
 |
 |
 |
 |
 |
Artigo
| Synthesis, characterization and application of ion imprinted poly(vinylimidazole) for zinc ion extraction/preconcentration with FAAS determination |
|
Queila O. dos SantosI; Marcos A. BezerraI; Giovana de Fátima LimaII; Kristiany M. DinizIII; Mariana G. SegatelliIII; Talitha O. GerminianoIII; Vivian da Silva SantosIV; César R. T. Tarley*, III, V
IDepartamento de Química e Exatas, Universidade Estadual do Sudoeste da Bahia, 45206-190 Jequié - BA, Brasil Recebido em 16/04/2013 *e-mail: ctarleyquim@yahoo.com.br In this paper, we describe the synthesis of an ion imprinted polymer (IIP) by homogeneous polymerization and its use in solid-phase to extract and preconcentrate zinc ions. Under optimal conditions (pH 5.0, preconcentration flow rate of 12.0 mL min-1, and eluted with 1.0 mol L-1 HNO3) this procedure allows the determination of zinc with an enrichment factor of 10.2, and with limits of detection and quantification of 1.5 and 5.0 µg L-1, respectively. The accuracy of our results was confirmed by analysis of tap water and certified reference materials: NIST 1570a (Spinach leaves) and NIST 1515 (Apple leaves). INTRODUCTION Zinc is an essential mineral nutrient for humans, plants and animals. A variety of biomolecules require zinc in order to maintain the normal structure, function and proliferation of cells. Both plant and animal growth require small quantities of zinc. Zinc has beneficial effects on cardio-circulatory function and prevents several diseases. Zinc can also play an important role in the progression of several bodily conditions, including disturbances in energy metabolism and increases in oxidative stress. Foods and drinking water are the main source of zinc for humans. However, this metal can be toxic for some biota in excessive amounts. Zinc is most toxic to microscopic organisms in aquatic environments. Water concentrations of zinc are reported to be a better indicator of fish contamination than zinc levels measured in sediments or invertebrates. Although toxic in excess, zinc is an essential component of thousands of proteins in plants. The determination of zinc concentrations in natural waters and vegetal matrices is very important, and can serve as a basis for characterizing regional pollution levels and for estimating how much zinc humans are ingesting. Zinc concentrations in unpolluted natural waters are very low.1-3 In natural surface waters, the concentration of zinc is usually below 10 µg L-1. The Brazilian environmental agency has established the maximum safe level of zinc in fresh waters to be 180 µg L-1 (Resolution CONAMA 357/2005). Very sensitive analytical techniques for detecting zinc levels are required, such as electrothermal atomic absorption spectrometry (ETAAS) or inductively coupled plasma-mass spectrometry (ICP-MS). Due to its inherent selectivity, simplicity and relatively low acquisition cost, flame atomic absorption spectrometry (FAAS) is widely employed for metal ion determination. In view of its reasonable sensitivity, combining solid-phase extraction with FAAS is also a promising tool for trace determination of metal ions.2,4 Separation/preconcentration procedures based on solid-phase extraction have been used to improve the analytical characteristics of zinc determination in several matrices.5 However, a disadvantage of this technique is its low metal selectivity, which can lead other species to interfere with the target metal ions when using techniques susceptible to lines superposition, such as inductively coupled plasma optical emission spectrometry (ICP OES). Ion imprinted polymers (IIPs) overcome these disadvantages in extraction procedures because they are capable of ion recognition, thus improving the selective separation/preconcentration of the analyte during solid-phase extraction (SPE). IIPs are obtained by the copolymerization of functional and cross-linking monomers in the presence of an ion template analyte, which traps the template in a highly cross-linked polymer matrix. Removing the template leaves back cavities having a size and shape corresponding to that of the ion template.6,7 These polymers have a high affinity and selectivity for the target ion, are easy to produce, are low cost and stable. Separation/preconcentration using IIPs has been employed, for example, in the separation of Fe3+ from plant and water samples with a posterior determination by ICP OES,7 of Dy3+ from synthesized seawater and spectrophotometric determination8, and of Cu2+, Ni2+, Pb2+ and Zn2+ from celery samples with determination by ICP-MS.9 In spite of the outstanding performance by IIPs for metal ion adsorption, a survey of the literature shows that few studies have been devoted to zinc preconcentration and further determination by FAAS. A Zn(II) IIP was recently prepared with acryloyloxyquinoline (8-AOQ) as the monomer.10 Shakerian et al.11 synthesized a new Zn(II) IIP using styrene as the monomer, ethylene glycol dimethacrylate (EGDMA) as the cross-linking agent and 8-hydroxyquinoline as the chelating agent for zinc. In these studies, auxiliary chelating agents must be used to form the binding sites for polymers and the synthesis procedures are very tedious. Therefore, in this study we propose a new method for synthesis, using homogeneous polymerization of a zinc ion-selective adsorbent with 1-vinylimidazole as the functional monomer, which is able to form a metal ion complex with zinc ions. We developed a preconcentration system for the enrichment and determination of zinc by FAAS using a mini-column loaded with this IIP and applied a Doehlert design to determine its best analytical characteristics.
