexos metálicos e reações de interesse biológico podem ser encontrados em compilações feitas por Meyer e colaboradores,64,65 bem como em outros trabalhos na literatura.66-69,33
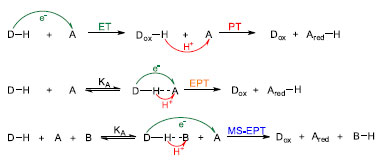 Esquema 2. Diferentes mecanismos para a transferência de elétron acoplada com prótons
O entendimento do mecanismo de oxidação da água no OEC tem permitido avanços consideráveis no desenvolvimento de catalisadores artificiais. Recentemente, Nocera e colaboradores descreveram um catalisador automontado composto por íons cobalto e fosfato com estrutura semelhante ao OEC natural e também capaz de gerar O2 a partir da água em pH neutro.42 Outros grupos de pesquisa ao redor do mundo têm descrito uma série de catalisadores baseados em complexos de Ru(II), Ir(III) e Mn(II) que serão brevemente discutidos a seguir. Catalisadores homogêneos para oxidação da água O primeiro catalisador homogêneo para oxidação da água foi descrito por Meyer e colaboradores em 1982. Trata-se do complexo cis,cis-[(bpy)2(H2O)RuIIIORuIII(OH2)(bpy)2]4+, bpy = 1,4-bipiridina, conhecido como blue dimer,70 Figura 2.
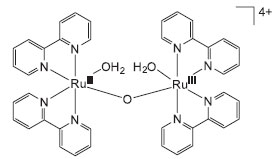 Figura 2. Estrutura do "blue dimer" - [(bpy)2(H2O)RuIIIORuIII(OH2)(bpy)2]4+
O mecanismo de atuação do blue dimer foi extensamente estudado71-74 por meio de técnicas eletroquímicas ou pela adição de agentes oxidantes fortes, como o Ce(IV), E0(CeIV/III) = 1,7 V a pH = 1,0. Da mesma forma que no OEC, os múltiplos equivalentes redox são formados a partir de transferências de elétrons acopladas com prótons, o que evita a formação de intermediários de alta energia. De fato, a química redox de aquo complexos de Ru(II) é fortemente dependente do pH, como pode ser visualizado em diagramas de Pourbaix dessas espécies.75-78 Para o blue dimer, a oxidação dos centros metálicos à Ru(III)/Ru(IV) leva a um aumento da acidez dos prótons dos ligantes aquos, com o primeiro pKa decrescendo de 6 para aproximadamente 0.72 Assim, as espécies com alto estado de oxidação, como [(bpy)2(O)RuVORuIV(O)(bpy)2]3+ e [(bpy)2(O)RuVORuV(O)(bpy)2]4+ existem como dioxo complexos e são termodinamicamente capazes de oxidar a água em uma ampla faixa de pH. Uma proposta de mecanismo para oxidação da água pelo blue dimer é apresentada no Esquema 3. Dados cinéticos de cada uma das etapas, a detecção espectroscópica dos intermediários, bem como dados sobre estudos de marcação isotópica podem ser encontrados no trabalho revisão de Liu e colaboradores.72
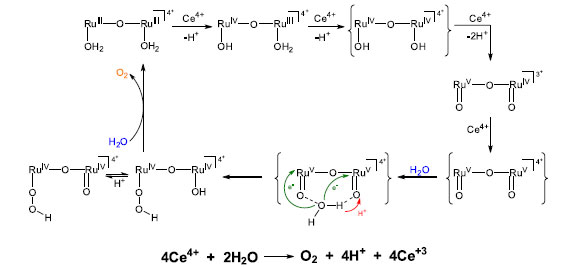 Esquema 3. Proposta de mecanismo de oxidação da água catalisada pelo "blue dimer". Adaptado das ref. 72 e 76
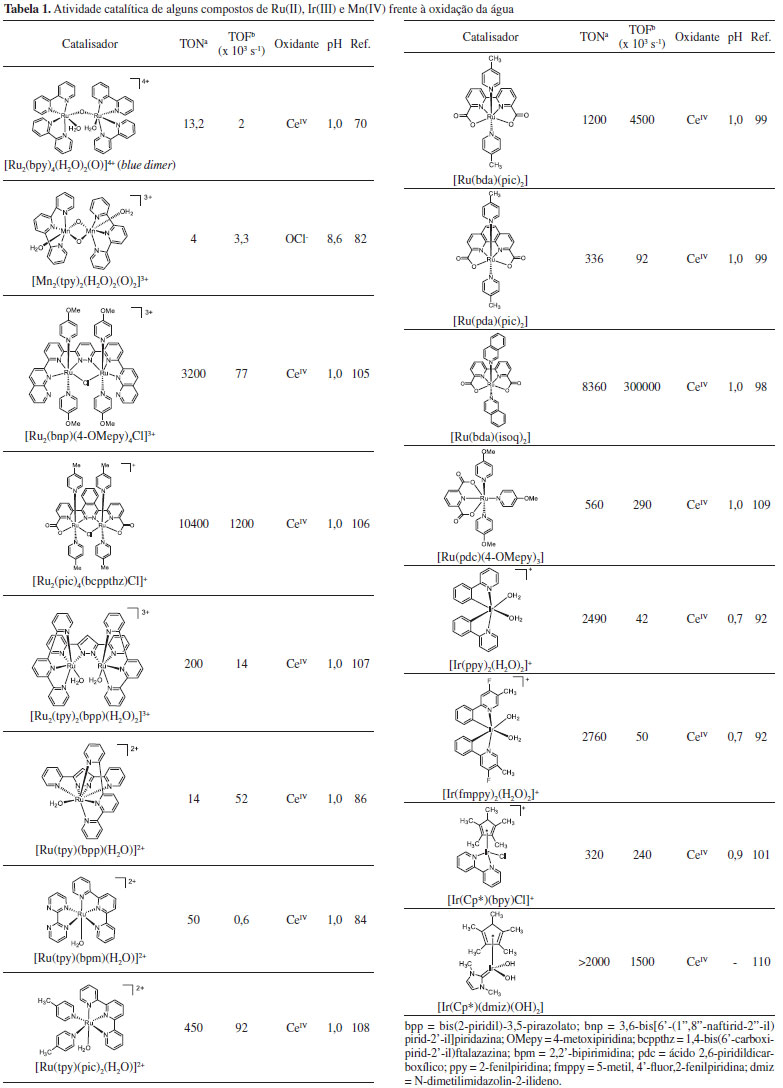
No Esquema 3, [(bpy)2(O)RuVORuV(O)(bpy)2]4+ é a espécie cataliticamente ativa e interage com a água por meio de ligações de hidrogênio com subsequente transferência de 2e-/1H+ por meio do mecanismo MS-EPT. Nesta etapa, a ligação O---O é formada. O ataque nucleofílico de uma segunda molécula de água ao centro metálico ligado ao grupo hidroperóxido leva à liberação de O2 e à regeneração do catalisador. A partir do blue dimer, outros aquocomplexos binucleares foram descritos como catalisadores frente à oxidação da água.75,79 Brudvig e colaboradores descreveram a evolução de O2 pelo composto [(tpy)(OH2)MnIV(O)2MnIV(OH2)(tpy)]3+, tpy = 2,2':6'2"-terpiridina, em que o grupo MnV= O é fundamental para a formação da ligação O---O.80-83 Uma nova geração de catalisadores envolve complexos mononucleares de Ru(II),71,84-88 Mn(II),89,48,90 ou Ir(III).91-94 A eficiência dos catalisadores é geralmente avaliada por três parâmetros, a saber: sobrepotencial para oxidação da água, Turn Over Number (TON), que expressa o número de mols de O2 produzido por mol de catalisador e o Turn Over Frequency (TOF), que expressa o número de ciclos catalíticos por unidade de tempo. Um bom catalisador apresenta baixo sobrepotencial, é estável (alto TON) e tem alta atividade catalítica (alto TOF). Concepcion e colaboradores descreveram uma série de aquocomplexos de fórmula geral [RuII(LLL)(LL)(OH2)]2+, (LLL = ligante tridentado; LL = ligante bidentado) que apresentaram atividade catalítica frente à oxidação da água em diferentes condições de pH.84 O mecanismo de reação envolve uma série de transferências de prótons e elétrons para a geração de espécies como [RuIV(LLL)(LL)(O)2]2+ ou [RuV(LLL)(LL)(O)2]3+, termodinamicamente capazes de oxidar a água. Como esperado, os potenciais eletroquímicos e a estabilidade