
 |
 |
 |
 |
 |
Nota Técnica
|
|
| Análise de fármacos em águas por SPE-UPLC-ESI-MS/MS Analysis of pharmaceutical compounds in water by SPE-UPLC-ESI-MS/MS |
|
Vanessa de Jesus GaffneyI; Vitor Vale Cardoso*, II; Alexandre RodriguesII; Elisabete FerreiraII; Maria Joao BenolielII; Cristina M. M. AlmeidaI
IInstitute for Medicines and Pharmaceutical Sciences, Faculdade de Farmácia da Universidade de Lisboa, Av. Prof. Gama Pinto, 1600-049 Lisboa, Portugal Recebido em 04/03/2013 *e-mail: vitorcar@epal.pt A method was developed for the analysis of 31 pharmaceutical compounds in Lisbon's drinking water system, using solid-phase extraction (SPE) and ultra-performance liquid chromatography coupled to tandem mass spectrometry (UPLC-MS/MS). The method was validated through estimation of the linearity range, method detection and quantification limits, matrix effects, precision and accuracy. The method detection and quantification limit ranges were 0.009-10 and 0.03-33 ng/L, respectively. Analytes were quantified in water samples collected from the EPAL (Empresa Portuguesa das Aguas Livres S.A.) supply system. Carbamazepine, atenolol, sulfadiazine, sulfamethazine, sulfapyridine, sulfamethoxazole, acetaminophen, caffeine and erythromycin were quantified in the analysed samples. INTRODUÇÃO Os meios hídricos são um meio receptor de esgotos domésticos, industriais e agropecuários e, consequentemente, são o principal veículo de dispersão ambiental dos poluentes químicos. Por este motivo, a problemática do ambiente e, mais concretamente, da água, continua a ser um tema actual a nível nacional e internacional. O desenvolvimento de novos equipamentos e métodos analíticos que permitem a identificação e quantificação de resíduos de compostos químicos em concentrações cada vez menores e os dados sobre os seus efeitos biológicos têm, no entanto, chamado a atenção da comunidade científica para novos contaminantes considerados emergentes. Os "Contaminantes Emergentes" são contaminantes não regulamentados, que se podem tornar candidatos para futuras regulamentações, dependendo dos resultados dos estudos sobre a sua toxicidade e/ou efeito nefasto sobre o meio ambiente e na saúde humana e animal e de dados de monitorização relativos à sua ocorrência no meio ambiente. Este grupo de compostos é essencialmente constituído por substâncias utilizadas no dia-a-dia, sendo que para a maioria ainda não se possui dados ecotoxicológicos que permitam prever os seus efeitos na saúde humana e animal.1 O termo "Contaminante Emergente" também se pode referir a compostos recentemente introduzidos no meio ambiente (por exemplo, medicamentos aprovados recentemente pelas autoridades competentes e que devido ao seu uso começam a entrar no ambiente) ou a compostos que se encontram presentes no meio ambiente há já algum tempo, mas cuja presença só recentemente tem sido detectada. O termo "emergente" pode também englobar um poluente convencional já legislado, mas que devido a novos dados referentes à sua ocorrência, destino ou efeitos adversos, tornou-se de novo foco de atenção.2 A descarga de contaminantes químicos emergentes para o meio aquático tem sido alvo de preocupação por parte da comunidade científica internacional, uma vez que, anualmente são sintetizados numerosos compostos químicos, os quais são lançados para o meio ambiente com consequências imprevisíveis, surgindo constantemente novas evidências sobre a sua toxicidade. Os contaminantes emergentes englobam uma diversidade de compostos, incluindo cianotoxinas, micotoxinas, aditivos de gasolina e industriais, retardantes de chama bromados, hormónios esteróides, surfactantes, produtos farmacêuticos e de cuidado pessoal, compostos polifluoretados, nanomateriais e subprodutos de desinfecção, bem como os produtos de transformação resultantes dos mesmos.3 No grupo dos contaminantes emergentes tem sido dada especial atenção aos fármacos, os quais são permanentemente libertados para o meio ambiente, tendo sido desenvolvidos vários estudos do seu impacto ambiental em nível mundial.4-9 Os estudos realizados até à data têm identificado, em diferentes matrizes, nomeadamente, águas residuais, águas superficiais e subterrâneas destinadas à produção de água para consumo humano, sedimentos e biota, vários fármacos de diferentes classes terapêuticas, incluindo analgésicos, anti-inflamatórios, antibióticos, antidepressores, antiepilépticos, bloqueadores beta, reguladores lipídicos, meios de contraste radiológico, contraceptivos orais, broncodilatadores, citotóxicos e medicamentos para uso veterinário.10 A presença de resíduos de fármacos no meio ambiente pode apresentar efeitos adversos em organismos aquáticos e terrestres. Este efeito pode ocorrer em qualquer nível da hierarquia biológica, tais como célula, órgão, organismo, população e ecossistema, e pode ser observado em concentrações na ordem de ng/L, para certos tipos de compostos.11,12 A presença destes contaminantes no meio ambiente pode representar um risco para os ecossistemas e direta ou indiretamente para o Homem, porque, contrariamente à maioria dos poluentes, eles foram concebidos para terem uma acção específica no corpo humano e actuarem em concentrações muito baixas. A exposição crónica a concentrações baixas de certas classes de fármacos existentes no meio ambiente, como os antineoplásicos, hormónios, antidepressores, antibióticos, analgésicos, anti-inflamatórios, antipiréticos e reguladores lipídicos, podem originar efeitos muito adversos na saúde humana, nomeadamente, lesão celular, desregulação endócrina, infertilidade, alteração comportamental, resistência aos antibióticos e alteração da pressão arterial, entre outros, o que torna relevante e necessário o estudo da ocorrência destes compostos no meio ambiente e estudos de avaliação do risco de exposição.13,14 Os fármacos que possam surgir nas águas brutas e na água de consumo humano possuem as seguintes propriedades específicas: elevada estabilidade química, baixa biodegradabilidade, elevada solubilidade em água e baixos coeficientes de absorção/adsorção. A maior fonte de entrada de fármacos no ambiente aquático é através das estações de tratamento de águas residuais (ETARs). Uma vez nas ETARs, os fármacos e respectivos metabolitos podem ser degradados, parcialmente degradados ou resistir aos processos de tratamento, permanecendo inalterados.11 Os fármacos podem ser degradados por meio de processos bióticos (filtros biológicos ou outros) e abióticos (oxidação, hidrólise ou fotólise). Os produtos de degradação são também compostos preocupantes, dado que podem ter uma toxicidade equivalente ou superior ao do fármaco original.15 A maioria dos estudos publicados tem sido realizada na Europa16-23 e Estados Unidos da América,24-27 existindo também estudos realizados na Ásia28 e na América do Sul.29,30 As matrizes monitorizadas são principalmente afluentes e efluentes de estações de tratamento de águas residuais16,23,26,31 e águas superficiais,6,16,17,22,23,26,31-37 sendo encontrados, em menor número, dados sobre a contaminação de águas subterrâneas37-39 e águas para consumo humano.4,5,30,40 A Tabela 1 apresenta um resumo dos fármacos detectados em águas para consumo humano em diversos estudos de ocorrência efectuados a nível mundial.4,5
