
 |
 |
 |
 |
 |
Revisão
|
|
| Monitorando atividades enzimáticas com sondas fluorogênicas Monitoring enzymatic activities with fluorogenic probes |
|
Caroline da Costa Silva Gonçalves; Anita Jocelyne Marsaioli*
Instituto de Química, Universidade Estadual de Campinas, Cidade Universitária Zeferino Vaz, 13081-970 Campinas - SP, Brasil Recebido em 27/06/2013 *e-mail: anita@iqm.unicamp.br Latent fluorogenic probes are essential tools for molecular and chemical biology, providing valuable information about enzymatic activity and occurrence. In this review, a brief outline of fluorophores and latent fluorogenic probes is given. Furthermore, advances and challenges in the development of fluorogenic chemical probes to visualize enzymatic activities (hydrolases and oxidoreductases) of biotechnological and biomedical interest are highlighted, including some methodologies for intracellular imaging. INTRODUÇÃO As enzimas catalisam as transformações químicas nos sistemas biológicos e estão relacionadas à sobrevivência e reprodução de todos os organismos vivos. Estas propriedades são apreciadas e empregadas na produção de intermediários sintéticos, nas indústrias de alimentos, de produtos de limpeza e de higiene. Além disso, alterações na atividade e/ou expressão enzimática estão diretamente relacionadas a diversas doenças humanas, fazendo do monitoramento das atividades enzimáticas um tópico de interesse na biotecnologia e na medicina.1 Dentre as estratégias de monitoramento da atividade enzimática podemos destacar as técnicas que detectam sinais fluorescentes, as quais são consideravelmente mais sensíveis do que as que detectam sinais colorimétricos. Metodologias baseadas na detecção de compostos fluorescentes podem auxiliar a visualização de proteínas de interesse em tempo real, fornecendo informações relevantes em processos biológicos relacionados a elas.2 Estas metodologias dependem de fluoróforos (compostos que emitem fluorescência após excitação a um comprimento de onda específico) e são importantes em engenharia genética, na identificação e seleção de novas enzimas, na triagem enzimática de coleções de micro-organismos, de coleções de clones metagenômicos e na triagem de candidatos a inibidores enzimáticos.3 Entre os fluoróforos mais utilizados podemos destacar os derivados de xantenos, como a rodamina (1) e a fluoresceína (2) e os derivados de cumarina como a 7-hidroxicumarina (3), que apresenta rendimento quântico elevado e comprimento de onda de excitação na região do UV (Figura 1).4 A intensa fluorescência destes compostos pode ser convenientemente mascarada por meio de modificações químicas, possibilitando o desenvolvimento de sondas fluorogênicas latentes nas quais o sinal fluorescente é detectado somente após uma série de reações desencadeadas pela ação enzimática de interesse.5
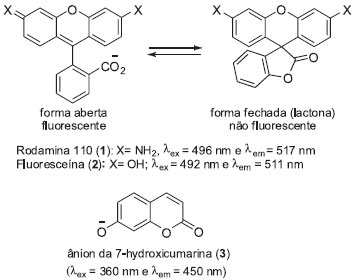 Figura 1. Estrutura química da Rodamina (1), da Fluoresceína (2) e do ânion da 7-hidroxicumarina (3)
O controle do sinal fluorescente (ligado/desligado - do inglês "ON/OFF") pode ainda envolver diversos mecanismos como, por exemplo, a transferência fotoinduzida de elétrons ( do inglês "photoinduced electron transfer", PET), a transferência de energia de ressonância por fluorescência (do inglês "fluorescence resonance energy transfer", FRET) e o aumento do sinal de fluorescência por quelação (do inglês "chelation-enhanced fluorescence", CHEF) (Figura 2).
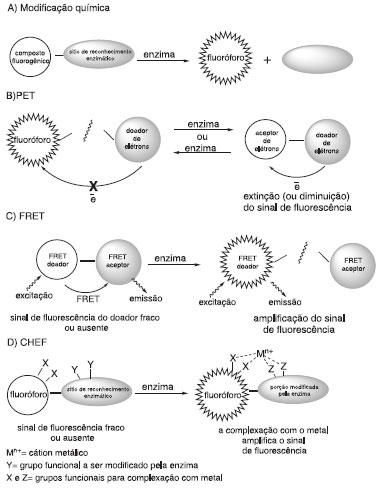 Figura 2. Representação esquemática de algumas estratégias de controle do sinal fluorescente (ligado/desligado)
O desenvolvimento de novas sondas fluorogênicas é fundamental para estudos de sistemas biológicos complexos, pois permitem visualizar e quantificar processos intracelulares em tempo real.6 Neste artigo de revisão serão abordados alguns aspectos do emprego e do desenvolvimento de sondas fluorogênicas latentes destinadas ao monitoramento intra e extracelular das atividades enzimáticas utilizando as estratégias de controle do sinal fluorescente descritas anteriormente.