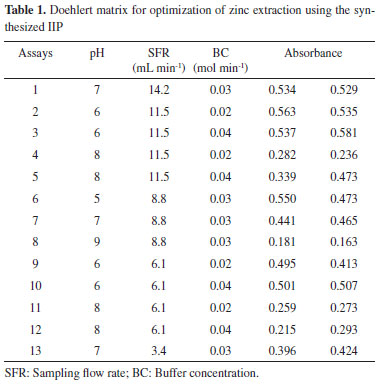
EXPERIMENTAL Instrumentation We used a Perkin Elmer (Norwalk, CT, USA, www.perkinelmer.com.br) AAnalyst model 200 flame atomic absorption spectrometer, equipped with a deuterium lamp for background correction to measure absorbance. In accordance with the recommendations of the manufacturer, we used a hollow cathode lamp for the zinc. The wavelength and spectral band pass used were 213.9 and 0.7 nm, respectively. The lamp current (5.0 mA) and the burner height (13.5 mm) we used had conventional values. The flame consisted of acetylene (flow rate 2.0 L min-1) and air (flow rate 13.5 L min-1). The flow rate used for the nebulizer was 5.0 mL min-1. Water for all applications was obtained from an Elga Purelab Classic purification system (High Wycombe, UK, www.elgalabwater.com) and its resistivity was equal to or higher than 18.2 MΩ cm. The IIP particle size was measured using an FEI Quanta 200 (Netherlands, www.fei.com) scanning electron microscope operating at 30 kV voltage. A manifold from a Baltec Scutt Coater SCD 050 (Germany, www.emsys.co.uk) was used to metalize the studied material. Infrared spectra were obtained in KBr pellets and then measured using a spectrophotometer Shimadzu (www.shimadzu.com.br) FTIR-8300 Infrared Fourier Transform with a resolution of 4 cm-1. Preconcentration was performed in a system consisting of a multi-channel peristaltic pump (Model BP 200, Milan, Colombo, Paraná/Brazil, www.longerpump.com), silicone (0.25 mm id) and Teflon tubing to propel the liquids, and a laboratory-made mini-column. A Digimed DM 20 (Santo Amaro, Brazil) pH meter was used to measure the pH of the solutions. Reagents, solutions and materials Ultrapure water was used to prepare all solutions. All the glassware was kept overnight in a 5% (v/v) nitric acid solution prior to use to remove any residual metals. All reagents used were of analytical grade. Working solutions of zinc were prepared daily by diluting 1000 µg mL-1 metal stock solutions (Merck, Darmstadt, Germany, www.merck.com.br). Hydrochloric acid solutions were prepared by direct dilution of the concentrated solution (Merck) with ultrapure water. An ammonia buffer solution (pH 8.0-10.0), sodium borate (pH 7.0-8.0) and sodium acetate (pH 4.5-5.7) were used to adjust the pH of metal solutions. For the accuracy studies, we analyzed certified reference materials-NIST 1570a (Spinach leaves) and NIST 1515 (Apple leaves) from the National Institute of Standards and Technology (www.nist.gov), NIST, USA. Synthesis of the IIP The IIP was prepared by bulk polymerization, according to the described procedure with modifications.12 The synthesis was carried out by dissolving 2.25 mmol ZnCl2 and 4.5 mmol of 1-vinylimidazole in 8.0 mL of acetonitrile and stirring for 30 minutes in order to form the Zn-vinylimidazole complex. Then, 42 mmol of the cross-linking reagent ethylene glycol dimethacrylate (EGDMA) and 20 mg of the radical indicator 2,2 '-azo-bis-iso-butyronitrile (AIBN) were added to mixture. The mixture was purged with nitrogen for 10 min and tightly enclosed in this atmosphere. The reaction was carried out at a temperature of 60 ºC for 12 hours. Subsequently the polymer was ground and sieved (particles smaller than 100 microns). Zinc ions retained in the polymer were removed by successive washings with 1.0 mol L-1 HNO3 and then monitored by FAAS. Finally, the polymers were dried for 12 h in an oven at 45 ºC. The imprinting effect, produced in the polymer, was evaluated by preparing the non-imprinted polymer (NIP), i.e. the blank