das espécies com alto estado de oxidação são fortemente influenciados pela capacidade σ doadora e π receptora dos ligantes.95 Kohl et al. descreveram a produção estequiométrica de H2 e O2 a partir da água por meio de etapas térmica e fotoquímica mediadas pelo complexo [(PNN)RuIIH(OH)(CO)], PNN = 2-(di-tert-butilfosfino-metil)-6-dietilaminometil)piridina.96 Sun et al. avaliaram a atividade catalítica de complexos de Ru(II) contendo ligantes polipiridínicos carboxilados e a 4-metilpiridina (pic) como ligante ancilar.97, 98 Observou-se que o mecanismo de oxidação da água é influenciado pela rigidez do ligante polipiridínico. Para complexos com o ligante ácido 2,2'-bipiridina-6,6'-dicarboxílico (bda), [Ru(bda)(pic)2], sugere-se um mecanismo bimolecular para evolução de O2, enquanto que para o complexo [Ru(pda)(pic)2], pda = ácido 1,10-fenantrolina-2,9-dicarboxílico, o mecanismo é unimolecular, resultando em melhor estabilidade.99 A diferença no mecanismo de oxidação dos dois complexos é atribuída à maior rigidez do ligante pda. O complexo com o ligante bda apresentou melhor TON, mas perdeu sua atividade catalítica em cerca de cinco minutos. Já o complexo com o ligante pda foi estável por até seis horas nas mesmas condições experimentais. Posteriormente, o mesmo grupo de pesquisa propôs a substituição do ligante ancilar 4-metilpiridina pela isoquinolina (isoq).98 A atividade catalítica do complexo [Ru(bda)(isoq)2] é comparável à do OEC das plantas, com TOF maior que 300 s-1 e, adicionalmente, o complexo mostrou-se bastante estável nas condições reacionais (TON = 8360). O aumento da atividade catalítica devido à introdução do ligante isoquinolina é atribuído à formação de interações atrativas não covalentes entre anéis aromáticos de isoquinolinas ligadas a diferentes centros metálicos. Essas interações diminuem a energia de ativação para o acoplamento entre duas espécies, favorecendo o mecanismo binuclear para evolução de O2. Bernhard et al. descreveram a síntese e atividade catalítica de aquocomplexos de Ir(III) ciclometalados.92 Os compostos se mostraram robustos frente à oxidação da água, contudo a evolução de O2 observada foi muito lenta, cerca de 100 milhões de vezes mais lenta que no OEC. Brudvig e Crabtree descreveram o uso de ligantes com maior caráter doador, como o pentametilciclopentadienil (Cp*), em complexos de Ir(III) tetracoordenados, o que levou a um aumento da atividade catalítica.100,101 Outros complexos de Ir(III) têm mostrado excelente resultados.26,102-104 Na Tabela 1 são listados alguns resultados sobre a atividade catalítica de compostos de coordenação de Ru(II), Mn(II) e Ir(III) frente à oxidação da água.
Os estudos de catalisadores para oxidação da água a partir de agentes oxidantes como o CeIV têm permitido grandes avanços no entendimento do mecanismo da produção de O2 e um maior controle sobre reações secundárias que desativam o catalisador. Contudo, para a realização da fotossíntese artificial, é preciso substituir os agentes oxidantes por fótons, que ao serem absorvidos, iniciarão uma série de transferências de elétrons para produzir os equivalentes redox necessários para a oxidação da água. Adicionalmente, é indispensável que seja produzido um dispositivo como uma célula fotoeletroquímica, em que o catalisador esteja imobilizado, de forma a permitir a aplicação em maior escala. As principais tentativas para produção de células fotoeletroquímicas para fotossíntese artificial têm se baseado na sensibilização de semicondutores de grande largura de banda,43,111-119 estratégia empregada com sucesso em dispositivos fotovoltaicos como as células solares sensibilizadas por corantes (DSCs).9,120-122 Esses dispositivos são conhecidos como DSPECs (do inglês, Dye-sensitized photoelectrosynthesis cells), Figura 3. Nas DSPECs, um catalisador para a oxidação da água, Catox, está ligado a um cromóforo, C, que por sua vez, está adsorvido na superfície de um filme de semicondutor com separação grande de bandas, como o TiO2. Ao absorver a luz, o cromóforo injeta um elétron na banda de condução do óxido e é rapidamente reduzido pelo Catox via transferência eletrônica acoplada com prótons. Dessa forma, o cromóforo pode absorver um segundo fóton e o ciclo se repetirá até que o Catox alcance o estado de oxidação cataliticamente ativo. No contraeletrodo, os elétrons gerados pela oxidação da H2O são utilizados para reduzir prótons gerando hidrogênio com o auxílio de um catalisador, Catred.