MONITORAMENTOS COM REAÇÕES QUIMIO-ENZIMÁTICAS EM CASCATA Avaliar a liberação do sinal fluorescente após uma série de reações químicas na sonda fluorogênica desencadeadas pela ação enzimática é uma estratégia bastante utilizada para o monitoramento das atividades de diferentes classes de enzimas. Por exemplo, Reymond e colaboradores desenvolveram sondas fluorogênicas latentes (4) baseadas na 7-hidroxicumarina (3) para a detecção de lipases e esterases em meio de cultura por triagem de alta eficiência (HTS).7 As esterases e as lipases são enzimas que catalisam, respectivamente, a hidrólise de ésteres solúveis em água e a hidrólise de triglicerídeos. Estas enzimas são biocatalisadores importantes, empregadas em diversos processos industriais.8 Na metodologia de triagem desenvolvida pelo grupo de Reymond, a hidrólise enzimática da função éster de 4 leva à formação do diol vicinal correspondente (5). A clivagem oxidativa de 5 com NaIO4 origina o aldeído 7 o qual, após uma reação de β-eliminação catalisada por BSA (albumina de soro bovino), libera o ânion da 7-hidroxicumarina (3), possibilitando a detecção da atividade enzimática (Esquema 1-A). Esta metodologia de triagem enzimática foi aplicada na avaliação da enantiosseletividade enzimática (Quick-E) empregando sondas quirais. Esta metodologia foi mais tarde adaptada por nosso grupo de pesquisas para ensaios com células integras de micro-organismos.9 A metodologia desenvolvida mostrou-se bastante robusta e eficiente para ensaios de triagem enzimática realizados em pH > 7 e foi expandida para o monitoramento das atividades de outras classes de enzimas, como fosfatases, álcool desidrogenases e epóxido-hidrolases.10 Recentemente, esta metodologia de triagem foi empregada para a caracterização funcional de uma epóxido-hidrolase clonada de Aspergillus brasiliensis CCT1435.11
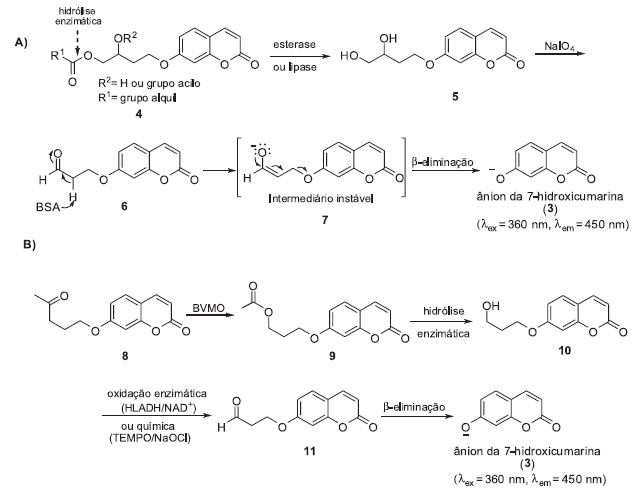 Esquema 1. Sistemas de monitoramento baseados em reações quimio-enzimáticas em cascata: A). Ensaios para detecção das atividades de esterases e lipases; B) Ensaios para o monitoramento de BVMO
Em um segundo exemplo descrevemos o monitoramento de Baeyer Villiger Monooxigenases (BVMOs) utilizando células íntegras de microrganismos e a sonda 11 (Esquema 1-B).12 As BVMOs são flavoenzimas que dependem de NAD(P)H e pertencem à classe das oxidorredutases, conhecidas por catalisar reações de Baeyer-Villiger, podendo ainda catalisar outras reações de oxidação como, por exemplo, sulfoxidações, oxidações de selenídeos e de aminas.13 Devido à seletividade e eficiência, as BMVOs são biocatalisadores importantes para a síntese orgânica e o desenvolvimento de metodologias que levam a uma maior compreensão destas enzimas é de grande importância para a descoberta e o desenvolvimento de novos biocatalisadores. No sistema de monitoramento desenvolvido, a oxidação da função cetona da sonda fluorogênica 11 por uma BVMO celular produz o éster 12, o qual após hidrólise enzimática (ou espontânea) origina o álcool primário 13. Oxidação química (TEMPO/NaOCl) ou enzimática (HLADH/NADH) de 13 fornece o correspondente aldeído o qual, após uma reação de β-eliminação catalisada por BSA, libera o ânion da 7-hidroxicumarina (3) (Esquema 1-B). Os melhores resultados foram obtidos utilizando-se o sistema de oxidação TEMPO/NaOCl. Sob estas condições o sinal de fluorescência foi detectado três vezes mais rápido do que na via de oxidação enzimática. Desta forma, processos de monitoramento de atividades enzimáticas baseadas em reações quimio-enzimáticas em cascata podem ser adaptados para a triagem de diferentes classes de enzimas. Entretanto, a necessidade da utilização de reagentes químicos dificulta o emprego destas sondas em ensaios enzimáticos in cellulo para o monitoramento das atividades de enzimas endógenas em tempo real.
SISTEMAS DE MONITORAMENTO BASEADOS EM REAÇÕES ESPONTÂNEAS EM CASCATA Um segundo sistema de monitoramento enzimático se baseia na utilização de conectores estáveis quando protegidos pelo substrato enzimático, mas que se tornam lábeis quando desprotegidos, podendo sofrer, após a transformação enzimática do substrato, uma série de reações espontâneas que culminam com a formação do composto fluorescente. Como primeiro exemplo citamos a sonda 14 desenvolvida pelo grupo de pesquisas de Kikuchi para monitoramento das atividades enzimáticas de histonas desacetilases (HDACs) (Esquema 2).14 HDACs catalisam a remoção de grupos acetila de resíduos de ε-N-acetil lisinas de histonas e outras proteínas. Estas enzimas representam um alvo molecular importante em processos de desenvolvimento de novos fármacos, sendo associadas a diversas doenças humanas como câncer e doenças neurodegenerativas.15 A sonda 14 utiliza como substrato enzimático um fragmento peptídico acetilado proveniente da região N-terminal da histona H3 e como fluoróforo um derivado cumarínico modificado com um éster carbonato na posição fenólica. A desacetilação da cadeia lateral da lisina de 14 pela HDAC gera uma amina primária, desencadeando um processo espontâneo de transesterificação intramolecular que culmina com a formação de um composto fluorescente (15). A eficiência da sonda 14 foi analisada frente à enzima Sirt 1 (histona desacetilase dependente de NAD+). Após 120 minutos de incubação foi observado um aumento considerável do sinal de fluorescência. Vale ressaltar que, sob as condições utilizadas no ensaio e na ausência da enzima (experimento de controle), foi observado um incremento do sinal de fluorescência devido à hidrólise espontânea do éster carbonato. Entretanto, a variação do sinal fluorescente causada pela hidrólise espontânea foi significativamente menor do que a observada nos ensaios com a Sirt 1, não interferindo de forma significativa na detecção da atividade enzimática. Finalmente, para avaliar o potencial da sonda 14 para triagem de inibidores de HDAC, os pesquisadores repetiram os ensaios enzimáticos na presença de um inibidor de Sirt 1. Na presença do inibidor não se observou a variação do sinal de fluorescência devido à hidrólise enzimática da sonda 14. Em conjunto, estes resultados demonstram que a sonda 14 é apropriada para observar a modulação das atividades de HDACs.