copolymer, which was submitted to the same protocol for synthesis, but without adding any ZnCl2. Mini-column preparation A cylindrical mini-column 3.0 cm in length, with an internal diameter of 4.0 mm, and containing about 100 mg of the synthesized IIP was used in the separation/preconcentration procedure, along with a syringe to put the sorbent inside the mini-column. Tissues (500 µm) were placed at the inlet and outlet of the mini-column to avoid any removal of the solid-phase material by the carrier stream. After the experiment, the mini-column was washed with ethanol 5% (v/v), nitric acid solution, and deionizer water, respectively at 2.5 mL min-1 flow rate. Washing with ethanol and nitric acid was done to prevent any metal or organic contamination, respectively. All mini-columns prepared in this way showed good reproducibility. Sample collection and preparation Samples of tap and river water were collected directly from the urban area of Jequié/Bahia. River water was collected by immersing bottles into the water stream. The polyethylene flasks used had been previously decontaminated with nitric acid solution (10% v/v) and rinsed with ultrapure water. Samples were filtered through a membrane (0.45 µm pore diameter) using a vacuum system after sampling to remove suspended particulate material. Later, 40.0 mL of these samples were immediately analyzed after the addition of the buffer and dilution to 50.0 mL in a volumetric flask. Plant samples (0.10 g) were digested by adding 2.0 mL of concentrated nitric acid and hydrogen peroxide to the sample and taking them to a hot plate. Blank samples were prepared analogously. Sodium hydroxide and an appropriate buffer solution were used to adjust the pH of the final digestion solution. The mixture was finally diluted to 100.0 or 200.0 mL (depending on the metal concentration of the sample) with ultrapure water. These solutions were analyzed immediately after preparation. Solid-phase extraction optimization using Doehlert design Doehlert designs, which are frequently used for analytical method optimization, provide a uniform distribution of points over an entire experimental region arranged in a rhomboidal figure. In the case of three variable designs, a cuboctahedron is geometrically produced. To optimize the solid-phase extraction method for zinc using the IIP method, we used a Doehlert matrix wherein experimental coordinates originate by the plane projection of a cuboctahedron on its squared face.13-16 The optimized variables were pH (from 5 to 9), sampling flow rate (from 3.4 to 14.2 mL min-1), and buffer concentration (BC, from 0.02 to 0.04 mol L-1). The experimental design and the responses obtained (in duplicate) from the Doehlert matrix are presented in the Table 1.
General procedure after the optimization process A 40.0 mL sample solution containing the analyte was buffered at a pH of 5.0 with an acetate buffer solution at 0.02 mol L-1 concentration and pumped at 12.0 mL min-1, percolating through the IIP mini-column that retained the Zn(II) ions. The remaining solution was discharged. The elution step was carried out by using 1.0 mL of 1.0 mol L-1 HNO3 at a flow rate of 12.0 mL min-1. The choice of HNO3 as the eluent was based on previous experiments in which we observed a better elution capacity for zinc from the mini-column using HNO3 than from HCl, both at 1.0 mol L-1 concentration and without memory effects. The eluates were taken into a 2 mL recipient and metal determination was carried out by FAAS. Signals were recorded as absorbance, using the instrument software.