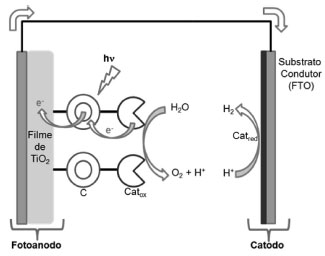 Figura 3. Esquema de uma DSPEC para foto-oxidação da água
A dinâmica de transferência eletrônica em sistemas TiO2-C-Catox tem sido extensamente estudada por Meyer e colaboradores por meio de técnicas fotoeletroquímicas e por absorção resolvida no tempo.113,117,119,123-125 Os estudos mostram que a acumulação de equivalentes redox no catalisador de oxidação depende dos potenciais de oxidação do cromóforo e do catalisador, que por sua vez, podem ser controlados pelos ligantes empregados. Adicionalmente, a cinética dos processos de transferência eletrônica e recombinação é fortemente influenciada pelo pH e pela composição do eletrólito utilizado. Youngblood e colaboradores descreveram uma DSPEC com um fotoanodo constituído por um filme de TiO2 sensibilizado por um complexo de Ru(II) com o ligante 4-metil-4'-dimetoximalonil-2,2'-bipiridina.126 O grupo dimetoximalonil é capaz de se ligar a nanopartículas de IrO2×nH2O, que atuam como catalisador de oxidação. A irradiação do fotoanodo leva à fotoexcitação do complexo de Ru(II) e, consequente injeção de elétrons na banda de condução do TiO2. O cromóforo foto-oxidado é rapidamente reduzido pelas nanopartículas de IrO2×nH2O (k = 103 s-1). A acumulação de equivalentes redox nas nanopartículas resulta na oxidação da água e evolução de O2. O rendimento quântico da célula fotoeletroquímica foi de aproximadamente 0,9% (λirr = 450 nm). Li e colaboradores utilizaram uma abordagem diferente para promover a interação entre cromóforo e catalisador nas DSPECs.112 Sobre o filme de TiO2 sensibilizado por um complexo de Ru(II), foi depositado uma membrana de Nafion, na qual o catalisador [Ru(bda)(pic)2] foi imobilizado. A membrana contém um número considerável de grupos sulfônicos, o que garante boa condutibilidade elétrica dependendo do pH. Os autores observaram a ocorrência de transferência eletrônica entre o cromóforo foto-oxidado e o catalisador. Observou-se também a produção de 140 nmol de O2 após uma hora de irradiação com luz na região do visível. Estratégia semelhante foi empregada por Brimblecombe e colaboradores, empregando como catalisador o cluster de manganês [Mn4O4L6]+, L = bis(metoxifenil)fosfinato.111 Os rendimentos e a estabilidade das DSPECs descritas até o momento ainda estão abaixo do esperado para se almejar uma aplicação em maior escala. Observa-se, porém, um avanço contínuo do entendimento sobre a dinâmica dos processos envolvidos e o desenvolvimento de catalisadores cada vez mais robustos. Desta forma, espera-se que as DSPECs possam se tornar, em um futuro próximo, uma opção para geração de combustíveis de forma limpa e renovável. Catalisadores homogêneos para redução do CO2 Outra reação de grande interesse na fotossíntese artificial é a redução do CO2.127,128 Por meio deste processo é possível produzir combustíveis como o metano, e também contribuir para o sequestro de CO2 da atmosfera, diminuindo assim o efeito estufa e o aquecimento global. Do ponto de vista termodinâmico, a redução do CO2 por um único elétron para gerar CO2•- é bastante desfavorável (E0 = -1,90 vs NHE). Desta forma, o processo precisa ser promovido por meio de transferências de elétrons múltiplas assistidas por prótons, EPT ou MS-EPT. As reações 2 e 3 ilustram, respectivamente, a produção de CO e CH4 por meio da redução do CO2.
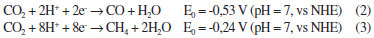
A utilização de compostos de coordenação para fotorredução de CO2 é bastante investigada pela alta atividade catalítica dos compostos e a seletividade dos produtos obtidos.127 Os principais sistemas descritos na literatura utilizam um cromóforo (C), um agente redutor de sacrifício (D) e um catalisador (Cat), Esquema 4A. Sobre irradiação, o cromóforo fotoexcitado é reduzido pelo agente de sacrifício. O catalisador reage com o cromóforo reduzido para gerar a espécie cataliticamente ativa, Cat-. Alternativamente, o cromóforo e o catalisador podem estar covalentemente ligados para formar sistemas supramoleculares, Esquema 4B.129
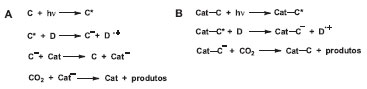 Esquema 4. Reações químicas envolvidas na fotorredução do CO2 em sistemas envolvendo um cromóforo (C), um catalisador (Cat) e um agente redutor de sacrifício (D)
Tipicamente, os cromóforos mais utilizados são compostos polipiridínicos de Ru(II), caracterizados por bandas intensas de transferência de carga do metal para o ligante (MLCT) na região do visível e estados excitados com tempos de vida relativamente longos.130 Como agentes redutores de sacrifício são utilizadas aminas terciárias (trietilamina, TEA ou trietanolamina, TEOA). Os catalisadores mais investigados envolvem compostos macrocíclicos de Ni(II) e Co(II),131,132 complexos carbonílicos de Ru(II),133,134 enzimas135 e complexos polipiridínicos de Re(I),129,136-142 sendo que estes últimos apresentam alto rendimento e podem atuar simultaneamente como cromóforos e catalisadores. Hawecker e colaboradores foram os primeiros a descrever complexos polipiridínicos de Re(I), fac-[ReI(CO)3(NN)(X)], NN = ligante polipiridínico, X = haletos, como fotocatalisadores para redução do CO2.143 A excitação da banda MLCT do complexo leva à formação da espécie fac-[ReII(CO)3(NN•-)(X)], que sofre supressão redutiva na presença de aminas terciárias para gerar a espécie fac-[ReI(CO)3(NN•-)(X)]-. Esta espécie é instável e o ligante haleto tende a ser substituído por uma molécula de solvente (S) para gerar a espécie cataliticamente ativa fac-[ReI(CO)3(NN•-)(S)]. Na presença de CO2, ocorre a produção de formiato (HCO2-) e/ou CO com consequente regeneração do catalisador. O mecanismo de formação do formiato é relativamente bem entendido e envolve a reação da espécie fac-[ReI(CO)3(NN•-)(S)] com prótons para formar ligações ReI-H (equação 4). O intermediário fac-[ReI(CO)3(NN)(H)] reage com o CO2 para formar o ânion formiato coordenado ao centro metálico (equação 5). Já o mecanismo de fotogeração de CO com catalisadores baseados em complexos de Re(I) ainda é tema de extenso debate e investigação. É sugerido que a geração de CO deve envolver um mecanismo bimolecular e a formação de um dímero de ReI contendo um CO2 como ligante ponte.127,144
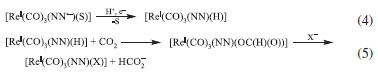
Ishitani e colaboradores estudaram a fotorreatividade de uma série de complexos de Re(I) na presença de CO2.140 Os autores observaram que a mistura dos complexos [ReI(CO)3(bpy)(CH3CN)]+ e [ReI(CO)3((MeO)2bpy)(P(OEt)3)]+, (MeO)2bpy = 4,4'-dimetoxi-2,2'-bipiridina e P(OEt)3 = trietilfosfito, na proporção 1:25 (mol/mol) levou a fotoprodução de CO com rendimento quântico de 0,59. O alto rendimento está associado à facilidade de substituição do ligante CH3CN no complexo [ReI(CO)3(bpy)(CH3CN)]+ e à alta eficiência de formação do intermediário [ReI(CO)3((MeO)2bpy•-)(P(OEt)3)]+ (Φ = 1,6). A Tabela 2 reúne alguns resultados para a fotorredução do CO2 empregando complexos de Re(I) e Ru(II).