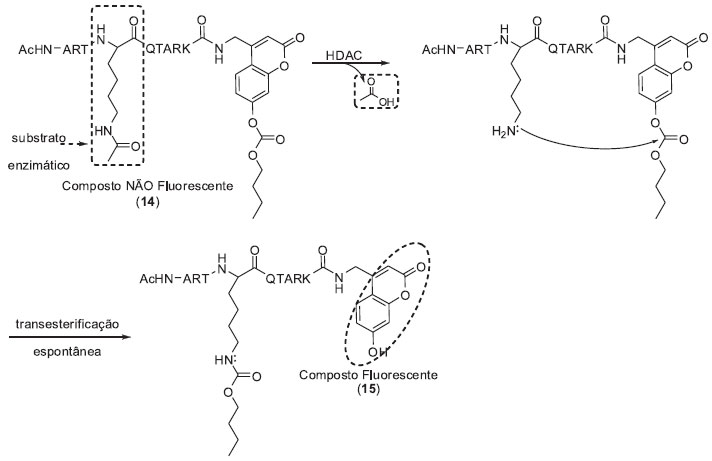 Esquema 2. Sonda fluorogênica (14) desenvolvida para o monitoramento de HDACs
Em um segundo exemplo, apresentamos a sonda fluorogênica (16) para o monitoramento de citocromo P450 intracelular (isoenzima CYP1A1) desenvolvida por Raines e colaboradores.16 Os citocromos P450 são hemeproteínas extremamente versáteis, responsáveis pelo metabolismo oxidativo de substâncias endógenas e de xenobióticos. Estas hemeproteínas oferecem oportunidades para o desenvolvimento de novas biotecnologias com potencial aplicação na solução de problemas relativos à saúde humana e a processos industriais,17 ressaltando a importância do desenvolvimento de metodologias para o monitoramento e/ou detecção da atividade destas proteínas. A sonda 16 apresenta o grupo trimetil-lock (17), o qual pode sofrer uma reação de lactonização levando à formação de uma di-hidrocumarina, devido a repulsão eletrônica entre os três grupos metílicos (Esquema 3). Assim, para evitar a reação de lactonização espontânea, a hidroxila fenólica de 17 foi protegida com um grupo etila e o etil-éter resultante foi utilizado como substrato para a CYP1A1. Para se evitar a necessidade de duas reações químicas para detecção do máximo de fluorescência, o que pode acarretar na diminuição do limite de detecção linear dos ensaios, foi utilizado um derivado morfolina-uréia da rodamina como fluoróforo (18). A sonda 16 foi inicialmente avaliada frente a CYP1A1 em ensaios in vitro. Posteriormente, foram realizados ensaios com a linhagem celular A549 de adenocarcinoma de pulmão, sendo detectado um sinal de fluorescência pouco intenso após 1 h de incubação com 16. A aplicabilidade de 16 em ensaios de modulação da atividade da P450 foi avaliada frente a dois substratos, o ativador 2,3,7,8-tetraclorodibenzo-p-dioxina (TCDD, 19) e um inibidor (resveratrol (20)) de CYP1A1. Nestes ensaios foi observado um aumento significativo do sinal de fluorescência após incubação das células (linhagem A549) com 16 e o TCDD (18). A incubação das células com uma mistura da sonda 16, TCDD (19) e do resveratrol (20) levou a uma diminuição significativa na intensidade do sinal de fluorescência. Estes ensaios demonstram a versatilidade da sonda fluorogênica desenvolvida, indicando que a mesmas poderiam ser utilizadas em ensaios de triagem de inibidores ou ativadores de P450 em processos de descobrimento de novos fármacos. Este sistema de latenciação foi utilizado pelo mesmo grupo no desenvolvimento de metodologias para a triagem de outras enzimas, como esterases e fosfatases alcalinas.18
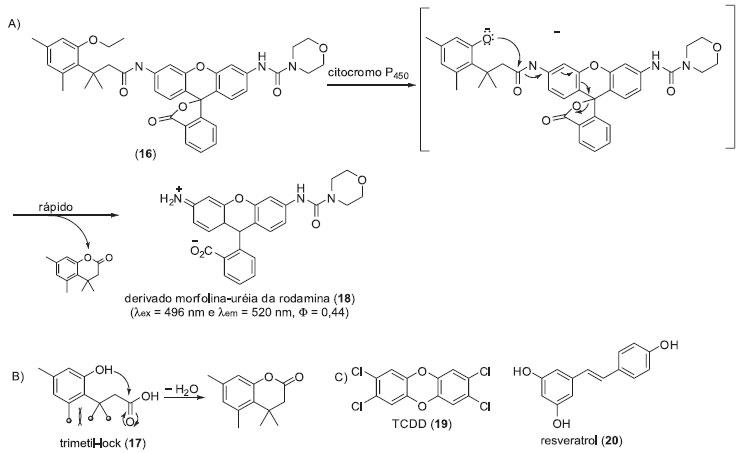 Esquema 3. Sonda fluorogênica para monitorar a atividade do citocromo P450 (isoenzima CYP1A1) intracelulares: A) Estratégia de liberação do fluoróforo iniciada pela ação do citocromo P450; B) Estrutura do grupo trimetil-lock (29) com destaque para interações estéreas entre as metilas; C) Estrutura do TCDD (19) e do resveratrol (20)