RESULTS AND DISCUSSION IIP characterization In addition to the analysis resulting from the scanning electron microscopy and infrared spectroscopy, we also characterized the synthesized IIP and NIP. Figure 1 verifies that the polymeric particles are very cohesive, presenting themselves as tightly-packed nanoparticles. The adsorbents also have a rough surface due to the pores formed in the IIP and NIP networks. The visual roughness of the IIP was slightly higher than the NIP, most likely due to the imprinting effect.
 Figure 1. Micrographs for the IIP (a) and the NIP (b). Magnification of 50000 times
The presence of pores in the adsorbent facilitates ion transfer, promotes a higher adsorption of the metal, and allows a better interaction of the ion with the active sites of the IIP. The polymer prepared by the bulk method yields particles with irregular shapes, but considering this study's solid-phase extraction, which works with low pressure, this feature is not considered a disadvantage, as observed on the packed chromatographic column. The Fourier transform-infrared (FT-IR) spectra of the NIP, the IIP-unleached and the IIP-leached are shown in Figure 2. A similar spectral profile was observed for the polymers. The signal at 3450 cm-1 is attributed to the absorption of OH stretching and at 2997 and 2952 cm-1 the signals are related to the CH stretch of the sp2 and sp3 bonds from the EGDMA. The absorption at 1635 cm-1 is assigned to the C=C and C=N (ring), and at 962 and 765 cm-1 to the C-H (ring) bending out-of-plane.12,17 Other important bands at 1726, 1458 and 1166 cm-1 are attributed to the C = O stretch, asymmetric CH deformation and C-O deformation, respectively. The peak at 1259 cm-1 is related to the ring vibration.18 The peak at 1380 cm-1 is attributed to C=N stretching modes from the ring. A very low intensity peak was observed for the loaded IIP, suggesting the interaction of zinc with the imidazole ring.
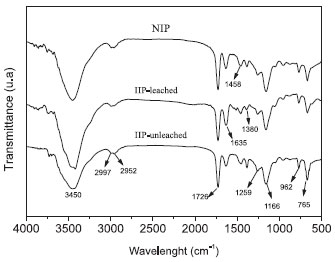 Figure 2. Infrared spectra for the NIP, the IIP-leached and the IIP-unleached
Solid-phase extraction optimization The performance of solid-phase extraction depends on some experimental variables-principally pH, sampling flow rate, and buffer concentration in the final solution. Thus, multivariate methodologies allow us to achieve the best conditions more efficiently than univariate methodologies, and they also allow the evaluation of the interaction effects. Table 1 presents the results of experiments carried out according to the level combinations imposed by the design application. Linear and quadratic functions were fitted to the experimental data to describe the experimental field. According to the analysis of variance (ANOVA) (Table 2), the linear model showed lack of fit (Fcalculated > Ftabled) and left large residues. The quadratic model showed no lack of fit (Fcalculated <Ftabled) so we chose it to describe the data behavior in the experimental region. Figures 3a and 3b show the response surfaces generated by the quadratic model.
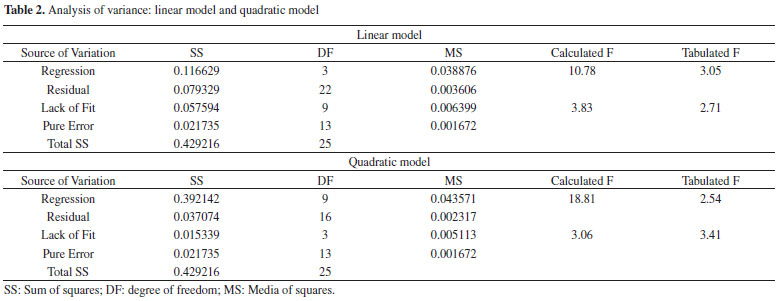
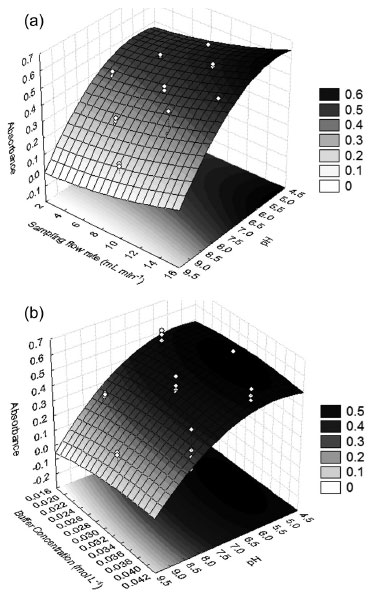 Figure 3. Response surfaces obtained after fitting a quadratic function to the experimental data: (a) sampling flow rate X pH and (b) buffer concentration X pH
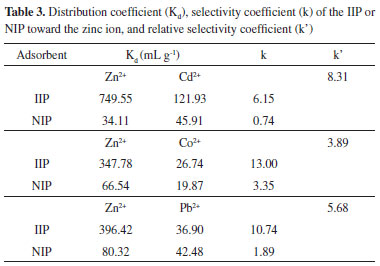