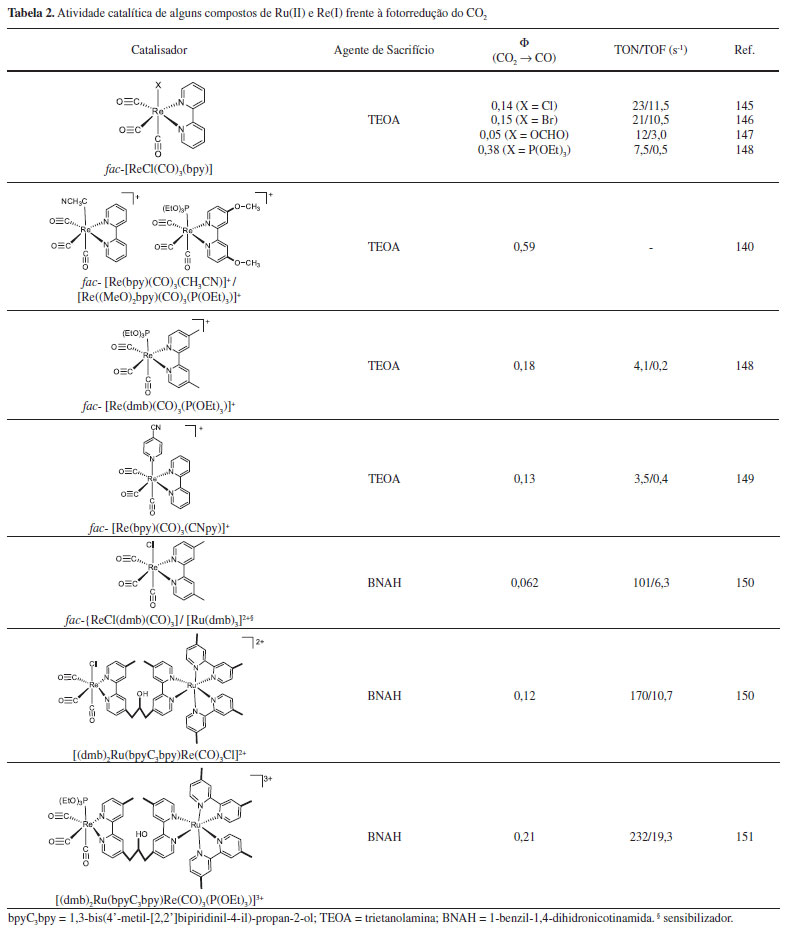
Os resultados listados na Tabela 2 mostram que os rendimentos quânticos para a geração de CO obtidos são relativamente altos, comprovando a eficiência dessa classe de compostos como fotocatalisadores para redução do CO2. Contudo, os valores de TON e TOF ainda são consideravelmente baixos, o que mostra a necessidade de mais estudos para o desenvolvimento de sistemas mais estáveis e rápidos.
CONCLUSÕES E PERSPECTIVAS A produção de combustíveis a partir da luz solar (fotossíntese artificial) é um tema multidisciplinar, desafiante e diretamente relacionado à busca da sociedade por fontes limpas de energia. Os compostos de coordenação se apresentam como catalisadores eficientes de reações fundamentais para a fotossíntese artificial, como a foto-oxidação da água e a fotorredução do CO2. O desenvolvimento de novas metodologias sintéticas permitiu o ajuste das propriedades fotoeletroquímicas dos compostos em função dos ligantes empregados e um avanço considerável no rendimento dos processos catalíticos foi alcançado. Paralelamente, o maior entendimento do mecanismo das reações a nível molecular tem permitido racionalizar diversos aspectos relacionados à estabilidade dos catalisadores. Alguns desafios que ainda precisarão ser superpostos envolvem o aumento da velocidade e da estabilidade dos catalisadores, o que está diretamente ligado ao melhor entendimento dos mecanismos de reação em diferentes condições de pH e da influência da composição do eletrólito na reatividade dos compostos. É necessário ainda o acoplamento eficiente destes catalisadores a dispositivos fotoeletroquímicos que possam ser reproduzidos em maior escala. Tais sistemas deverão ser ativos na região do infravermelho próximo, de forma a maximizar o aproveitamento da luz solar. Outros desafios envolvem o uso de metais mais abundantes como cobalto e cobre, e o desenvolvimento de novos catalisadores para produção de H2, de forma a se evitar o uso de metais nobres como a platina. Para superpor estes desafios, cientistas de diferentes áreas têm estabelecido grandes redes multidisciplinares, capazes de desenvolver trabalhos coordenados nos diferentes aspectos que envolvem o tema. Dado o grande investimento humano e financeiro, espera-se para um futuro próximo, uma nova geração de células fotoeletroquímicas capazes de converter eficientemente a luz solar em combustíveis limpos, o que muito contribuirá para a geração de energia limpa e sustentável.