Num terceiro exemplo, Rao e colaboradores desenvolveram uma metodologia para monitorar a atividade da β-lactamase TEM-1 (Bla) empregando sondas fluorogênicas que utilizam cefalosporinas como substrato enzimático (sonda 21, Esquema 4).19 As β-lactamases são enzimas responsáveis pela clivagem da penicilina e cefalosporinas e são consideradas as principais causas da resistência bacteriana.20
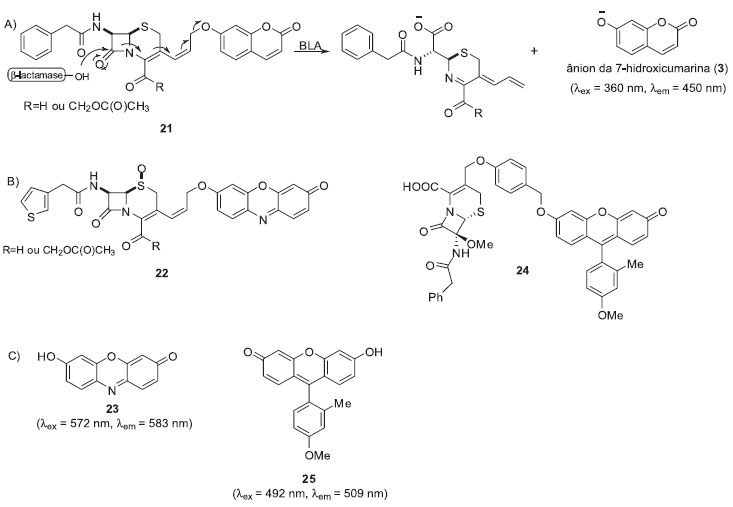 Esquema 4. Sondas fluorogênicas para monitorar BLA. A) Mecanismo de liberação do fluoróforo; B) Estrutura da sonda para ensaios intracelulares 22 e da sonda 24, específica para detecção da enzima BlaC; C) Estrutura dos fluoróforos resorufina (23) e Tokyo Green 25
A clivagem do anel β-lactâmico da cefalosporina de 21 pela enzima Bla desencadeia um processo de fragmentação que termina com a liberação do ânion da 7-hidroxicumarina (3). A sonda apresentou uma boa estabilidade química e foi empregada com sucesso em ensaios com a enzima isolada. Entretanto, devido à sobreposição do sinal de fluorescência do ânion da 7-hidroxicumarina (3) (λex = 360 nm and λem = 460 nm) com alguns componentes celulares (λem(NADH) = 360 nm; λem(NADH) = 460 nm e λex(FAD) = 360/450 nm; λem(FAD) = 520 nm),21 este fluoróforo não é muito apropriado para ensaios in cellulo. Desta forma, para o monitoramento de Bla intracelular empregou-se a sonda fluorogênica 22, a qual utiliza a resorufina (23) como núcleo fluorescente. Os ensaios foram realizados com uma linhagem de células Jurkat transfectadas com o gene BLA de citomegalovirus. A partir destes ensaios foi possível detectar a atividade enzimática de BLA intracelular em tempo real utilizando técnicas de microscopia confocal de fluorescência. Recentemente, o mesmo grupo de pesquisas desenvolveu sondas fluorogênicas específicas para detecção da enzima BlaC, expressa pela Mycobacterium tuberculosis, bactéria causadora da tuberculose.22 A maior seletividade frente à enzima BlaC foi alcançada pela introdução de substituintes volumosos no anel lactâmico da cefalosporina. Dentre os vários derivados analisados a sonda 24 (-OMe na posição C-7 da lactama e um -Bn na cadeia lateral do grupo 7-amino) apresentou maior seletividade frente a BlaC. A detecção do patógeno vivo em solução foi alcançada com a utilização fluoróforo Tokyo Green (25).23 Para se aumentar a estabilidade química da sonda foi adicionado um conector entre o fluoróforo e o substrato enzimático. Assim, as sondas desenvolvidas permitiram a detecção de β-lactamases isoladas e/ou em solução de forma rápida e eficiente. Estes sistemas poderão eventualmente facilitar o diagnóstico de tuberculose bacteriana e contribuir com processos de triagem de inibidores de β-lactamases. Ao contrário do observado com os sistemas quimioenzimáticos, a adaptação dos processos de monitoramento das atividades enzimáticas baseados em cascatas de reações espontâneas para o monitoramento in cellulo é mais apropriada, possibilitando, por exemplo, a determinação da sublocalização celular destas enzimas.