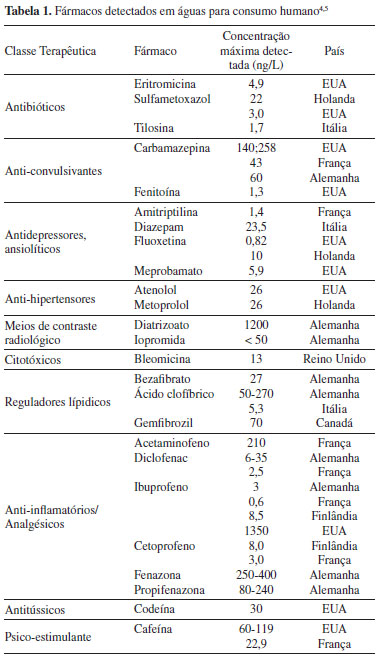
A curto e médio prazo poderão vir a ser tomadas medidas legislativas relativas aos limites de descarga de medicamentos no meio ambiente por parte da indústria, agropecuária e hospitais e desenvolver uma política de sensibilização do cidadão para a eliminação correcta dos medicamentos excedentes e/ou fora de prazo. Na área das estações de tratamento da água de consumo e da água residual existem novas tecnologias na área do tratamento da água, que permitem a remoção destes compostos de modo mais eficaz. No entanto, estas novas tecnologias exigem investimentos e custos de exploração bastante elevados às Entidades Gestoras de águas de consumo e de águas residuais. Em termos de legislação comunitária aplicável ao meio aquático, a Directiva 2000/60/CE de 23 de Outubro,41 estabelece um quadro de acção comunitária no domínio da política da água, designada resumidamente por Directiva-Quadro da Água (DQA). A transposição da DQA para o direito nacional é assegurada pela Lei nº 58/2005,42 de 29 de Dezembro, e pelo Decreto-Lei nº 77/200643 de 30 de Março, que estabelecem as bases para a gestão sustentável das águas e definem o novo quadro institucional para o sector. A Directiva 2008/105/CE44 transposta para o direito nacional pelo Decreto-Lei 103/2010 de 24 de Setembro de 201045 estabelece uma lista de substâncias prioritárias em vigor, que deverão ser monitorizadas nas águas superficiais, bem como normas de qualidade ambiental (NQA). Nesta lista não se encontra incluído qualquer fármaco. No entanto, de acordo com o ponto 4º do artigo 16º da DQA, a Comissão Europeia tem de rever a lista de substâncias prioritárias referidas no Anexo X da Directiva 2008/105/CE,44 de quatro em quatro anos. Desta forma, foi elaborada uma proposta para uma nova Directiva, datada de 31 de Janeiro 2012, que visa alterar as Directivas 2000/60/CE41 e 2008/105/CE44 no que diz respeito às substâncias prioritárias no domínio da política da água.46 Esta proposta refere os hormónios 17-α-etinilestradiol e 17-β-estradiol e o anti-inflamatório não-esteróide diclofenac como substâncias prioritárias, estabelecendo como normas de qualidade ambiental valores médios anuais (NQA-MA) de 3,5x10-5 µg/L, 4x10-4 µg/L e 0,1 µg/L em águas de superfície interiores e valores médios anuais de 7x10-6 µg/L, 8x10-5 µg/L e 0,01 µg/L nos outros tipos de águas de superfície para 17-α-etinilestradiol, 17-β-estradiol e diclofenac, respectivamente.46 A Directiva 98/83/CE47 de 3 de Novembro e o actual Decreto-Lei 306/2007 de 27 de Agosto48 relativos à qualidade da água para consumo humano não fazem referência à monitorização de quaisquer fármacos. No entanto, independentemente de quaisquer riscos, deve ser normalmente aplicado o princípio da precaução, e é desejável que os micropoluentes orgânicos, qualquer que sejam as suas fontes, não devam estar presentes na água de consumo humano em concentrações que possam ser prejudiciais para a saúde humana. A aceitação pública e a confiança na qualidade da água para consumo humano também terão de ser tidas em consideração. Actualmente, as técnicas instrumentais de análise permitem a concentração de alguns litros de amostras de água para um extracto de alguns mL ou µL, permitindo detectar compostos com concentrações na ordem dos ng/L ou menores. A associação da cromatografia gasosa (GC) ou líquida (LC) à espectrometria de massas (MS) permitiu a aplicação de técnicas instrumentais hifenadas (GC-MS, LC-MS, GC-MS/MS, LC-MS/MS), na análise ambiental, que, pelas suas capacidades de medição, permitiram a detecção de novos contaminantes emergentes no meio ambiente.13,49,50 Em simultâneo com o avanço da instrumentação analítica, em termos de sensibilidade e de especificidade nos sistemas de detecção, têm sido desenvolvidas novas técnicas de preparação de amostra que permitem extrair e concentrar os compostos de interesse a partir de diversas matrizes, como águas de consumo, águas naturais, ou ainda mais complexas, como águas residuais, solos, sedimentos e biota. Em alternativa às técnicas tradicionais de extracção líquido-líquido (LLE - Liquid-Liquid Extraction) ou de extracção sólido-líquido por Soxhlet, existem técnicas de extracção mais rápidas e amigas do ambiente, que requerem o uso de um menor volume de solventes, como a extracção em fase sólida (SPE - Solid Phase Extraction), a microextracção em fase sólida (SPME - Solid Phase Microextraction), a extracção assistida por micro-ondas (MAE - Microwave-Assisted Extraction) ou a extracção pressurizada com solvente (PLE - Pressurized Liquid Extraction). A EPAL - Empresa Portuguesa das Águas Livres, SA é responsável pela gestão do sistema de captação, tratamento, transporte e abastecimento de água a cerca de 2,9 milhões de pessoas em trinta e cinco municípios da região da Grande Lisboa. A monitorização da qualidade da água em toda a extensão do sistema de abastecimento da EPAL, desde os recursos hídricos até ao ponto de entrega ao consumidor, constitui uma das grandes preocupações da empresa. Desta forma, a política da empresa passa pelo desenvolvimento de métodos analíticos que permitam a pesquisa de diversos compostos não legislados, permitindo assim uma caracterização pormenorizada da água nas suas origens, a avaliação dos compostos que poderão passar a barreira das estações de tratamento de água e um controlo periódico destes compostos na água para consumo humano. Assim, este trabalho teve por objectivo a otimização e validação do método de SPE-UPLC-(ESI)-MS/MS para a análise de 35 fármacos, de forma a caracterizar as águas captadas e distribuídas pela Entidade Gestora de Abastecimento Público em Lisboa, assim como implementar, a curto prazo, a monitorização destes Contaminantes Emergentes na rotina do controlo de qualidade da empresa. O método foi validado de acordo com as normas nacionais ou internacionais adoptadas para este tipo de métodos de ensaio. Os fármacos foram seleccionados de acordo com vários critérios, nomeadamente, dados de consumo, avaliação da massa excretada, representatividade das classes terapêuticas, dados de toxicidade e de ocorrência no meio ambiente, compostos propostos para inclusão na proposta de alteração da DQA e persistência no meio ambiente. Deste modo, o estudo incluiu fármacos de diversas classes terapêuticas, como antibióticos (amoxicilina, doxiciclina, oxitetraciclina, eritromicina, sulfadiazina, sulfamerazina, sulfametazina, sulfametoxazol, sulfapiridina, sulfatiazol, ciprofloxacina), anti-diabético (metformina), anti-convulsivante (carbamazepina), antidepressivo (fluoxetina), anti-hipertensores (atenolol, metoprolol, propranolol), anti-inflamatórios (ibuprofeno, cetoprofeno, naproxeno, diclofenac, indometacina, nimesulida, cortisona, hidrocortisona, prednisona, prednisolona), analgésicos/antipiréticos (acetaminofeno, ácido salícilico), anti-dislipidémicos (ácido clofíbrico, bezafibrato, gemfibrozil), hormónios sexuais (gestodeno, testosterona) e estimulantes do sistema nervoso central (cafeína) (Tabela 1S).