Surface and regression coefficients analysis reveals, as expected, that pH is the variable that most influences the extraction of zinc by the IIP. The buffer concentration and flow rate sampling do not have any significant effects in the experimental region studied. Since the response surface is characterized by a saddle point, optimum conditions were obtained by visual inspection of this surface. Thus, we chose the following values as optimum conditions for zinc extraction: a pH of 5.0, a buffer concentration of 0.02 mol L-1, and a sampling flow rate of 12.0 mL min-1. Evaluation imprinting effect on selectivity of ion imprinted poly(vinylimidazole) To perform studies dedicated to the selectivity of ion imprinted poly(vinylimidazole), batch adsorption experiments were performed. For this task, 100 mg of the IIP or NIP adsorbents were stirred with a binary mixture of 25.0 mL of 1.5 mg L-1 Zn2+ solution in the presence of Cd2+, Pb2+, or Co2+ ions at the same concentration. An acetic/acetate buffer solution at 0.02 mol L-1 maintained the pH of the solution at 5.0. The mixture was equilibrated by stirring at 2000 rpm for 60 min at room temperature. Then, the supernatants were collected into polyethylene vials and analyzed by FAAS, and the content of the zinc and co-existing ions retained on the adsorbents were determined according to literature reports.17 In addition, we determined the distribution coefficient (Kd), the selectivity coefficient (k) of the IIP or NIP toward the zinc ion, and the relative selectivity coefficient (k').17 From Table 3, we observe a higher selectivity coefficient (k) for the IIP to the detriment of the NIP with the consequent relative selectivity coefficient (k') greater than unity. These results reveal that the IIP for the Zn2+/Cd2+, Zn2+/Co2+, and Zn2+/Pb2+ systems was 8.31, 3.89, and 5.68, respectively; showing clearly that the IIP is more selective than non-imprinted polymer.
Analytical characteristics and application of the preconcentration method The analytical features of the proposed procedure were assessed after optimization. The concentrations of the analytical standards for the preconcentration procedure ranging between 5.0 and 50.0 µg L-1 yielded the follow equation Abs = 0.0225 CZn + 0.037 (R=0.9988). Without this preconcentration step, the concentrations of the calibration standards ranged between 200 and 1000 µg L-1 and the equation was Abs = 0.0022 CZn + 0.061. The limits of detection (LOD) and quantification (LOQ) are defined as LOD = 3 σ / S and LOQ = 10σ / S, respectively, where S is the slope of the analytical curve, and σ is the standard deviation of ten consecutive measurements of the blank signal. For 50.0 mL of sample solution used in the preconcentration step, we obtained an LOD of 0.88 µg L-1, an LOQ of 3.0 µg L-1, and a precision (repeatability) of 2.8%, expressed as relative standard deviation (n = 10, 20.0 µg L-1). The enrichment factor, defined as the ratio of the slopes of the analytical curves with and without the pre-concentration, was 10.2. A comparison of the slopes of the analytical curves built for the IIP and for NIP (Abs = 0.0017 CZn + 0.0016) shows an increase by a factor of 13 in the sensitivity, demonstrating the improved efficiency in the imprinting process using the solid phase extractor. Direct determination of zinc without a preconcentration step offers values of 39.0 and 130.0 µg L-1 for the LOD and LOQ, respectively, showing that an enrichment step is necessary for zinc determination at low concentrations. The accuracy of the proposed procedure was assessed by addition/recovery tests and analysis of the following certified reference materials: NIST 1570 (Spinach leaves) and NIST 1515 (Apples leaves). Results are presented in the Table 4.