SONDAS FLUOROGÊNICAS BASEADAS EM FRET O processo de FRET envolve a transferência não radioativa de energia entre duas moléculas, na qual uma atua como doador e a outra como aceptor. A energia de ressonância de fluorescência é transferida do doador para o aceptor, resultando na diminuição (ou extinção) da emissão de fluorescência do doador e, quando for o caso, no aumento do sinal referente ao aceptor.24 Diversos sistemas de monitoramento de proteases utilizam FRET para o controle da intensidade do sinal de fluorescência (Esquema 5-A).25 Essa metodologia é bastante utilizada para monitorar proteases que catalisam a hidrólise de ligações peptídicas e realizam funções fundamentais em todos os organismos vivos. Alterações nas atividades destas enzimas estão associadas a diversas doenças dos humanos e, atualmente, seus inibidores são empregados no tratamento de hipertensão, câncer, diabetes e infecção por HIV.26 Neste contexto, o desenvolvimento de sistemas de monitoramento (inibição/ativação) das atividades das proteases pode contribuir no descobrimento de novos inibidores e auxiliar na melhor compreensão do papel biológico destas enzimas. Como exemplo apresentamos o trabalho de Hudecz e colaboradores,27 os quais desenvolveram uma metodologia de monitoramento intracelular por FRET de calpaínas, cisteína-proteases associadas a diversas desordens humanas (sonda fluorogênica 26, Esquema 5-B). A penetração de 26 no citosol foi alcançada ligando-se um decapeptídeo específico de reconhecimento enzimático (TPLKSPPSPR)28 ao grupo diretor (poliarginina, CPP).29 O ácido 5-[(2-aminoetil)amino]naftaleno-1-sulfônico (EVANS, 27) foi utilizado como doador e o ácido 4-((4-(dimetilamino)fenil) benzóico (Dabcyl, 28) como aceptor de energia de ressonância por fluorescência. Uma unidade de glutamato foi adicionada entre o peptídeo TPLKSPPSPR e o CPP para conexão do grupo EVANS (27). Este arranjo colocou o par receptor/doador a uma distância apropriada para a manutenção do processo de FRET. Após clivagem enzimática do fragmento peptídico foi observado um aumento de quatro vezes na intensidade do sinal de fluorescência. A sonda 26 foi posteriormente utilizada pelo mesmo grupo para avaliar o potencial de inibição de uma série de aza-peptídeos sobre a caspase B.30
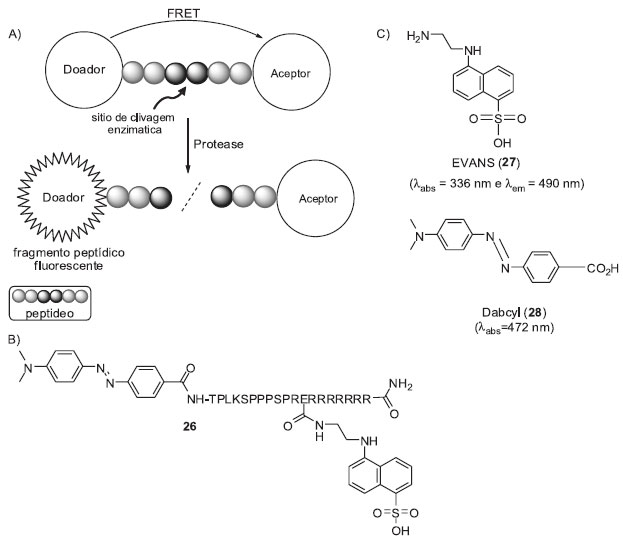 Esquema 5. Sistema de monitoramento de proteases por FRET. A) Princípio geral dos ensaios. B) Sonda fluorogênica desenvolvida para o monitoramento da atividade intracelular de calpaínas
FRET é também utilizado na triagem de outras enzimas como lipases,31 transferases,32 celulases33 e quinases.34 Além disso, os princípios do processo de FRET são amplamente aplicados em sistemas de marcação de proteínas, em estudos de mudanças conformacionais e de regulação espacial de proteínas.35 Entretanto, vale ressaltar que para a variação de sinal de FRET seja efetiva os cromóforos (doador e aceptor) devem estar espacialmente próximos, tipicamente entre 10-100 Å, o que pode representar um problema para o desenvolvimento de sistemas de monitoramento baseados nesta tecnologia.36
EXEMPLOS ADICIONAIS Vários outros sistemas de monitoramento de diferentes atividades enzimáticas intra e extracelular podem ser encontrados na literatura, dentre os quais citamos a sonda 29 desenvolvida por Imperiali e colaboradores para o monitoramento de quinases (Esquema 6).37 O processo de fosforilação reversível de proteínas é uma das modificações pós-traducionais mais importantes das células eucariotas, estando envolvido na regulação de uma grande variedade de eventos celulares, como expressão gênica, divisão celular, diferenciação, apoptose e transporte. Os níveis de fosforilação das proteínas são controlados pelo balanço das atividades de quinases e fosfatases. As fosfatases catalisam a desfosforilação de proteínas e as quinases catalisam as reações de fosforilação via um mecanismo envolvendo a transferência de um grupo γ-fosfato do ATP (adenosina trifosfato) para os resíduos de aminoácidos específicos. Alterações nas atividades destas enzimas estão associadas ao desenvolvimento de inúmeras doenças humanas como câncer, problemas vasculares, doença de Alzheimer e diabetes.38 Como consequência da importância destas enzimas, um grande número de pesquisas de desenvolvimento de novos fármacos utilizam as quinases e, mais recentemente, as fosfatases como alvos terapêuticos.39 Face ao exposto, metodologias de monitoramento da modulação (ativação/inibição) das atividades de quinases por compostos naturais ou sintéticos contribuem para o desenvolvimento de novos fármacos e para a compreensão das vias biológicas controladas por estas enzimas. O mecanismo de sinalização das atividades de quinases pela sonda 29 é fundamentado no aumento da intensidade do sinal de fluorescência do amino ácido sulfonamido-oxina (Sox, 30a) após fosforilação e quelação com íons Mg2+ (processo de aumento da intensidade do sinal de fluorescência por CHEF). A fosforilação dos resíduos Ser, Thr ou Tyr (posicionados no N- ou C-terminal do fragmento peptídico) aumenta a afinidade de 29 pelo metal Mg2+. A quelação do metal com os grupos fosfatos acarreta um aumento da emissão de fluorescência do Sox (30a) e, consequentemente, possibilita o monitoramento da atividade de quinases. Uma melhora na sensibilidade enzimática foi obtida empregando-se o derivado Sox-cisteína (C-Sox, 30b) como fluoróforo.