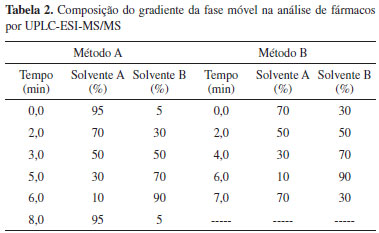
PARTE EXPERIMENTAL Equipamento Cromatógrafo líquido de ultra eficiência, modelo Waters UPLC Acquity System (Waters), associado a um espectrómetro de massa (MS/MS), Mass Spectrometer Acquity TQD com sonda de ionização por electrospray (ESI) e analisador de triplo quadrupolo (Waters). Coluna cromatográfica, Acquity BEH C18 (2,1 x 50 mm, 1,7 µm dp) (Waters). Solventes e padrões Os padrões utilizados apresentam um grau de pureza superior ou igual a 97% e são pró-análise (p.a.). Os solventes utilizados são adequados para análise por cromatografia gasosa (GC) ou para análise por cromatografia líquida (LiChrosolv® ou EMPROVE®). Água ultra pura (Millipore), acetato de amónio (CH3COONH4) p.a. (98%), acetona (CH3COCH3) (GC), ácido fórmico (HCOOH) EMPROVE®, ácido clorídrico fumante 37% p.a. (HCl), dimetilsulfóxido p.a.(CH3)2SO), metanol (CH3OH) LiChrosolv® (Merck, Alemanha). Todos os padrões de fármacos são de qualidade analítica adequada à análise por cromatografia e, como são fármacos, alguns obedecem às especificações das farmacopeias (Americana - USP, Inglesa - BP), embora pertencentes a marcas diferentes: (Sigma-Aldrich, Espanha) acetaminofeno (C8H9NO2) 98-101%, ácido clofíbrico (C10H11ClO3) > 97%, ácido salicílico (C7H6O3) > 99%, atenolol (C14H22N2O3) > 98%, bezafibrato (C19H20ClNO4) > 98%, carbamazepina (C15H12N2O) > 98%, (S)-(+)-cetoprofeno (C16H14O3) > 99%, cortisona (C21H28O5) >98%, diclofenac (C22H24Cl2NNaO2) > 98%, eritromicina (C37H67NO13) > 98%, fluoxetina (C17H18F3NO) > 98%, gemfibrozil(C15H22O3) > 99%, hidrocortisona (C21H30O5) > 98%, (S)- (+)-ibuprofeno (C13H18O2) > 98%, indometacina (C19H16ClNO4) > 99%, metformina (C4H11N5.HCl) > 97%, naproxeno (C14H14O3) > 98%, nimesulida (C13H12N2O5S)> 99%, prednisolona (C21H28O5) >98%, prednisona (C21H26O5)>98%, (R)- (+)-propranolol (C16H21NO2.HCl) >99%, sulfadiazina (C10H10N4O2S) >99%, sulfamerazina (C11H12N4O2S) >99%, sulfametazina (C12H14N4O2S) > 99%, sulfapiridina (C11H11N3O2S) >99% e (Fluka, Espanha) amoxicilina (C16H19N3O5S.3 H2O) >99,3%, cafeína (C8H10N4O2) > 99%, ciprofloxacina (C17H18FN3O3) >98%, doxiciclina (C14H10N2O8.1/2 H2O.1/2 C2H6O) > 99,5%, oxitetraciclina (C22H25ClN2O9) >98.1%, sulfametoxazol (C10H11N3O3S) > 98%, sulfatiazol (C9H9N3O2S2) > 99% e testosterona (C19H28O2) > 99%, (LGC, Espanha) metoprolol [(C15H25NO3)2.C4H4O4] 100% e gestodeno (C21H26O2)> 98%. As soluções padrão individuais a 200 mg/L foram preparadas em metanol, à excepção da amoxicilina, a qual foi dissolvida em dimetilsulfóxido, e da ciprofloxacina, que foi preparada numa mistura água/metanol (1:1) contendo 0,2 % de ácido clorídrico, dado a sua fraca solubilidade em metanol. A solução padrão conjunta (0,25 mg/L de cada fármaco) foi preparada em metanol e as soluções padrão de calibração foram preparadas numa mistura de água/metanol (95:5 v/v). As soluções padrão individuais e a solução padrão conjunta foram armazenadas ao abrigo da luz e a uma temperatura entre -20ºC ± 5ºC. Amostragem e conservação de amostras As amostras foram colhidas e analisadas entre Dezembro de 2011 e Janeiro de 2012. As amostras foram recolhidas em frascos de vidro âmbar de 1000 mL e armazenadas a 5 ± 3 ºC. Para as amostras de água para consumo humano, contendo cloro residual, as amostras foram tratadas in situ com ácido ascórbico, de forma a eliminar eventuais reacções de oxidação. Adicionou-se 10 mg de ácido ascórbico a 1 L de amostra. As amostras foram analisadas nas 48 horas subsequentes à amostragem. Otimização do método analítico A - UPLC-ESI-MS/MS As condições de operação são otimizadas em duas etapas principais, MS/MS e UPLC-ESI-MS/MS. A primeira fase tem por objectivo a obtenção do ião precursor, resultante da ionização do analito, e a obtenção dos iões produto. Nesta fase vão ser otimizados os seguintes parâmetros: modo de ionização, voltagem de cone e energia de colisão. Numa segunda fase é avaliado o comportamento dos compostos no sistema completo UPLC-ESI-MS/MS, sendo definidos os parâmetros cromatográficos e otimizada a voltagem do capilar. Algumas condições do espectrómetro de massas foram mantidas constantes, nomeadamente, voltagem do extractor (2 V), radiofrequência (0,1 V), temperatura da fonte (150 ºC), temperatura de dessolvatação (500 ºC), fluxo do gás de dessolvatação (900 L/h) e fluxo do gás de cone (25 L/h). O argônio foi utilizado como gás de colisão e o nitrogênio como gás de nebulização, dessolvatação e de cone. B - Condições do triplo quadrupolo O ião precursor é obtido através da ionização do composto, por electrospray, sendo utilizado o azoto como gás de nebulização, dessolvatação e de cone, e por aplicação de voltagem na fonte de ionização (voltagem do capilar) e no cone de extracção (voltagem de cone). Mantendo constante a voltagem do capilar, mantém-se a energia de ionização. A voltagem do capilar foi mantida constante a 3,5 kV. Para otimizar as condições operacionais de obtenção do ião precursor, são analisados isoladamente cada um dos compostos em estudo. A otimização baseia-se essencialmente na selecção do modo de ionização (negativo ou positivo) e da voltagem de cone. A injecção é realizada por infusão directa no espectrómetro de massa em modo de aquisição full scan para o primeiro quadrupolo. A voltagem de cone foi testada, em modo de ionização positivo e negativo, entre 5 e 50 V, em incrementos de 5 V. Uma vez definidas as condições para o primeiro quadrupolo é avaliado o efeito da energia de colisão fazendo variar a energia de colisão entre 5 e 40 eV em incrementos de 5 eV. A avaliação dos espectros de massa obtidos obedece a vários requisitos, nomeadamente: os iões produto com melhor resultado em termos de intensidade de sinal que resultem da fragmentação do ião precursor, quando os iões produto são seleccionados deve haver a presença do ião precursor e, só em casos excepcionais, fragmentação extensa, devem ser utilizados iões produto resultantes de outros fragmentos. Otimizados os parâmetros operacionais para obtenção dos iões precursores e seleccionados os respectivos iões produto e respectivas energias de colisão, estão reunidas as condições para definir as duas transições para cada composto alvo. A primeira transição (MRM1) é usada na quantificação e a segunda transição (MRM2) é usada na qualificação de cada composto. Uma vez que a energia de ionização é um parâmetro constante ao longo da análise por UPLC-ESI-MS/MS, a sua influência só foi avaliada após otimização das condições cromatográficas. C - Condições cromatográficas Por forma a aumentar a sensibilidade e o perfil cromatográfico de alguns compostos foi decidido utilizar dois métodos cromatográficos fazendo variar o pH da fase móvel, a qual levou à selecção e optimização de duas fases móveis, solvente A e solvente B. Para a maioria dos compostos básicos (método A), o solvente A da fase móvel é constituído por água com 0,01 mM de acetato de amónio e 0,5% ácido fórmico (v/v) e o solvente B por metanol com 0,01 mM de acetato de amónio (v/v). Para a maioria dos compostos ácidos (método B), o solvente A da fase móvel é constituído por água com 0,05% de hidróxido de amónio (v/v) e o solvente B por metanol. Para ambos os métodos, a coluna foi mantida a 40 ºC e o volume de injecção foi de 20 µL. Para o método A utilizou-se um fluxo de eluente de 0,3 mL/min e para o método B um fluxo de 0,5 mL/min. Os gradientes de eluição para os dois métodos encontram-se na Tabela 2.