AGRADECIMENTOS Os autores agradecem o apoio da Fundação de Amparo à Pesquisa de Minas Gerais (FAPEMIG), do Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), da Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) e da Rede Mineira de Química (RQ-MG).
REFERÊNCIAS 1. Lewis, N. S.; Chem. Eng. News 2001, 79, 278. 2. Serrano, E.; Rus, G.; Garcia-Martinez, J.; Renewable Sustainable Energy Rev. 2009, 13, 2373. 3. Meyer, T. J.; Nat. Chem. 2011, 3, 757. 4. Zhou, H.; Fan, T.; Zhang, D.; ChemCatChem 2011, 3, 513. 5. Styring, S.; Faraday Discuss. 2012, 155, 357. 6. Friedlingstein, P.; Nature 2008, 451, 297. 7. Vichi, F. M.; Mansor, M. T. C.; Quim. Nova 2009, 32, 757. 8. Garcia, C. G.; Iha, N. Y. M.; Argazzi, R.; Bignozzi, C. A.; J. Braz. Chem. Soc. 1998, 9, 13. 9. Grätzel, M.; Nature 2001, 414, 338. 10. De Paoli, M. A.; Gazotti, W. A.; J. Braz. Chem. Soc. 2002, 13, 410. 11. Longo, C.; De Paoli, M. A.; J. Braz. Chem. Soc. 2003, 14, 889. 12. Kroon, J. M.; Bakker, N. J.; Smit, H. J. P.; Liska, P.; Thampi, K. R.; Wang, P.; Zakeeruddin, S. M.; Grätzel, M.; Hinsch, A.; Hore, S.; Wurfel, U.; Sastrawan, R.; Durrant, J. R.; Palomares, E.; Pettersson, H.; Gruszecki, T.; Walter, J.; Skupien, K.; Tulloch, G. E.; Prog. Photovoltaics Res. Appl. 2007, 15, 1. 13. Yella, A.; Lee, H.-W.; Tsao, H. N.; Yi, C.; Chandiran, A. K.; Nazeeruddin, M. K.; Diau, E. W.-G.; Yeh, C.-Y.; Zakeeruddin, S. M.; Grätzel, M.; Science 2011, 334, 629. 14. Lin, Y.; Li, Y.; Zhan, X.; Chem. Soc. Rev. 2012, 41, 4245. 15. Burschka, J.; Pellet, N.; Moon, S. J.; Humphry-Baker, R.; Gao, P.; Nazeeruddin, M. K.; Gratzel, M.; Nature 2013, 499, 316. 16. Barber, J.; Chem. Soc. Rev. 2009, 38, 185. 17. Ciamician, G.; Science 1912, 36, 385. 18. Scandola, F.; Bignozzi, C. A.; Balzani, V.; Quim. Nova 1997, 20, 423. 19. Toma, H. E.; J. Braz. Chem. Soc. 2003, 14, 845. 20. Polo, A. S.; Itokazu, M. K.; Murakami Iha, N. Y.; Coord. Chem. Rev. 2004, 248, 1343. 21. Polo, A. S.; Itokazu, M. K.; Frin, K. M.; Patrocinio, A. O. T.; Murakami Iha, N. Y.; Coord. Chem. Rev. 2006, 250, 1669. 22. Carlos, R. M.; Quim. Nova 2007, 30, 1686. 23. Elliott, P. I. P.; Annu. Rep. Prog. Chem. Sect. A: Inorg. Chem. 2011, 107, 399. 24. Moore, G. F.; Blakemore, J. D.; Milot, R. L.; Hull, J. F.; Song, H.-e.; Cai, L.; Schmuttenmaer, C. A.; Crabtree, R. H.; Brudvig, G. W.; Energy Environ. Sci. 2011, 4, 2389. 25. Winter, A.; Newkome, G. R.; Schubert, U. S.; ChemCatChem 2011, 3, 1384. 26. Blakemore, J. D.; Schley, N. D.; Kushner-Lenhoff, M. N.; Winter, A. M.; D'Souza, F.; Crabtree, R. H.; Brudvig, G. W.; Inorg. Chem. 2012, 51, 7749. 27. Happ, B.; Winter, A.; Hager, M. D.; Schubert, U. S.; Chem. Soc. Rev. 2012, 41, 2222. 28. Hintermair, U.; Hashmi, S. M.; Elimelech, M.; Crabtree, R. H.; J. Am. Chem. Soc. 2012, 134, 9785. 29. Young, K. J.; Martini, L. A.; Milot, R. L.; Snoeberger, R. C.; Batista, V. S.; Schmuttenmaer, C. A.; Crabtree, R. H.; Brudvig, G. W.; Coord. Chem. Rev. 2012, 256, 2503. 30. Blakemore, J. D.; Mara, M. W.; Kushner-Lenhoff, M. N.; Schley, N. D.; Konezny, S. J.; Rivalta, I.; Negre, C. F. A.; Snoeberger, R. C.; Kokhan, O.; Huang, J.; Stickrath, A.; Lan Anh, T.; Parr, M. L.; Chen, L. X.; Tiede, D. M.; Batista, V. S.; Crabtree, R. H.; Brudvig, G. W.; Inorg. Chem. 2013, 52, 1860. 31. Crabtree, R. H.; Dalton Trans. 2013, 42, 4104. 32. Luca, O. R.; Crabtree, R. H.; Chem. Soc. Rev. 2013, 42, 1440. 33. Wenger, O. S.; Acc. Chem. Res. 2013, 46, 1517. 34. http://www.esf.org/coordinating-research/eurocores/running-programmes/eurosolarfuels.html, acessada em Setembro 2013. 35. http://www.perspect-h2o.eu/, acessada em Setembro 2013. 36. http://www.hydrosol3d.org/, acessada em Setembro 2013. 37. http://www.solarfuelshub.org/, acessada em Setembro 2013. 38. http://www.k-cap.or.kr/eng/info/index.html?sidx=2, acessada em Setembro 2013. 39. http://llchemical.com/, acessada em Setembro 2013. 40. http://www.suncatalytix.com/index.html, acessada em Setembro 2013. 41. Kok, B.; Forbush, B.; Mcgloin, M.; Photochem. Photobiol. 1970, 11, 45. 42. Kanan, M. W.; Nocera, D. G.; Science, 2008, 321, 1072. 