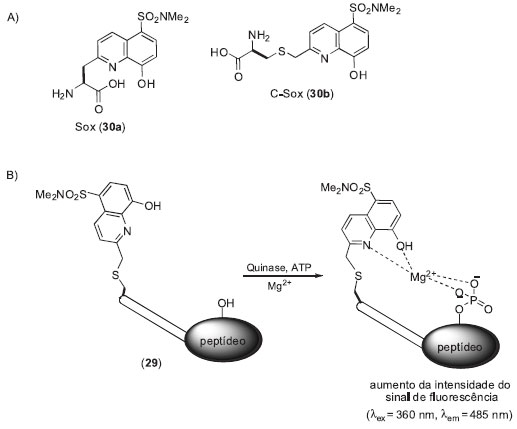 Esquema 6. Sonda para o monitoramento de quinases baseada no processo de CHEF. A) Estrutura do fluoróforo Sox (30a) e C-Sox (30b). B) O mecanismo de sinalização das atividades de quinases pela sonda 29
Como último exemplo citamos o trabalho realizado por Hong, Park e colaboradores,40 que utilizaram o processo de extinção de fluorescência por PET(do inglês: fluorescence quencher) no desenvolvimento de uma sonda fluorescente para o monitoramento de proteínas quinases (31, Esquema 7). O processo de PET envolve a transferência de um elétron para um fluoróforo no seu estado excitado a partir de um doador de elétrons introduzido próximo ao fluoróforo. O caminho inverso (doação de elétrons do fluoróforo para um aceptor de elétrons) também pode ocorrer.41 A sonda 31 contém uma molécula de fenotiazina (PTZ - composto extintor de fluorescência) conectada a um complexo Zn-DPA (bis(Zn2+-dipicolamina)), o qual se liga fortemente a grupos fosfatos em solução aquosa. Os ensaios enzimáticos são realizados com sonda 31 na presença de um substrato peptídico (32a) marcado com o fluoróforo tetrametilrodamina (TMR 33). Assim, após a fosforilação de 32a pela quinase tem-se a formação do fosfopeptídeo correspondente (32b), o qual se complexa a 31, levando ao decaimento drástico do sinal de fluorescência e, consequentemente, possibilita o monitoramento das atividades da proteína quinase de interesse.
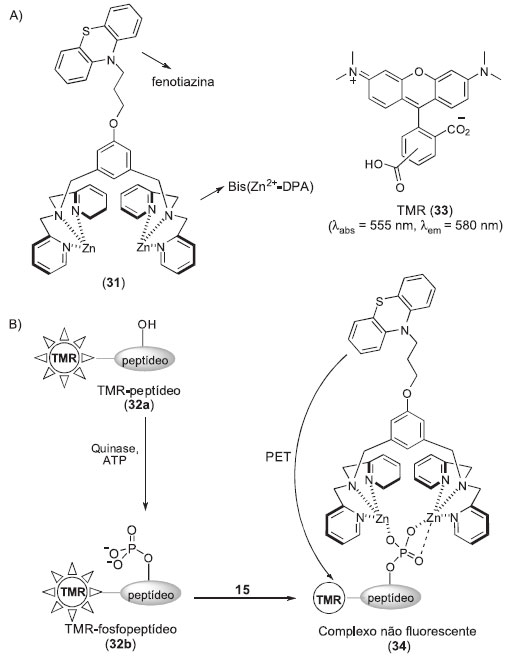 Esquema 7. Sonda para detecção da atividade de quinases pelo processo de extinção de fluorescência do por PET (transferência de elétron fotoninduzida). A) Estrutura da sonda 31 e do fluoróforo TMR (33). B) Mecanismo de extinção de fluorescência por PET
CONCLUSÕES E PERSPECTIVAS Os sistemas de monitoramento das atividades enzimáticas baseados em sondas fluorescentes contribuem de forma significativa com estudos relacionados a aspectos fundamentais da biologia, biotecnologia e medicina. O esforço conjunto de químicos e biólogos resulta na implementação de novas metodologias de sinalização que possibilitam a melhor compreensão de processos biológicos complexos. Entretanto, apesar do grande número de sondas descritas, muitas proteínas e vias de sinalização ainda não possuem um sistema de monitoramento adequado ou específico, apontando para a necessidade crescente de novas pesquisas neste campo. Um dos maiores desafios é a construção de sondas específicas para uma dada enzima, uma vez que elas podem aceitar diferentes substratos, ou seja, mais de uma enzima pode ser responsável pela reação que dá início a liberação do fluoróforo. Outro grande obstáculo é a obtenção de sondas capazes de penetrar nas células e tecidos e soluções para este problema incluem, por exemplo, a utilização de CPPs e de tranportadores. Finalmente, as estratégias de marcação de proteínas com compostos fluorogênicos é um desafio atual na interface da química e biologia.42 Acreditamos que estas estratégias poderão ter um impacto significativo na compreensão das vias de sinalização no contexto intracelular.