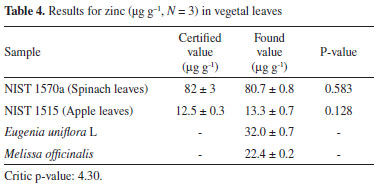
The values obtained in this study agree well with the certified values reported for these certified reference materials. A paired t-test was applied to evaluate these results. At a 95% confidence level, the difference in the population means is not significantly different for the certified and our experimental results. We also checked the accuracy of the analysis of the spiked tap water samples, which also showed good recoveries values of 102 and 111% (Table 5).
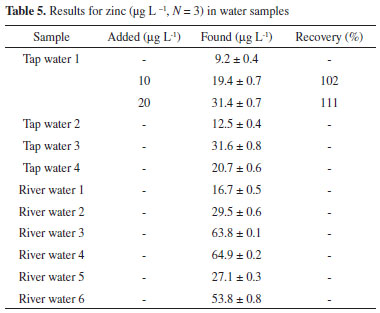
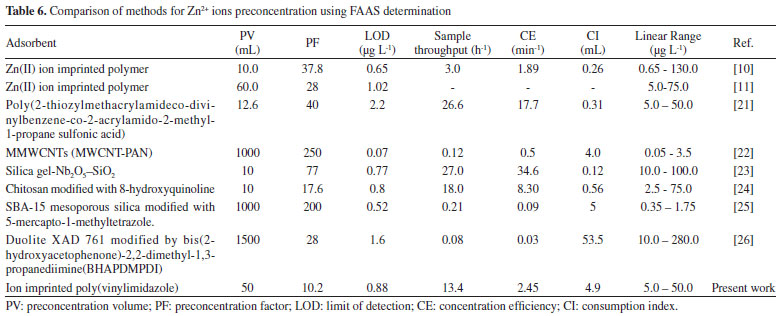
In summary, our proposed method was successfully applied for the determination of zinc in tap water, river water and in two vegetal leaves used for tea preparation (Melissa officinalis, and Eugenia uniflora L). All samples were collected in the city of Jequié, Bahia, Brazil. The results are given in Tables 4 and 5, and the content of the zinc ions found in these samples are similar to those found in previous publications.19,20 Table 6 shows a comparison of a number of methods for zinc ion preconcentration using FAAS determination. As observed, our developed method exhibits better or comparable figures of merit, primarily when considering the LOD, sample consumption and sample throughput. Furthermore, this preconcentration method also corroborates the concept of Clean Chemistry, since it requires no auxiliary chelating agent prior to the preconcentration step or toxic organic solvent as an eluent.
CONCLUSIONS In this study, a new ion imprinted poly(vinylimidazole) synthesized via a bulk procedure was successfully applied to preconcentrate zinc ions from water and vegetal leaves samples. The application of a Doehlert matrix allowed the efficient optimization of the preconcentration procedure, making it suitable for zinc determination by FAAS with a low LOD, low sample consumption and high sample throughput. The increment in the zinc selectivity was assured from the relative selectivity coefficient (k') obtained. The achieved zinc concentrations in the certified reference materials were consistent with those reported, confirming the method's accuracy. Results from the analysis of real samples have shown that the developed method can be successfully applied to determine zinc in these kinds of samples.
ACKNOWLEDGEMENTS The authors gratefully acknowledge the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), the Fundação Araucária de Apoio ao Desenvolvimento Científico e Tecnológico do Paraná, the Instituto Nacional de Ciência e Tecnologia em Bioanalítica (INCT-Bio), and the Fundação de Amparo à Pesquisa do Estado da Bahia (FAPESB) for financial support and fellowships.