43. Youngblood, W. J.; Lee, S. H. A.; Maeda, K.; Mallouk, T. E.; Acc. Chem. Res. 2009, 42, 1966. 44. Meyer, T. J.; Acc. Chem. Res. 1989, 22, 163. 45. Alstrum-Acevedo, J. H.; Brennaman, M. K.; Meyer, T. J.; Inorg. Chem. 2005, 44, 6802. 46. Ferreira, K. N.; Iverson, T. M.; Maghlaoui, K.; Barber, J.; Iwata, S.; Science 2004, 303, 1831. 47. Yano, J.; Kern, J.; Sauer, K.; Latimer, M. J.; Pushkar, Y.; Biesiadka, J.; Loll, B.; Saenger, W.; Messinger, J.; Zouni, A.; Yachandra, V. K.; Science 2006, 314, 821. 48. Sproviero, E. M.; Gascon, J. A.; McEvoy, J. P.; Brudvig, G. W.; Batista, V. S.; Coord. Chem. Rev. 2008, 252, 395. 49. Chen, Z. F.; Concepcion, J. J.; Hu, X. Q.; Yang, W. T.; Hoertz, P. G.; Meyer, T. J.; Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 7225. 50. Shevela, D.; Koroidov, S.; Najafpour, M. M.; Messinger, J.; Kurz, P.; Chem. Eur. J. 2011, 17, 5415. 51. Gagliardi, C. J.; Vannucci, A. K.; Concepcion, J. J.; Chen, Z.; Meyer, T. J.; En. Energy Environ. Sci. 2012, 5, 7704-7717. 52. Pace, R. J.; Jin, L.; Stranger, R.; Dalton Trans. 2012, 41, 11145. 53. Pace, R. J.; Stranger, R.; Petrie, S.; Dalton Trans. 2012, 41, 7179. 54. Yamanaka, S.; Kanda, K.; Saito, T.; Umena, Y.; Kawakami, K.; Shen, J. R.; Kamiya, N.; Okumura, M.; Nakamura, H.; Yamaguchi, K.; Em Advances in Quantum Chemistry, Vol 64; J.R. Sabin; E.J. Brandas, eds.; San Diego, 2012, pp. 121-187. 55. Cox, N.; Messinger, J.; Biochim. Biophys. Acta, Bioenerg. 2013, 1827, 1020. 56. Cox, N.; Pantazis, D. A.; Neese, F.; Lubitz, W.; Acc. Chem. Res. 2013, 46, 1588. 57. Krewald, V.; Neese, F.; Pantazis, D. A.; J. Am. Chem. Soc. 2013, 135, 5726. 58. Lohmiller, T.; Ames, W.; Lubitz, W.; Cox, N.; Misra, S. K.; Appl. Magn. Reson. 2013, 44, 691. 59. Siegbahn, P. E. M.; Biochim. Biophys. Acta, Bioenerg. 2013, 1827, 1003. 60. Ono, T.; Noguchi, T.; Inoue, Y.; Kusunoki, M.; Matsushita, T.; Oyanagi, H.; Science 1992, 258, 1335. 61. Meyer, T. J.; Huynh, M. H. V.; Thorp, H. H.; Angew. Chem. 2007, 46, 5284. 62. Proshlyakov, D. A.; Pressler, M. A.; Babcock, G. T.; Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 8020. 63. Tommos, C.; Babcock, G. T.; Acc. Chem. Res. 1998, 31, 18. 64. Huynh, M. H. V.; Meyer, T. J.; Chem. Rev. 2007, 107, 5004. 65. Weinberg, D. R.; Gagliardi, C. J.; Hull, J. F.; Murphy, C. F.; Kent, C. A.; Westlake, B. C.; Paul, A.; Ess, D. H.; McCafferty, D. G.; Meyer, T. J.; Chem. Rev. 2012, 112, 4016. 66. Fukuzumi, S.; Coord. Chem. Rev. 2013, 257, 1564. 67. Herzog, W.; Bronner, C.; Loeffler, S.; He, B.; Kratzert, D.; Stalke, D.; Hauser, A.; Wenger, O. S.; ChemPhysChem 2013, 14, 1168. 68. Kuss-Petermann, M.; Wenger, O. S.; J. Phys. Chem. A 2013, 117, 5726. 69. Kuss-Petermann, M.; Wenger, O. S.; J. Phys. Chem. Lett. 2013, 4, 2535. 70. Gersten, S. W.; Samuels, G. J.; Meyer, T. J.; J. Am. Chem. Soc. 1982, 104, 4029. 71. Concepcion, J. J.; Jurss, J. W.; Templeton, J. L.; Meyer, T. J.; Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 17632. 72. Liu, F.; Concepcion, J. J.; Jurss, J. W.; Cardolaccia, T.; Templeton, J. L.; Meyer, T. J.; Inorg. Chem. 2008, 47, 1727. 73. Cape, J. L.; Lymar, S. V.; Lightbody, T.; Hurst, J. K.; Inorg. Chem. 2009, 48, 4400. 74. Xu, Y. H.; Akermark, T.; Gyollai, V.; Zou, D. P.; Eriksson, L.; Duan, L. L.; Zhang, R.; Akermark, B.; Sun, L. C.; Inorg. Chem. 2009, 48, 2717. 75. Lebeau, E. L.; Adeyemi, S. A.; Meyer, T. J.; Inorg. Chem. 1998, 37, 6476. 76. Yang, X.; Baik, M.-H.; J. Am. Chem. Soc. 2006, 128, 7476. 77. Concepcion, J. J.; Tsai, M. K.; Muckerman, J. T.; Meyer, T. J.; J. Am. Chem. Soc. 2010, 132, 1545. 78. Tong, L.; Inge, A. K.; Duan, L.; Wang, L.; Zou, X.; Sun, L.; Inorg. Chem. 2013, 52, 2505. 79. Xu, Y.; Duan, L.; Åkermark, T.; Tong, L.; Lee, B.-L.; Zhang, R.; Åkermark, B.; Sun, L.; Chem. Eur. J 2011, 17, 9520. 80. Tagore, R.; Chen, H. Y.; Zhang, H.; Crabtree, R. H.; Brudvig, G. W.; Inorg. Chim. Acta 2007, 360, 2983. 81. Concepcion, J. J.; Jurss, J. W.; Brennaman, M. K.; Hoertz, P. G.; Patrocinio, A. O. T.; Murakami Iha, N. Y.; Templeton, J. L.; Meyer, T. J.; Acc. Chem. Res. 2009, 42, 1954. 