D - Voltagem do capilar Para a otimização da voltagem de capilar, foi injectada a solução padrão conjunta em modo MRM, utilizando as condições anteriormente otimizadas. Foi estudada a relação entre a intensidade do sinal dos diferentes compostos alvo e a variação da voltagem de capilar entre 0 e 4 kV. E - Validação do método UPLC-ESI-MS/MS A validação do método UPLC-ESI-MS/MS foi iniciada com a realização de vários testes de forma a avaliar o intervalo de linearidade, os limiares analíticos instrumentais e a precisão instrumental em condições de repetibilidade. O intervalo de linearidade foi avaliado por meio da injecção de soluções padrão com diferentes concentrações preparadas em água ultra pura. Os resultados obtidos foram tratados através da realização de uma regressão linear pelo método dos mínimos quadrados. Os critérios internos estabelecidos para aceitação dos resultados foram: coeficiente de Determinação (r2) >0,995, coeficiente de variação do método (CVm) <10% e o intervalo de aceitação do teste de análise de resíduos de ±10%. Ao longo da gama de concentrações do intervalo de linearidade foi ainda estudada a variabilidade da razão MRM1/MRM2, usada como critério de qualificação juntamente com o tempo de retenção. Os limites de detecção e de quantificação instrumentais foram calculados por meio da razão sinal/ruído. A precisão instrumental foi estudada em condições de repetibilidade. Para a avaliação da repetibilidade instrumental, as soluções conjuntas correspondentes ao limite inferior, intermédio e superior da gama de trabalho foram analisadas 10 vezes durante a mesma série de trabalho. F - Preparação da amostra A preparação da amostra foi realizada num sistema automatizado de SPE, Caliper Life Sciences, modelo Autotrace (Hopkinton, USA). O efeito de matriz foi avaliado através de estudos de recuperação em amostras fortificadas de água ultra pura, água para consumo humano, superficiais e subterrâneas. As amostras foram concentradas em cartuchos OASIS HLB 200 mg (Waters, Milford, MA). A cada 500 mL de amostra adicionou-se 2 mL de uma solução 5,0 g/L de Na-EDTA e processar a pH neutro. Os cartuchos foram condicionados com 4 mL de metanol e 4 mL de água ultra pura. De seguida, as amostras foram passadas pelos cartuchos a uma velocidade de fluxo de 10ml/min. Os cartuchos foram de seguida secos sob um fluxo de azoto durante 30 minutos. Os analitos foram depois eluídos com 8 mL de metanol. Após eluição, os extratos foram levados à secura num sistema de evaporação Turbovap (Zymark), sob fluxo de azoto, dissolvidos em 0,5 mL de uma mistura água:metanol (95:5 v/v) e analisados por UPLC-ESI-MS/MS. G - Efeitos de matriz e limiares analíticos do método Os limiares analíticos do método global foram determinados após os estudos de recuperação em amostras de água para consumo humano, superficiais e subterrâneas, fortificadas a uma concentração correspondente ao limite inferior da gama de linearidade. Para uma correcta quantificação, foram construídas curvas de calibração utilizando os extratos das diferentes matrizes obtidos após o passo de preparação da amostra. Estas curvas de calibração foram também utilizadas para avaliar a ocorrência de efeitos de supressão/enriquecimento iónico através da sua comparação com as curvas de calibração construídas com as intensidades de sinal dos padrões preparados em água ultra-pura. Dado que se detectou a ocorrência de efeitos de matriz decorrentes da utilização do electrospray como fonte ionização, a linearidade do método foi avaliada para os diferentes tipos de matriz, recorrendo aos critérios referidos anteriormente no ponto E. Os limites de detecção e quantificação do método foram determinados por meio da análise de brancos de amostra e estimados para razões sinal/ruído de 3 e 10 respectivamente. Se a amostra apresenta sinal, os limites são estimados recorrendo aos cromatogramas dos "brancos amostra" sem fortificação. Se o "branco amostra" não apresenta sinal, a amostra é fortificada com uma concentração mínima que produza sinal e é determinada a razão sinal/ruído. Os resultados obtidos são divididos pelo factor de concentração do SPE (factor concentração = volume de amostra/volume do extracto =500/0,5=1000) para se obter os limites do método global (SPE-UPLC-MS/MS). Foram também realizados estudos de precisão do método (repetibilidade e precisão intermédia) e estudo de exatidão por participação num ensaio interlaboratorial. H - Análise de amostras De forma a testar a aplicabilidade do método SPE-UPLC-MS/MS optimizado analisaram-se amostras de diversos pontos de amostragem do sistema de abastecimento da EPAL e também das principais fontes de água bruta da empresa (Rio Tejo, rio Zêzere e poços de Alenquer e Lezírias). A identificação dos analitos foi realizada por comparação dos tempos de retenção e da razão entre a intensidade do sinal da transição MRM1 e a intensidade do sinal da razão da transição MRM2 nas amostras com os obtidos com as soluções padrão. A quantificação dos analitos foi realizada utilizando o sinal do ião produto MRM1 de quantificação e as respectivas taxas de recuperação.
RESULTADOS E DISCUSSÃO Otimização das condições UPLC-ESI-MS/MS A Tabela 3 apresenta as condições óptimas para a análise dos fármacos em estudo por UPLC-ESI-MS/MS. A Figura 1 apresenta o efeito da variação da energia de colisão na fragmentação do ião precursor da ciprofloxacina e os respectivos espectros de massas são apresentados no material suplementar (Figura 1S).