REFERÊNCIAS 1. Gonçalves, C. C. S.; Marsaioli, A. J.; Quim. Nova no prelo; Buchhovz, K.; Kasche, V.; Bornscheuer, U. T.; Biocatalyst and Enzyme Technology, 2nd ed., Wiley: New York, 2005. 2. Haugland, R. P.; Handbook of Fluorescence probes and Research chemicals, 6th ed., Molecular Probes: Eugene, 1996. 3. Reymond, J.-L.; Fluxà, V. S.; Maillard, N.; Chem. Commun. 2008, 46. 4. Liu, J.; Liu, C.; He, W.; Curr. Org. Chem. 2013, 17, 564; Laves, L. D.; Raines, R. T.; ACS Chem. Biol. 2008, 3, 142. 5. Drake, C. R.; Miller, D. C.; Jones, E. F.; Curr. Org. Synth. 2011, 8, 498; Laves, L. D.; Chao, T.-Y.; Raines, R. T.; ACS Chem. Biol. 2006, 1, 252. 6. Razgulin, A.; Ma, N.; Rao, J.; Chem Soc. Rev. 2011, 40, 4186; Zhang, J.; Campbell, R. E.; Ting, A. Y.; Tsien, R. Y.; Nat. Rev. Moll. Cell Biol. 2002, 3, 906. 7. Nyfeler, E.; Grognux, J.; Wahler, D.; Helv. Chim. Acta 2003, 86, 2919 ; Badalassi, F.; Wahler, D.; Klein, G.; Crottti, P.; Reymond, J.-L.; Angew. Chem. Int. Ed. 2000, 39, 4067. 8. Para uma revisão sobre lipases e esterases ver: Hasan, F.; Shah, A. A.; Hameed, A.; Enzyme Microb. Technol. 2006, 26, 235; Sharma, R.; Chisti, Y.; Banerjee, U. C. Biotechnol. Adv. 2001, 19, 627. 9. de Vasconcellos, S. P.; Angolini, C. F. F.; García, I. N. S.; Dellagnezze, B. M.; da Silva, C. C.; Marsaioli, A. J.; Santos Neto, E. V.; de Oliveira, V. M.; Org. Geochem. 2010, 41, 675; da Cruz, G.; Angoline, C. F. F.; de Oliveira, L. G.; Lopes, P. F.; de Vasconcellos, S. P.; Crespim, E.; de Oliveira, V. M.; Santos neto, E. V.; Marsaioli, A. J.; Appl. Microbiol. Biotechnol. 2010, 87, 319; Mantovani, S. M.; de Oliveira, L. G.; Marsaioli, A. J.; J. Braz. Chem. Soc. 2010, 21, 1484; Mantovani, S. M.; de Oliveira, L. G.; Marsaioli, A. J.; J. Mol. Catal. B: Enzym. 2008, 52- 53, 173; Pinheiro, L.; Marsaioli, A. J.; J. Mol. Catal. B: Enzym. 2007, 44, 78; Sicard, R.; Chen, L. S.; Marsaioli, A. J.; Reymond, J.-L.; Adv. Synth. Catal. 2005, 347, 1041; Bicalho, B; Chen, L. S.; Grognux, J.; Reymond, J.-L.; Marsaioli, A. J.; J. Braz. Chem. Soc. 2004, 15, 911; Gonçalves, R. A. C.; Porto, A. L. M.; Pinheiro, L.; Cagnon, J. R.; Manfio, G. P.; Marsaioli, A. J.; Food Technol. Biotechnol. 2004, 4, 355. 10. Reymond, J. -L.; Food Technol. Biotechnol. 2004, 42, 265. 11. Beloti, L.; Costa, B. Z.; Toledo, M. A. S.; Santos, C. A.; Crucello, A.; Fávaro, T. P.; Santiago, A. S.; Mendes, J. S.; Marsaioli, A. J.; Souza, A. P.; Protein Expression Purif. 2013, 91, 175. 12. Gutiérrez, M. C.; Sleegers, A.; Simpson, H. D.; Alphand, V.; Furstoss, R.; Org. Biomol. Chem. 2003, 1, 3500. 13. Kamerbeek, N. M.; Janssen, D. B.; van Berkel, W. J. H.; Fraaije, M. W.; Adv. Synth. Catal. 2003, 345, 667. 14. Baba, R.; Hori, Y.; Mizukami, S.; Kikuchi, K.; J. Am. Chem. Soc. 2012, 134, 14310. 15. Para uma revisão sobre histonas deacetilases ver: Li, Y.; Shin, D.; Kwon, S. H.; FEBS J. 2013, 280, 775; Bolden, J. E.; Peart, M. J.; Johnstone, R. W.; Nat. Rev. Drug Discovery 2006, 5, 769. 16. Lesine, M. N.; Raines, R. T.; Chem. Sci. 2012, 3, 2412; Yatzack, M. M.; Lavis, L. D.; Chao, T. -Y.; Chandran, S. S.; Raines, R. T.; Bioorg. Med. Chem. 2008, 18, 5864. 17. Para uma revisão sobre P450 ver: Guengerich, F. P.; Munro, A. W.; J. Biol. Chem. 2013, 288, 17065; Lewis, D. F. V.; Ito, Y.; Goldfarb, P. S.; Drug Dev. Res. 2005, 66, 19; Guengerich, F. P.; Chem. Res. Toxicol. 2001, 14, 611. 18. Levine, M. N.; Raines, R. T.; Anal. Biochem. 2011, 418, 247; Lavis, L. D.; Chao, T. -Y.; Raines, R. T.; ACS Chem. Biol. 2011, 1, 252. 19. Gao, W.; Xing, B.; Tsien, R. Y.; Rao, J.; J. Am. Chem. Soc. 2003, 125, 11146. 20. Para uma revisão sobre β-lactamases ver: Drawz, S. W.; Bonomo, R. A.; Clinical Microbiol. Rev. 2010, 160; Wilke, M. S.; Lovering, A. L.; Strynadka, N. C. J.; Curr. Opin. Microbiol. 2005, 8, 525; Livermore, D. M.; Brown, D. F. J.; J. Antimicrob. Chemother. 2001, 48, 59. 21. Andersson, H.; Baechi, H. T.; Hoechl, M.; Richter, C.; J. Microsc. 1998, 191, 1. 