82. Cady, C. W.; Shinopoulos, K. E.; Crabtree, R. H.; Brudvig, G. W.; Dalton Trans. 2010, 39, 3985. 83. Zhang, F.; Cady, C. W.; Brudvig, G. W.; Hou, H. J. M.; Inorg. Chim. Acta 2011, 366, 128. 84. Concepcion, J. J.; Jurss, J. W.; Templeton, J. L.; Meyer, T. J.; J. Am. Chem. Soc. 2008, 130, 16462. 85. Radaram, B.; Ivie, J. A.; Singh, W. M.; Grudzien, R. M.; Reibenspies, J. H.; Webster, C. E.; Zhao, X.; Inorg. Chem. 2011, 50, 10564. 86. Roeser, S.; Farras, P.; Bozoglian, F.; Martinez-Belmonte, M.; Benet-Buchholz, J.; Llobet, A.; ChemSusChem 2011, 4, 197. 87. Yagi, M.; Tajima, S.; Komi, M.; Yamazaki, H.; Dalton Trans. 2011, 40, 3802. 88. Maji, S.; Lopez, I.; Bozoglian, F.; Benet-Buchholz, J.; Llobet, A.; Inorg. Chem. 2013, 52, 3591. 89. Depaula, J. C.; Beck, W. F.; Brudvig, G. W.; New J. Chem. 1987, 11, 103. 90. Brimblecombe, R.; Bond, A. M.; Dismukes, G. C.; Swiegers, G. F.; Spiccia, L.; Phys. Chem. Chem. Phys. 2009, 11, 6441. 91. Flamigni, L.; Collin, J. P.; Sauvage, J. P.; Acc. Chem. Res. 2008, 41, 857. 92. McDaniel, N. D.; Coughlin, F. J.; Tinker, L. L.; Bernhard, S.; J. Am. Chem. Soc. 2008, 130, 210. 93. Gust, D.; Moore, T. A.; Moore, A. L.; Acc. Chem. Res. 2009, 42, 1890. 94. Yagi, M.; Syouji, A.; Yamada, S.; Komi, M.; Yamazaki, H.; Tajima, S.; Photochem. Photobiol. Sci. 2009, 8, 139. 95. Concepcion, J. J.; Jurss, J. W.; Norris, M. R.; Chen, Z. F.; Templeton, J. L.; Meyer, T. J.; Inorg. Chem. 2010, 49, 1277. 96. Kohl, S. W.; Weiner, L.; Schwartsburd, L.; Konstantinovski, L.; Shimon, L. J. W.; Ben-David, Y.; Iron, M. A.; Milstein, D.; Science 2009, 324, 74. 97. Duan, L. L.; Fischer, A.; Xu, Y. H.; Sun, L. C.; J. Am. Chem. Soc. 2009, 131, 10397. 98. Duan, L.; Bozoglian, F.; Mandal, S.; Stewart, B.; Privalov, T.; Llobet, A.; Sun, L.; Nat. Chem. 2012, 4, 418. 99. Tong, L.; Duan, L.; Xu, Y.; Privalov, T.; Sun, L.; Angew. Chem. 2011, 50, 445. 100. Hull, J. F.; Balcells, D.; Blakemore, J. D.; Incarvito, C. D.; Eisenstein, O.; Brudvig, G. W.; Crabtree, R. H.; J. Am. Chem. Soc. 2009, 131, 8730. 101. Blakemore, J. D.; Schley, N. D.; Balcells, D.; Hull, J. F.; Olack, G. W.; Incarvito, C. D.; Eisenstein, O.; Brudvig, G. W.; Crabtree, R. H.; J. Am. Chem. Soc. 2010, 132, 16017. 102. Savini, A.; Bellachioma, G.; Ciancaleoni, G.; Zuccaccia, C.; Zuccaccia, D.; Macchioni, A.; Chem. Commun. 2010, 46, 9218. 103. Parent, A. R.; Blakemore, J. D.; Brudvig, G. W.; Crabtree, R. H.; Chem. Commun. 2011, 47, 11745. 104. Graeupner, J.; Brewster, T. P.; Blakemore, J. D.; Schley, N. D.; Thomsen, J. M.; Brudvig, G. W.; Hazari, N.; Crabtree, R. H.; Organometallics 2012, 31, 7158. 105. Zong, R.; Thummel, R. P.; J. Am. Chem. Soc. 2005, 127, 12802. 106. Xu, Y. H.; Fischer, A.; Duan, L. L.; Tong, L. P.; Gabrielsson, E.; Akermark, B.; Sun, L. C.; Angew. Chem. 2010, 49, 8934. 107. Bozoglian, F.; Romain, S.; Ertem, M. Z.; Todorova, T. K.; Sens, C.; Mola, J.; Rodriguez, M.; Romero, I.; Benet-Buchholz, J.; Fontrodona, X.; Cramer, C. J.; Gagliardi, L.; Llobet, A.; J. Am. Chem. Soc. 2009, 131, 15176. 108. Duan, L.; Xu, Y.; Tong, L.; Sun, L.; ChemSusChem 2011, 4, 238. 109. An, J. X.; Duan, L. L.; Sun, L. C.; Faraday Discuss. 2012, 155, 267. 110. Hetterscheid, D. G. H.; Reek, J. N. H.; Chem. Commun. 2011, 47, 2712. 111. Brimblecombe, R.; Koo, A.; Dismukes, G. C.; Swiegers, G. F.; Spiccia, L.; ChemSusChem 2010, 3, 1146. 112. Li, L.; Duan, L. L.; Xu, Y. H.; Gorlov, M.; Hagfeldt, A.; Sun, L. C.; Chem. Commun. 2010, 46, 7307. 113. Song, W. J.; Glasson, C. R. K.; Luo, H. L.; Hanson, K.; Brennaman, M. K.; Concepcion, J. J.; Meyer, T. J.; J. Phys. Chem. Lett. 2011, 2, 1808. 114. Hanson, K.; Brennaman, M. K.; Luo, H.; Glasson, C. R. K.; Concepcion, J. J.; Song, W.; Meyer, T. J.; ACS Appl. Mater. Interfaces 2012, 4, 1462. 115. Giokas, P. G.; Miller, S. A.; Hanson, K.; Norris, M. R.; Glasson, C. R. K.; Concepcion, J. J.; Bettis, S. E.; Meyer, T. J.; Moran, A. M.; J. Phys. Chem. C 2013, 117, 812. 116. Luo, H.; Song, W.; Hoertz, P. G.; Hanson, K.; Ghosh, R.; Rangan, S.; Brennaman, M. K.; Concepcion, J. J.; Binstead, R. A.; Bartynski, R. A.; Lopez, R.; Meyer, T. J.; Chem. Mater. 