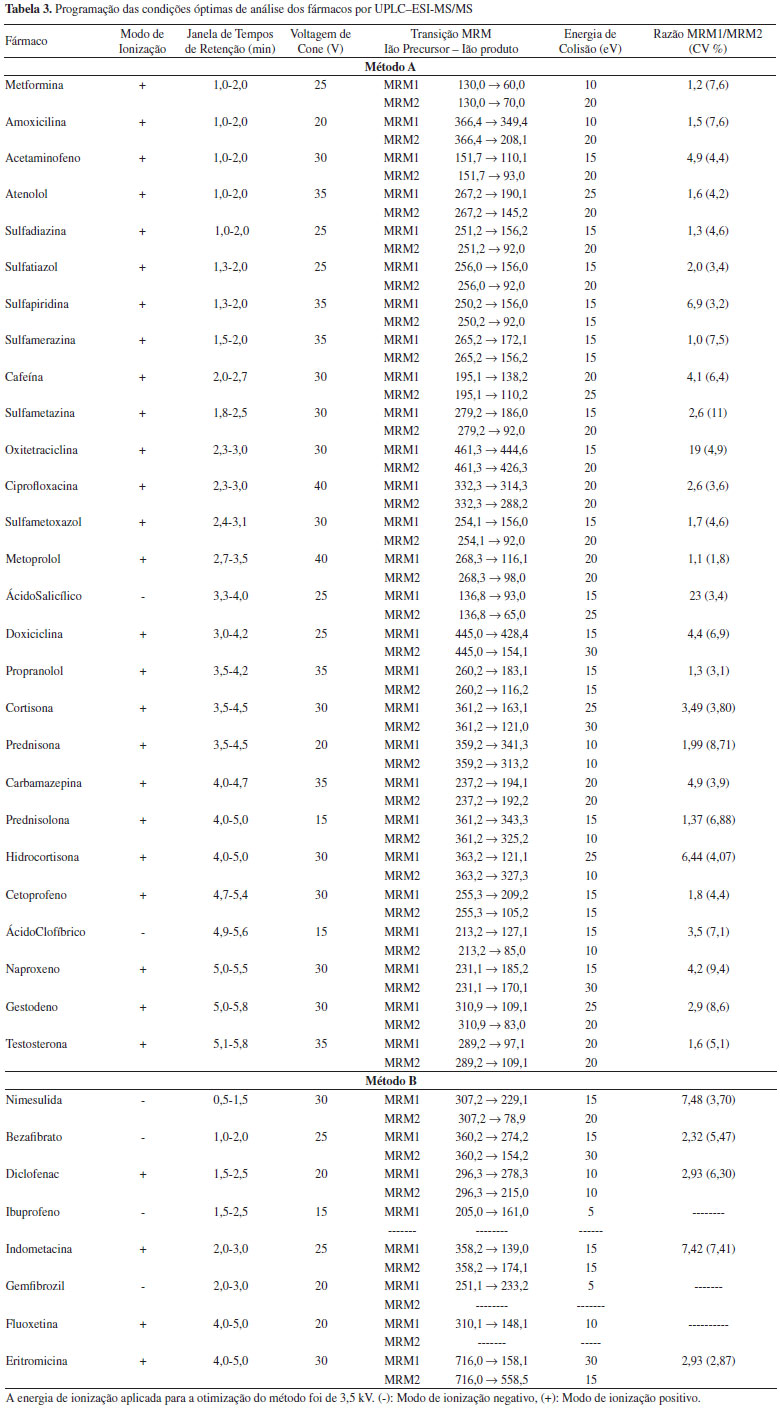 | |
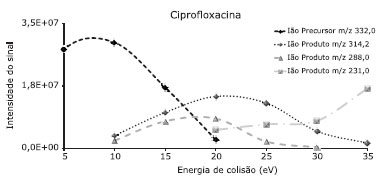 Figura 1. Efeito da variaçao da energia de colisao na fragmentaçao do iao precursor da ciprofloxacina
À medida que a energia de colisão aumenta, observa-se um aumento da fragmentação do ião precursor, detectando-se dois iões produtos resultantes da dissociação directa do ião precursor, os quais, representam dois fragmentos óptimos para a definição das duas transições. A energia de colisão óptima é a de 20 eV, uma vez que permite a formação dos dois iões produto com uma boa intensidade, detectando-se ainda a presença vestigial do ião precursor (Figura 1S). A aplicação de uma voltagem de capilar de 0,5 kV favorece a sensibilidade da maioria dos fármacos, tendo sido esta a voltagem escolhida como valor a utilizar na implementação do método analítico (Figura 2S). Condições cromatográficas De forma a conseguir-se a melhor resolução cromatográfica possível e um perfil adequado do pico cromatográfico para todos os compostos-alvo, decidiu-se utilizar duas corridas cromatográficas diferentes baseadas no pH da fase móvel. Desta forma, para a maioria dos compostos que ionizam em modo positivo foi utilizado um pH inicial de fase móvel de 2. Para os compostos que ionizam em modo negativo foi utilizado um gradiente básico. Estes dois métodos também permitem a análise dos compostos-alvo com a maior sensibilidade possível. No método A verificou-se que alguns compostos, nomeadamente a doxiciclina, oxitetraciclina e a ciprofloxacina, apresentavam um mau perfil cromatográfico. Estes compostos possuem vários valores de pka. No caso das tetraciclinas, os valores de pKa são aproximadamente 3, 7 e 9 e, no caso da ciprofloxacina 6 e 9, pelo que se decidiu verificar a influência da diminuição do pH no perfil cromatográfico destes compostos. O pH da fase móvel do método A altera o perfil cromatográfico destes três compostos. À medida que o pH diminui, a cauda do pico cromatográfico diminui e a forma do pico cromatográfico melhora, conseguindo-se a um pH inicial da fase móvel de 2,0, picos satisfatórios e que permitem a análise destes compostos (Figura 3S). A Tabela 3 apresenta as janelas de tempo de retenção para cada composto, nas quais são definidas as condições de monitorização otimizadas anteriormente para ambos os métodos: ião precursor, voltagem de cone, transições MRM1 e MRM2 e respectivas energias de colisão, assim como, a variabilidade da razão MRM1/MRM2. Validação do Método UPLC-ESI-MS/MS Estudos de linearidade Para determinar o intervalo de linearidade, trabalhou-se numa gama de concentrações entre 0,02 e 600 µg/L, aproximadamente. Para alguns compostos a gama não se inicia na concentração inferior (0,02 µg/L) devido à ausência de sinal cromatográfico do ião produto MRM1 de quantificação e/ou do ião produto MRM2 de qualificação. A Tabela 4 apresenta os resultados do estudo da linearidade após o cumprimento dos critérios internos de aceitação.
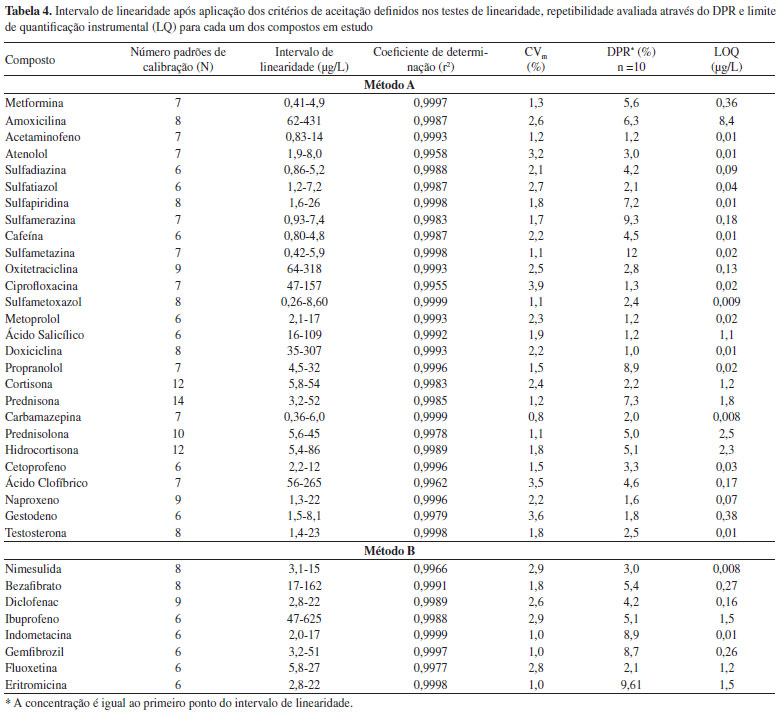
O método permite obter bons coeficientes de determinação (r2) para todos os compostos (o mínimo de 0,9959 para a ciprofloxacina e o máximo de 0,9999 para o sulfametoxazol e carbamazepina) e excelentes coeficientes de variação do método, inferiores a 4,0%. Precisão A precisão do método foi avaliada em termos de repetibilidade e de precisão intermédia, expresso em termos de desvio padrão relativo (DPR). Na Tabela 4 apresentam-se os resultados obtidos para a concentração inferior do intervalo de linearidade. Extração em fase sólida, SPE Foi realizado um estudo de algumas das principais condições que afectam a recuperação dos analitos alvo em água ultra pura - pH da amostra e condições de eluição. O efeito do pH foi avaliado realizando estudos de recuperação a pH ácido, neutro e básico. A extração realizada a pH neutro foi a que resultou em resultados mais satisfatórios para a maioria dos compostos e que permite a extracção conjunta de fármacos ácidos, básicos e neutros com diferentes propriedades físico-químicas. Uma vez selecionado o tipo de adsorvente e o pH da amostra, diferentes tipos de solventes de eluição foram testados: metanol, acetonitrilo, acetona e diclorometano. As eluições com metanol e acetonitrilo foram as que resultaram em melhores recuperações, verificando-se que as taxas de recuperação eram significativamente melhores utilizando metanol. O volume de eluição foi também testado, tendo-se verificado que a utilização de dois passos de eluição com 4 mL de metanol era a opção mais favorável. Desta forma, após optimização das condições referidas optou-se por utilizar o procedimento descrito no ponto F deste artigo, uma vez que as taxas de recuperação (entre 75 e 125 %) cumprem os requisitos da legislação nacional48 no que se refere à exactidão dos métodos para a análise de compostos orgânicos (em águas de consumo humano) para a maioria dos compostos (Tabela 5). Na ausência de requisitos para os critérios de aceitação das recuperações dos fármacos nas águas, definiu-se o critério de aceitação para a exactidão de métodos analíticos aplicados à análise de compostos orgânicos já legislados. Os 25% de critério de aceitação é o menor dos valores legislados, uma vez que de acordo com o DL 236/9851 (águas subterrâneas ou superficiais destinadas à produção de água para consumo humano) são aceitáveis 30% e 50%, de acordo com a natureza dos compostos orgânicos e respectivos métodos de análise. No entanto, é importante referir que não se conseguiu extrair os fármacos amoxicilina, metformina, doxiciclina e oxitetraciclina com este procedimento.