22. Xie, H.; Mire, J.; Kong, Y.; Chang, M.; Hassounah, H. A.; Thornton, C. N.; Scchettini, J. C.; Cirillo, J. D.; Rao, J.; Nat. Chem. 2012, 4, 802. 23. Urano, Y.; Kamiya, M.; Kanda, K.; Ueno, T.; Hirose, K.; Nagano, T.; J. Am. Chem. Soc. 2005, 127, 4888. 24. Para uma revisão sobre o processo de FRET ver: Kiyokawa, E.; Hara, S.; Nakamura, T.; Matsuda, M.; Cancer Sci. 2006, 97, 8; Jares-Erijman, E. A.; Jovin, T. M.; Nat. Biotechnol. 2003, 21, 1387; Föster, T. Discuss. Faraday Soc. 1959, 27, 7. 25. Karnmüller, E. K.; Ollé, E. P.; Bannwarth, W.; Chem. Commum. 2005, 5459; Mittoo, S.; Sundstrom, L. E.; Bradley, M.; Anal. Biochem. 2003, 319, 234; Matayoshi, E. O.; Wang, G.; T.; Krafft, G. A.; Erickson, J.; Science 1990, 247, 954. 26. Para uma revisão sobre proteases ver: Merdanovic, M.; Mönig, T.; Ehrmann, M.; Kaiser, M.; ACS Chem. Biol. 2013, 8, 19; Turk, B.; Turk, D.; Turk, V.; EMBO J. 2012, 31, 1630; Choi, K. Y.; Swierczwska, M.; Lee, S.; Chen, X.; Theranostics 2012, 2, 156; Deu, E.; Verdoes, M.; Bogyo, M.; Nat. Struct. Mol. Biol. 2012, 19, 9; Scott, C. J.; Taggart, C. C.; Biochimie 2010, 92, 1681; Turk, B. Nat. Rev. 2006, 5, 785. Puente, X. S.; Sánchez, L. M.; Overall, C. M.; López-Otin, C.; Nat. Rev. Genet. 2003, 4, 544. 27. Bánóczi, Z.; Alexa, A.; Farkas, A.; Friedrich, P.; Hudecz, F.; Bioconjugate Chem. 2008, 19, 1375. 28. Abreviações: A: Alanina (Ala); I: Isoleucina (Ile); L: Leucina (Leu); V: Valina (Val); F: Fenilalanina (Phe); W: triptofan (Trp); Y: tirosina (Try); N: Asparagina (Asn); C: Cisteína (Cys); Q: Glutamina (Gln); M: Metionina (Met); S: Serina (Ser); T: Treonina (Thr); D: Àcido Aspártico (Asp); E: Àcido Glutâmico (Glu); R: Arginina (Arg); H: Histidina (His); K: Lisina (Lys); G: Glicina (Gly); P: prolona (Pro). 29. Para uma revisão sobre CPP ver: Bechara, C.; Sagan, S. FEBS Lett. 2013, 587, 1693; Milletti, F. Drug Discovery Today 2012, 17, 850. 30. Bánóczi, Z.; Alexa, A.; Farkas, A.; Friedrich, P.; Hudecz, F.; J. Pept. Sci. 2010, 16 (S1), 197. 31. Yang, Y. Z.; P. Babiak, P.; Reymond, J. -L.; Org. Biomol. Chem. 2006, 4, 1746. 32. Maeda, T.; Nishimura, S. I.; Chem. Eur. J. 2008, 14, 478; Li, J. -J.; Bugg, T. D. H.; Chem. Commun. 2004, 182. 33. Boyer, V.; Fort, S.; Frandsen, T. P.; Schülein, M.; Cottaz, S.; Driguez, H.; Chem. Eur. J. 2002, 2, 1389. 34. Vaasa, A.; Lust, M.; Terrin, A.; Uri, A.; Zaccolo, M.; Biochem. Biophys. Res. Commun. 2010, 397, 750. 35. Chen, S.; Fahmi, N. E.; Wang, L.; Bhattacharya, C.; Benkovic, S. J.; Hecht, S. M.; J. Am. Chem. Soc. 2013, 135, 12924; Mizukami, S.; Watanabe, S.; Hori, Y.; Kikuchi, K.; J. Am. Chem. Soc. 2009, 131, 5016; Yudushkin, I. A.; Schleifenbaum, A.; Kinkhabwala, A.; Neel, B. G.; Schultz, C.; Bastiaens, P. I. H.; Science 2007, 315, 115. 36. Lemke, E. A.; Schultz, C.; Nat. Chem. Biol. 2007, 7, 480. 37. Gonzáles-Vera, J. A.; Lukovic, E.; Imperiali, B.; Bioorg. Chem. Lett. 2009, 1258; Lukovic, E.; González-Vera, J. A.; Imperiali, B.; J. Am. Chem. Soc. 2008, 130, 12821. 38. Para uma revisão sobre quinases e fosfatases ver: Guo, M.; Huang, B. X.; Proteomics 2013, 13, 424; Sacco, F.; Perfetto, L.; Castagnoli, L.; Cesareni, G.; FEBS Lett. 2012, 586, 2732; Schreiber, T. B.; Mäusbacher, N.; Breitkopf, S. B.; Grundner-Culemann, K.; Daub, H.; Proteomics 2008, 8, 4416; Jia, Z.; Biochem. Cell. Biol. 1997, 75, 17. 39. Cohen, P.; Alessi, D. R.; ACS Chem. Biol. 2013, 8, 96; Knight, Z. A.; Lin, H.; Shokat, K. M.; Nat. Rev. Cancer 2010, 10, 130. 40. Rhee, H. -W.; Lee, S. H.; Shin, I. -S.; Choi, S. J.; Park, H. H.; Park, T. H.; Hong, J. -I.; Angew. Chem. Int. Ed. 2010, 49, 4919. 41. Para uma revisão sobre PET ver: De Silva, P.; Moody, T. S.; Wright, G. D.; Analyst 2009, 134, 2385; Pearson, A. j.; Xiao, W.; J. Org. Chem. 2003, 13, 5361. 42. Crivat, G.; Taraska, J. W.; Trends Biotechnol. 2012, 30, 8; Romanini, D. W.; Cornish, V. W.; Nat. Chem. 2012, 4, 248; Chen, I.; Ting, A. Y.; Curr. Opin. Biotechnol. 2005, 16, 35. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access