2013, 25, 122. 117. Norris, M. R.; Concepcion, J. J.; Harrison, D. P.; Binstead, R. A.; Ashford, D. L.; Fang, Z.; Templeton, J. L.; Meyer, T. J.; J. Am. Chem. Soc. 2013, 135, 2080. 118. Song, W.; Ito, A.; Binstead, R. A.; Hanson, K.; Luo, H.; Brennaman, M. K.; Concepcion, J. J.; Meyer, T. J.; J. Am. Chem. Soc. 2013, 135, 11587. 119. Song, W. J.; Luo, H. L.; Hanson, K.; Concepcion, J. J.; Brennaman, M. K.; Meyer, T. J.; Energy Environ. Sci. 2013, 6, 1240. 120. Grätzel, M.; Acc. Chem. Res. 2009, 42, 1788. 121. Patrocinio, A. O. T.; Murakami Iha, N. Y.; Quim. Nova 2010, 33, 574. 122. Patrocinio, A. O. T.; Paterno, L. G.; Murakami Iha, N. Y.; J. Phys. Chem. C 2010, 114, 17954. 123. Brennaman, M. K.; Patrocinio, A. O. T.; Song, W.; Jurss, J. W.; Concepcion, J. J.; Hoertz, P. G.; Traub, M. C.; Murakami Iha, N. Y.; Meyer, T. J.; ChemSusChem 2011, 4, 216. 124. Song, W. J.; Brennaman, M. K.; Concepcion, J. J.; Jurss, J. W.; Hoertz, P. C.; Luo, H. L.; Chen, C. C.; Hanson, K.; Meyer, T. J.; J. Phys. Chem. C 2011, 115, 7081. 125. Ashford, D. L.; Stewart, D. J.; Glasson, C. R.; Binstead, R. A.; Harrison, D. P.; Norris, M. R.; Concepcion, J. J.; Fang, Z.; Templeton, J. L.; Meyer, T. J.; Inorg. Chem. 2012, 51, 6428. 126. Younpblood, W. J.; Lee, S. H. A.; Kobayashi, Y.; Hernandez-Pagan, E. A.; Hoertz, P. G.; Moore, T. A.; Moore, A. L.; Gust, D.; Mallouk, T. E.; J. Am. Chem. Soc. 2009, 131, 926. 127. Morris, A. J.; Meyer, G. J.; Fujita, E.; Acc. Chem. Res. 2009, 42, 1983. 128. Windle, C. D.; Perutz, R. N.; Coord. Chem. Rev. 2012, 256, 2562. 129. Bian, Z. Y.; Chi, S. M.; Li, L.; Fu, W. F.; Dalton Trans. 2010, 39, 7884. 130. Lumpkin, R. S.; Kober, E. M.; Worl, L. A.; Murtaza, Z.; Meyer, T. J.; J. Phys. Chem. 1990, 94, 239. 131. Fujita, E.; Coord. Chem. Rev. 1999, 185-6, 373. 132. Grodkowski, J.; Dhanasekaran, T.; Neta, P.; Hambright, P.; Brunschwig, B. S.; Shinozaki, K.; Fujita, E.; J. Phys. Chem. A 2000, 104, 11332. 133. Wang, C.; Ma, X.-X.; Li, J.; Xu, L.; Zhang, F.-x.; J. Mol. Catal. A: Chem. 2012, 363, 108. 134. Zhu, Y.; Zhang, S.; Ye, Y.; Zhang, X.; Wang, L.; Zhu, W.; Cheng, F.; Tao, F.; ACS Catal. 2012, 2, 2403. 135. Woolerton, T. W.; Sheard, S.; Reisner, E.; Pierce, E.; Ragsdale, S. W.; Armstrong, F. A.; J. Am. Chem. Soc. 2010, 132, 2132. 136. Koike, K.; Hori, H.; Ishizuka, M.; Westwell, J. R.; Takeuchi, K.; Ibusuki, T.; Enjouji, K.; Konno, H.; Sakamoto, K.; Ishitani, O.; Organometallics 1997, 16, 5724. 137. Hayashi, Y.; Kita, S.; Brunschwig, B. S.; Fujita, E.; J. Am. Chem. Soc. 2003, 125, 11976. 138. Tsubaki, H.; Sekine, A.; Ohashi, Y.; Koike, K.; Takeda, H.; Ishitani, O.; J. Am. Chem. Soc. 2005, 127, 15544. 139. Sato, S.; Sekine, A.; Ohashi, Y.; Ishitani, O.; Blanco-Rodriguez, A. M.; Vicek, A.; Unno, T.; Koike, K.; Inorg. Chem. 2007, 46, 3531. 140. Takeda, H.; Koike, K.; Inoue, H.; Ishitani, O.; J. Am. Chem. Soc. 2008, 130, 2023. 141. Doherty, M. D.; Grills, D. C.; Fujita, E.; Inorg. Chem. 2009, 48, 1796. 142. Tamaki, Y.; Watanabe, K.; Koike, K.; Inoue, H.; Morimoto, T.; Ishitani, O.; Faraday Discuss. 2012, 155, 115. 143. Hawecker, J.; Lehn, J. M.; Ziessel, R.; J. Chem. Soc., Chem. Commun. 1983, 536. 144. Agarwal, J.; Fujita, E.; Schaefer, H. F.; Muckerman, J. T.; J. Am. Chem. Soc. 2012, 134, 5180. 145. Lehn, J. M.; Ziessel, R.; J. Organomet. Chem. 1990, 382, 157. 146. Kutal, C.; Weber, M. A.; Ferraudi, G.; Geiger, D.; Organometallics 1985, 4, 2161. 147. Kitamura, N.; Tazuke, S.; Chem. Lett. 1983, 1109. 148. Koike, K.; Hori, H.; Ishizuka, M.; Westwell, J. R.; Takeuchi, K.; Ibusuki, T.; Enjouji, K.; Konno, H.; Sakamoto, K.; Ishitani, O.; Organometallics 1997, 16, 5724. 149. Hori, H.; Ishihara, J.; Koike, K.; Takeuchi, K.; Ibusuki, T.; Ishitani, O.; J. Photochem. Photobiol., A 1999, 120, 119. 150. Gholamkhass, B.; Mametsuka, H.; Koike, K.; Tanabe, T.; Furue, M.; Ishitani, O.; Inorg. Chem. 2005, 44, 2326. 151. Sato, S.; Koike, K.; Inoue, H.; Ishitani, O.; J. Photochem. Photobiol., A 2007, 6, 454. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access