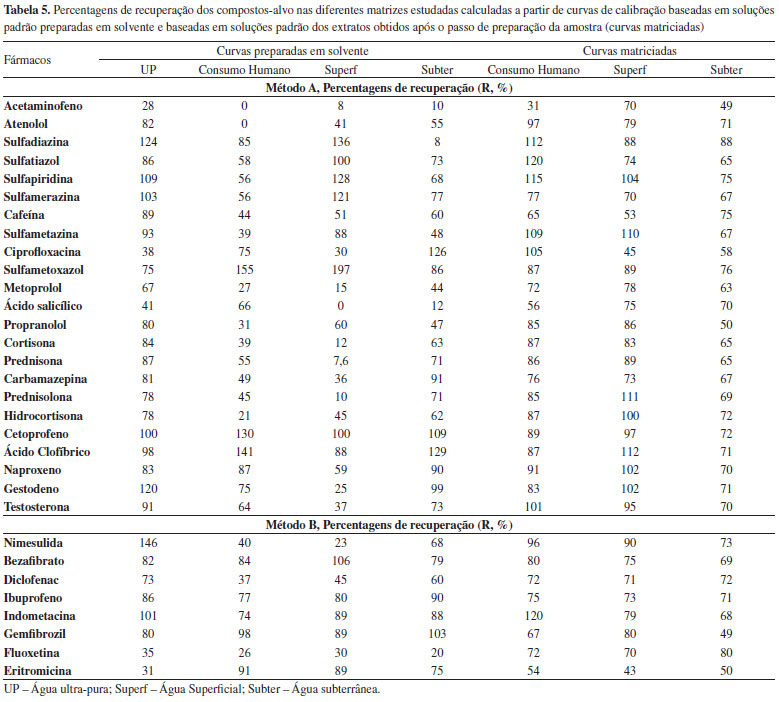
Efeitos de matriz e limites de deteção e quantificação do método O procedimento de SPE otimizado foi depois testado utilizando águas para consumo humano, superficial e subterrânea por forma a avaliar a ocorrência de efeitos de matriz. Neste tipo de matrizes verificou-se a ocorrência de efeitos de matriz, o que resultou em taxas de recuperação significativamente mais baixas quando comparadas com os resultados obtidos com água ultra pura, para a maioria dos compostos (Tabela 5). Considerando que estes efeitos de matriz estão principalmente relacionados com a utilização do electrospray como fonte de ionização, optou-se por realizar a calibração com padrões preparados nos extratos das diferentes matrizes obtidos após o passo de preparação da amostra. Os resultados obtidos encontram-se na Tabela 5. Pode-se verificar que a utilização de curvas de calibração matriciadas corrige os efeitos de matriz, obtendo-se taxas de recuperação superiores a 50% para todas as matrizes, sendo que para a maioria dos compostos as taxas de recuperação se situam entre 75 e 125%. Na Tabela 6 são referidas as percentagens de supressão/enriquecimento iónico resultantes da utilização do electrospray como fonte de ionização. Pode-se também verificar que compostos pertencentes à mesma classe terapêutica possuem diferentes comportamentos ao serem ionizados por electrospray, tornando a utilização de padrões internos por classe terapêutica um método pouco fiável. Desta forma, prepararam-se os extratos de cada uma das matrizes em estudo (água superficial, água subterrânea e água de consumo humano) por SPE, os quais foram fortificados de forma a obter cada um dos padrões de calibração, corrigindo o efeito de matriz em ESI.
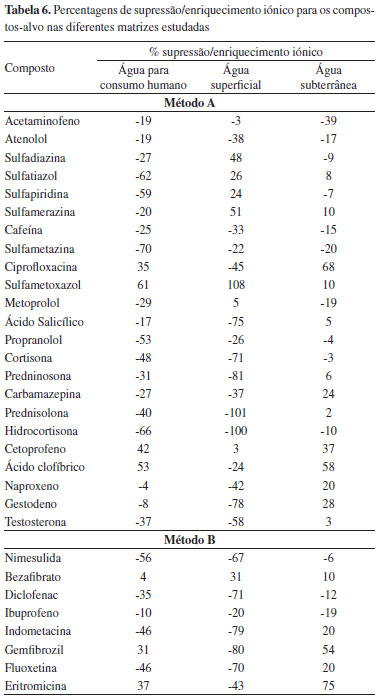
Os limites de detecção e quantificação do método obtidos com base na razão sinal/ruído para os diferentes tipos de matriz encontram-se na tabela 7. Verifica-se que para a maioria dos compostos os LOD e LOQ correspondentes à matriz da água subterrânea são inferiores aos das outras matrizes. De uma forma geral, pode-se também concluir que os limiares analíticos correspondentes às águas para consumo humano e superficiais não apresentam diferenças significativas, apresentando valores da mesma ordem de grandeza. Os valores obtidos, entre 0,009 e 10 ng/L para os LOD e 0,03 e 33 ng/L para os LOQ, permitem a aplicação deste método a amostras reais, dado serem da mesma ordem de grandeza ou inferiores às concentrações de fármacos detectados neste tipo de amostras. Os LOD e LOQ mais elevados dizem respeito aos fármacos da classe dos corticosteróides, facto que pode ser explicado não só devido à menor sensibilidade do método para estes compostos, mas também ao elevado grau de supressão iónica a que estão sujeitos, à semelhança do reportado por Batt et al.,52 que também desenvolveram um método de análise de fármacos por SPE-UPLC-MS/MS.
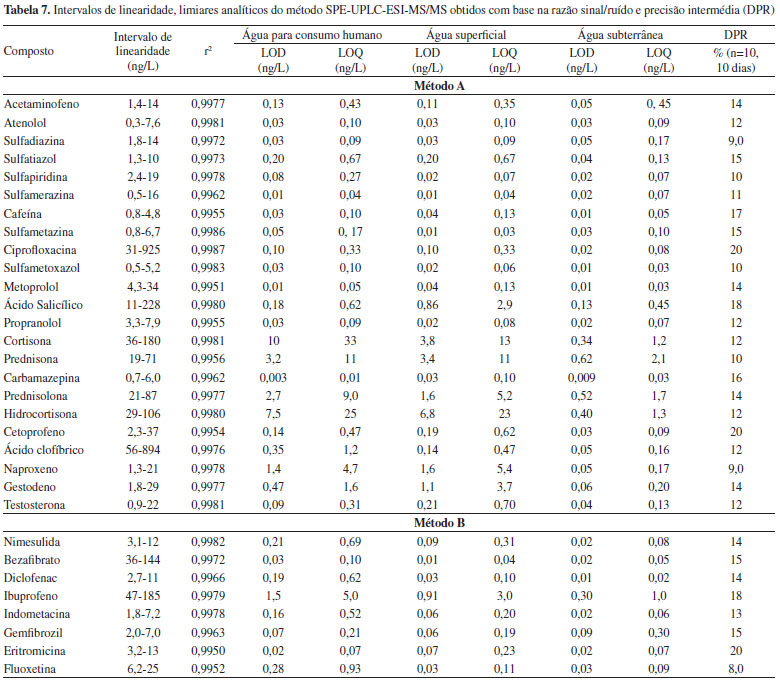
Estudos de precisão e exatidão A aplicabilidade do método validado foi avaliada para alguns compostos (atenolol, metoprolol, carbamazepina, diclofenac, ibuprofeno e cetoprofeno) através da participação num ensaio interlaboratorial (AGLAE - Association Générale des Laboratoires d'Analyse de l'Environnement). Os resultados obtidos encontram-se na Figura 2. Pode-se verificar, com base nos valores Z-score, que existe uma boa concordância entre o resultado obtido e o valor de referência.