REFERENCES 1. Kaim, W.; Schwederski. B.; Bioinorganic Chemistry: Inorganic Elements in the Chemistry of Life, Wiley: New York, 1994. 2. Lemos, V. A.; Santos, W. N. L.; Santos, J. S.; Carvalho, M. B.; Anal. Chim. Acta 2003, 481, 283. 3. Dong, Y. G.; Tsugufumi, O.; Lin D. Z., Koh, H. G.; Hiroshi, K. M.; Mitsuhiro, S. H.; Field Crop. Res. 2006, 95, 420. 4. Vandecasteele C.; Block, C. B.; Modern methods for trace element determination, John Wiley & Sons: Chichester, 1993. 5. Lemos, V. A.; Teixeira, L. S. G.; Bezerra, M. A.; Costa, A. C. S.; Castro, J. T.; Cardoso, L. A.; Jesus, D. S.; Santos, E. S.; Baliza, P. X.; Santos, L. N.; Appl. Spectrosc. 2008, 43, 303. 6. Prasada, R. T.; Kala, R.; Daniel, S.; Anal. Chim. Acta 2006, 578, 105. 7. Chang, X.; Na, J.; Zheng, H.; He, Q.; Hu, Z. ; Zhai, Y. ; Cui, Y.; Talanta 2007, 71, 38. 8. Biju, V. M.; Gladis, J. M.; Rao, T. P.; Anal. Chim. Acta 2003, 478, 43. 9. Otero-Romaní, J.; Moreda-Piñeiro, A.; Bermejo-Barrera, P.; Martin-Esteban, A.; Talanta 2009, 79, 723. 10. Zhao, J.; Han, B.; Zhang, Y.; Wang, D.; Anal. Chim. Acta 2007, 603, 87. 11. Shakerian, F.; Dadfarnia, S.; Mohammad, A.; Shabani, H.; Food Chem. 2012, 134, 488. 12. Segatelli, M. G.; Santos, V. S.; Presotto, A. B. T.; Yoshida, I. V. P.; Tarley, C. R. T.; React. Funct. Polym. 2010, 70, 325. 13. Tarley, C. R. T.; Silveira, G.; dos Santos, W. N.; Matos, G. D.; da Silva, E. G.; Bezerra, M. A.; Miró, M.; Ferreira, S. L. C.; Microchem. J. 2009, 92, 58. 14. Ferreira, S. L. C.; Bruns, R. E.; Silva, E. G. P.; Santos, W. N. L.; Quintella, C. M.; David, J. M.; Andrade, J. B.; Breitkreitz, M. C.; Jardim, I. C. S. F.; Neto, J. B. B.; J. Chromatogr. A 2007, 1158, 2. 15. Bezerra, M. A.; Santelli, R. E.; Oliveira, E. P. ; Villar, L. S.; Talanta 2008, 76, 965. 16. Santos, W. J. R.; Lima, P. R.; Tarley, C. R. T.; Kubota, L. T.; Anal. Bioanal. Chem. 2007, 389, 1919. 17. Tarley, C. R. T.; Andrade, F. N.; Santana, H.; Zaia, D. A. M.; Beijo, L. A.; Segatelli, M. G.; React. Funct. Polym. 2012, 72, 83. 18. Wu, K. H.; Chang, T. C. ; Wang, Y. T.; Hong, Y. S.; Wu, T. S.; Eur. Polym. J. 2003, 39, 239. 19. Tarley, C. R. T.; de Ávila, T. C.; Segatelli, M. G.; Lima, G. F.; Peregrino, G.; Scheeren, C. W.; Dias, S. L.; Ribeiro, E. S.; J. Braz. Chem. Soc. 2010, 21, 1106. 20. Pylakowskaa, K.; Kitaa, A.; Jonoska, P.; Polowniaka, M.; Kozik, V.; Food Chem. 2012, 135, 494. 21. Yilmaz, S.; Tokalioglu S.; Sahan, S.; Ulgen, A.; Sahan, A.; Soylak, C.; J. Trace Elem. Med. Biol. 2013, 27, 85. 22. Tajik, S.; Taher, M. A.; Desalination 2011, 278, 57. 23. Dutra, R. L.; Maltez, H. F.; Carasek, E.; Talanta 2006, 69, 488. 24. Carletto, J. S. ; Roux, K. C.; Maltez, H. F.; Martendal, E.; Carasek, E.; J. Hazard. Mater. 2008, 157, 88. 25. Pérez-Quintanilla, D.; Sánchez, A.; Del Hierro, I.; Fajardo, M.; Sierra, I.; J. Hazard. Mater. 2009, 166, 1449. 26. Ghaedi, M.; Mortazavi, K.; Montazerozohori, M.; Shokrollahi, A.; Soylak, M.; Mat. Sci. Eng. C 2013, 33, 2338. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access