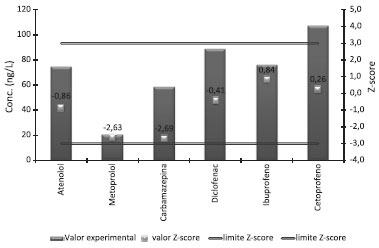 Figura 2. Concentraçao dos fármacos no ensaio interlaboratorial do atenolol, metoprolol, carbamazepina, diclofenac, ibuprofeno e cetoprofeno e respectivos valores de z-score
A precisão foi avaliada em condições de repetibilidade sendo inferior a 15% para todos os compostos. Análise de amostras Foram recolhidas 22 amostras de diferentes pontos da rede de abastecimento da empresa e também das suas fontes de água bruta. Na Tabela 8 encontram-se os resultados das amostras para os compostos que possuem intensidade de sinal superior ao correspondente ao LOQ ao tempo de retenção previsto (tR ± 0,5%) e para os quais a razão MRM1/MRM2 cumpre os requisitos definidos pelo laboratório (razão MRM1/MRM2 ± 20%).
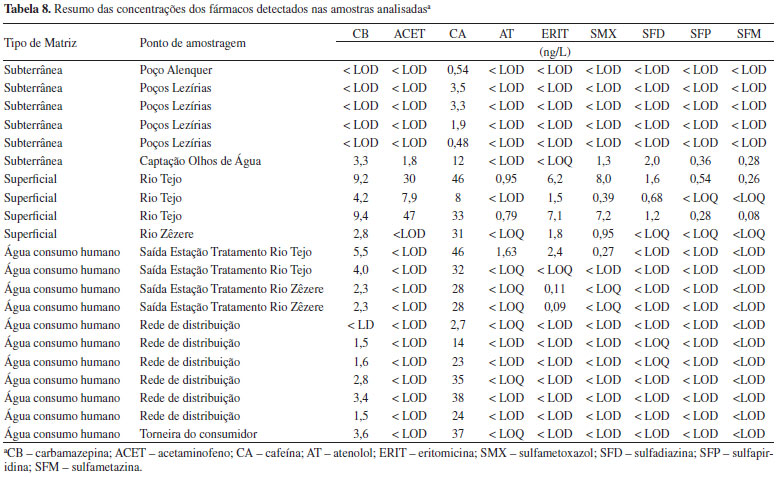
Dos 31 fármacos analisados, apenas 9 foram detetados em concentrações superiores aos respectivos limites de quantificação nas diferentes amostras analisadas. As concentrações dos diferentes compostos variam entre 0,08 e 46 ng/L nas águas brutas e entre 1,6 e 46 ng/L nas águas tratadas, sendo que nestas últimas só 5 fármacos, a cafeína, a carbamazepina, o atenolol, o sulfametoxazol e a eritromicina estão presentes em níveis quantificáveis. De notar também que a cafeína foi detectada em todas as amostras e a carbamazepina em 15 das 22 amostras analisadas. Estes resultados podem ser explicados tendo em conta o uso generalizado da cafeína em bebidas e não como fármaco e ao facto de a carbamazepina ser um composto quimicamente estável, resistindo aos processos de tratamento utilizados nas estações de tratamento, quer de águas residuais quer de produção de água para consumo humano. Estes resultados estão de acordo com os valores reportados noutros estudos.4,5,52 Em relação à carbamazepina, atenolol e sulfametoxazol, os valores máximos presentes na água de consumo foram de 5,5, 1,63 e 0,27 ng/L, respectivamente, valores bastante inferiores aos valores máximos reportados em estudos realizados nos EUA, França, Alemanha e Holanda (entre 43-258 ng/L para a carbamazepina,4 26 ng/L para o atenolol4 e 22 ng/L para o sulfametoxazol5). Relativamente à cafeína e eritromicina os valores máximos detectados na água de consumo foram de 46 e 2,4 ng/L, respectivamente, sendo da mesma ordem de grandeza dos valores obtidos em estudos realizados nos EUA e França (23-119 ng/L para a cafeína4 e 4,9 ng/L para a eritromicina5). As concentrações detectadas referentes a estes 5 fármacos não apresentam qualquer risco para a saúde do consumidor.52 Verifica-se que nas amostras da rede de abastecimento e águas subterrâneas os compostos-alvo encontram-se na sua maioria abaixo dos limites de detecção do método, sendo as amostras de águas superficiais correspondentes a pontos de amostragem do Rio Tejo e Rio Zêzere, as que apresentam concentrações em níveis quantificáveis. De notar também que a amostra correspondente à captação de Olhos de Água (fonte de água bruta actualmente desativada), ao contrário das outras amostras de matrizes subterrâneas, apresenta níveis quantificáveis de fármacos utilizados em animais. Este facto pode ser explicado dado que esta captação situa-se numa zona onde existem várias agropecuárias, podendo os fármacos administrados aos animais contaminar as águas do subsolo por lixiviação. Face a estes resultados preliminares, será posteriormente realizado um estudo de monitorização a uma escala maior e mais representativa, abrangendo um maior número de amostras recolhidas em diferentes alturas do ano.
CONCLUSÕES O método de SPE otimizado mostrou ser eficiente para a extração simultânea dos compostos-alvo nas matrizes estudadas. Este passo de extração combinado com a técnica de UPLC-MS/MS fornece um método rápido, sensível, preciso e com uma boa exactidão para a quantificação vestigial de diversos fármacos. Como o limite de quantificação para a maioria dos compostos é geralmente inferior a 1 ng/L e, por vezes, inferior a 0,1 ng/L, a técnica de SPE-UPLC-MS/MS é o método de excelência para a monitorização dos fármacos em concentrações vestigiais nas águas brutas (superficiais e subterrâneas) e de consumo humano. Fortes efeitos de supressão/enriquecimento iónico foram detectados para a maioria dos compostos, embora se verifique que fármacos pertencentes à mesma classe terapêutica não apresentem o mesmo grau de supressão/enriquecimento iónico, o que faz com que a utilização de padrões internos não seja uma forma fiável de corrigir a ocorrência destes efeitos. A utilização de curvas de calibração baseadas em soluções preparadas nos extratos das amostras (curvas matriciadas) é, no nosso entender, o melhor método para corrigir este problema. O método optimizado provou ser útil na análise de amostras reais de águas para consumo humano, superficiais e subterrâneas. No seguimento do trabalho realizado até à data estão previstas as seguintes actividades: a) estudos adicionais de otimização do passo de preparação da amostra para os fármacos amoxicilina, metformina, doxiciclina e oxitetraciclina; b) estudo de formas de diminuir/corrigir os efeitos de matriz detetados e c) análise de amostras durante um longo período de tempo de forma a caracterizar a qualidade da água captada e distribuída pela EPAL S.A..
MATERIAL SUPLEMENTAR As Figuras 1S, 2S e 3S e a Tabela 1S encontram-se disponíveis na forma de arquivo pdf, com acesso livre, http://quimicanova.sbq.org.br.
AGRADECIMENTOS À Empresa Portuguesa das Águas Livres S.A. (EPAL) e à Fundação para a Ciência e a Tecnologia (FCT), os autores agradecem o financiamento no âmbito da Bolsa de Doutoramento em Empresa (SFRH/BDE/33804/2009), o qual tornou possível a realização deste trabalho de investigação. Ao Infarmed, os autores agradecem a disponibilização dos dados de consumo dos fármacos.
REFERÊNCIAS 1. Petrovic, M.; Elijarrat, E.; Lopez de Alda, M. J.; Barceló, D.; Anal. Bioanal. Chem. 2004, 378, 549. 2. Daughton, C.; Renewable Resources Journal 2005, 23, 6. 3. Farré, M.; Pérez, S.; Kantiani, L.; Barceló, D.; Trends Anal. Chem. 2008, 27, 991. 4. Mompelat, S.; Le Bot, B.; Thomas, O.; Environ. Int. 2009, 35, 803. 5. Daughton, C. G. em Contaminants of Emerging Concern in the Environment: Ecological and Human Health Considerations; Halden, R., ed. American Chemical Society: Washington, 2010, cap. 2. 6. Alonso, S. G.; Catalá, M.; Maroto, R. R.; Gil, J. L. R.; Miguel, A. G.; Valcárcal, Y.; Environ. Int. 2010, 36, 195. 7. Gros, M.; Petrovic, M.; Barceló, D.; Talanta 2006, 70. 8. Schultz, M.; Furlong, E.; Kolpin, D.; Werner, S.; Schoenfus, H.; Barber, L.; Blazer, V.; Norris, D.; Vajda, A.; Environ. Sci. Technol. 2010, 44, 1918. 9. Peysson, W.; Vulliet, E.; J. Chromatogr. A 2013, 1290, 46. 10. Bialk-Bielinska, A.; Kumirska, J.; Palavinskas, R.; Stepnowski, P.; Talanta 2009, 80, 947. 11. Billa, D. M.; Dezotti, M.; Quim. Nova 2003, 26, 523. 12. Jørgensen, S. E.; Halling-Sørensen, B.; Chemosphere 2000, 40, 691. 13. Henriques, M.; Cardoso, V. V.; Rodrigues, A. M.; Ferreira, E.; Benoliel, M. J.; Almeida, C. M. M.; J. Water Resour. Prot. 2010, 2, 818. 14. Kummerer, K.; Annual Review of Environment and Resources 2010, 35, 57. 15. Andreozzi, R.; Raffaele, M.; Nicklas, P.; Chemosphere 2003, 50, 319. 16. Gros, M.; Petrovic, M.; Barceló, D.; Anal. Chem. 2009, 81, 898. 17. Demeestere, K.; Petrovic, M.; Gros, M.; Dewulf, J.; Van Langenhove, H.; Barceló, D.; Anal. Bioanal. Chem. 2010, 825. 18. Garcia-Lor, E.; Sancho, J. V.; Hernández, F.; J. Chromatogr. 2010, 1217, 622. 19. Fernández, C.; González-Doncel, M.; Pro, J.; Carbonell, G.; Tarazona, J. V.; Sci. Total Environ. 2010, 408, 543. 20. Ternes, T. A.; Water Res. 1998, 32, 3245. 21. Sousa, M. A.; Gonçalves, C.; Cunha, E.; Hajšlová, J.; Alpendurada, M. F.; Anal. Bioanal. Chem. 2011, 399, 807. 22. Pena, A.; Lino, C. M.; Solich, P.; J. Sep. Sci. 2007, 30, 2924. 23. Seifrtová, M.; Pena, A.; Lino, C. M.; Solich, P.; Anal. Bioanal. Chem. 2008, 391, 799. 24. Koplin, D.W.; Furlong, E. T.; Meyer, M. T.; Thurman, E. M.; Zaugg, S. D.; Barber, L. B.; Environ. Sci. Technol. 2002, 36, 1202. 25. Schultz, M.; Furlong, E.; Koplin, D.; Werner, S.; Schoenfus, H.; Barber, L.; Environ. Sci. Technol. 2010, 44, 1918. 26. Brown, K. D.; Kulis, J.; Thomson, B.; Chapman, T. H.; Sci. Total Environ. 2006, 366, 772. 27. Brooks, B. W.; Chambliss, C. K.; Stanley, J. K.; Ramirez, A.; Banks, K. E.; Johnson, R. D.; Environ. Toxicol. Chem. 2005, 24, 464. 28. Takada, N.; Tanishima, T.; Shinohara, H.; Kiri, K.; Takada, H.; Water Res. 2006, 40, 3297. 29. Kuster, M.; Azevedo, D. A.; Alda, M. J., Aquino Neto, F. R. A., Barceló, D.; Environ. Int. 2009, 35, 997. 30. Verbinnem, R. T.; Nunes, G. S.; Vieira, E. M.; Quim. Nova 2010, 33, 1837. 31. Ternes, T.; Hirsch, R.; Mueller, J.; Haberer, K.; Fresenius J. Anal. Chem. 1998, 62, 329. 32. Miao, X. S.; Koenig; B. G.; Metcalfe, C. D.; J. Chromatogr A. 2002, 952, 139. 33. Bendz, D.; Paxéus; N. A.; Ginn, T. R.; Loge, F. J.; J. Hazard. Mater. 2005, 122, 195. 34. Calamari, D.; Zuccato, E.; Castiglioni, S.; Bagnati, R.; Fanelli, R.; Environ. Sci. Technol. 2003, 37. 35. Roberts, P.H.; Thomas, K. V.; Sci. Total Environ. 2006, 356, 143. 36. Rabiet, M.; Togola, A.; Brissaud, F.; Seidel, J. L.; Budzinski, H.; Elbaz-Poulichet, F.; Environ. Sci. Technol. 2006, 40, 5282. 37. Scheurell, M.; Franke, S.; Shah, R. M.; Huhnerfuss, H.; Chemosphere 2009, 77, 870. 38. Redderson, K.; Heberer, T.; Dunnbier, U.; Chemosphere 2002, 49, 539. 39. Heberer, T.; Mechlinski, A.; Fanck, B.; Knappe, A.; Massmann, G.; Pekdeger, A.; Groundwater Monitoring & Remediation 2004, 24, 70. 40. Kozisek, F.; Pomykacova, I.; Jeligova, H.; Cadek, V.; Svobodova, V.; J. Water Health 2013, 11, 84. 41. Directiva 2000/60/CE, do Parlamento Europeu e do Conselho de 23 de Outubro, Jornal Oficial das Comunidades Europeias, L 327, 2000. 42. Lei n.º 58/2005 de 29 de Dezembro, Diário da República, I Série A, n.º 249, 2005. 43. Decreto-Lei n.º 77/2006 de 30 de Março, Diário da República, I Série A, n.º 64, 2006. 44. Directiva 2008/105/CE do Parlamento Europeu e do Conselho de 16 de Dezembro, Jornal Oficial das Comunidades Europeias, L 348/84, 2008. 45. Decreto-Lei n.º 103/2010 de 24 de Setembro, Diário Da República, 1.ª Série, n.º 187, 2010. 46. Comissão das Comunidades Europeias, Proposta de Diretiva do Parlamento Europeu e do Conselho que altera as Diretivas 2000/60/CE e 2008/105/CE no que respeita às substâncias prioritárias no domínio da política da água, Relatório de Progresso, COM 876, 2011. 47. Directiva 98/83/CE do Conselho de 3 de Novembro de 1998 relativa à qualidade da água destinada ao consumo humano, Jornal Oficial das Comunidades Europeias, L 330/32, 1998. 48. Decreto-Lei n.º 306/2007, de 27 de Agosto, Diário da República, 1.ª série, n.º 164, 2007. 49. Zwiener, C.; Frimmel, F. H.; Anal. Bioanal. Chem. 2004, 378, 862. 50. Petrovic, M.; Gonzalez, S.; Barceló, D.; Trends Anal. Chem. 2003, 22, 685. 51. Decreto-Lei n.º 236/98 de 1 de Agosto, Diário da República, I Série A, n.º 176, 1998. 52. Batt, A. L.; Kostich, M. S.; Lazorchak, J. M.; Anal. Chem. 2004, 80, 5021. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access