
 |
 |
 |
 |
 |
Educação
|
|
| Síntese de cristais líquidos derivados do nitroazobenzeno: uma proposta de síntese multi-etapas aplicada às aulas de Química Orgânica Experimental Synthesis of liquid crystals derived from nitroazobenzene: a proposed multistep synthesis applied to organic chemistry laboratory classes |
|
Rodrigo Cristiano*; Marília Gabriela B. Cabral; Rafael B. de Aquino; Cláudia M. Z. Cristiano
Departamento de Química, Universidade Federal da Paraíba - UFPB, Campus I, 58051-900 João Pessoa - PB, Brasil Recebido em 29/05/2013 *e-mail: rcristiano@quimica.ufpb.br We describe a synthetic route consisting of five steps from aniline to obtain liquid crystal compounds derived from nitroazobenzene. Syntheses were performed during the second half of the semester in organic chemistry laboratory classes. Students characterized the liquid crystal phase by the standard melting point techniques, differential scanning calorimetry and polarized optical microscopy. These experiments allow undergraduate students to explore fundamentally important reactions in Organic Chemistry, as well as modern concepts in Chemistry such as self-assembly and self-organization, nanostructured materials and molecular electronics. INTRODUÇÃO Cristais líquidos (CLs) constituem importantes materiais eletrônicos moleculares, que combinam as propriedades de ordem do estado sólido e a fluidez do estado líquido.1-2 Esta combinação única de mobilidade molecular e anisotropia ótica, elétrica e magnética propicia aos CLs um vasto número de aplicações tecnológicas, destacando-se como componentes ativos em mostradores planos (Liquid Crystal Displays, LCDs) de celulares, tablets, laptops e outros dispositivos. As fases líquido-cristalinas, chamadas de mesofases, são fases condensadas exibidas por determinadas moléculas, que possuem anisotropia de forma (alongadas, achatadas, biaxiais, etc.), em temperaturas intermediárias à fase cristal e à fase líquida. Os CLs são divididos basicamente em duas classes: os termotrópicos, cuja unidade fundamental geradora da mesofase é a molécula, e como o nome sugere, suas propriedades dependem da temperatura; e os liotrópicos, onde a unidade fundamental geradora da mesofase é um agregado de moléculas em um solvente. Os CLs termotrópicos são comumente classificados de acordo com sua forma molecular, sendo os CLs calamíticos constituídos por moléculas alongadas em forma de bastão e os CLs discóticos com a forma de disco. Os CLs calamíticos, objeto deste trabalho de educação, exibem normalmente mesofases nemática e esméticas. Em termos de ordenamento molecular, a fase nemática (N), aproxima-se mais do estado líquido isotrópico (Iso) e é caracterizada por apresentar apenas ordem orientacional, onde o eixo mais longo das moléculas está alinhado em uma direção comum (Figura 1). Nas fases esméticas (Sm), além da ordem orientacional, as moléculas possuem algum grau de ordem posicional, arranjadas em camadas. Possuem interações laterais mais fortes e maior viscosidade quando comparadas com a mesofase N, aproximando-se estruturalmente mais do estado sólido, ou cristal (Cr). Essas mesofases Sm são designadas por letras (A, B, C... K) de acordo com o seu ordenamento molecular. As mais normalmente observadas em CLs são a SmA e SmC (Figura 1). Na fase SmA, as moléculas estão alinhadas perpendicularmente ao plano das camadas e, na SmC, o arranjo molecular é levemente inclinado em relação a este plano (Figura 1).3
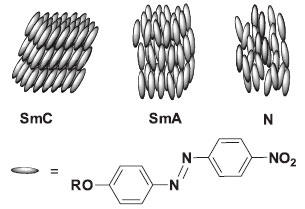 Figura 1. As mesofases esméticas (SmC e SmA) e nemática (N) de CLs calamíticos. No contexto de nosso trabalho, os elipsoides representam as moléculas dos nitroazobenzenos
Apesar de sua importância nas tecnologias atuais, bem como em Ciência básica, CLs são geralmente omitidos dos currículos de graduação em Química e áreas afins. Embora tenha sua natureza íntima ligada à Química Orgânica, pouca ou nenhuma atenção a esta classe de materiais é dada nas disciplinas da área no Brasil, especialmente no que diz respeito às aulas experimentais. Os cursos atuais de Química Orgânica Experimental são, em geral, divididos em dois níveis. No primeiro são abordados conceitos e técnicas fundamentais de separação, purificação e determinação de propriedades físicas e algumas transformações. No segundo nível, o programa envolve sempre uma síntese multietapas ou sequência de reações, onde o objetivo é fazer com que o aluno desenvolva as habilidades de laboratório e entenda a importância de se obter um bom rendimento, quando o reagente usado é aquele preparado por ele na reação anterior. Como um intrigante estado físico da matéria, constituído por moléculas orgânicas, CLs podem fazer parte das aulas, desde a sua síntese e caracterização estrutural até o estudo de suas propriedades mesomórficas por microscopia ótica de luz polarizada (MOLP) e calorimetria diferencial de varredura (DSC). Além disso, os experimentos com CLs tornam o ambiente propício para o ensino de conceitos fundamentais da Química moderna, tais como processos de automontagem e auto-organização, nanoestruturas, materiais avançados e eletrônica molecular.4 Existem na literatura textos acessíveis, em Língua Portuguesa, aos alunos de graduação sobre os conceitos e propriedades de CLs.4,5 Além disso, CLs já foram explorados em aulas laboratoriais envolvendo síntese orgânica em única etapa. Exemplos que se destacam são a síntese do benzoato de colesterila,6 e de ácidos p-alcoxibenzóicos.7 Também foram explorados em laboratórios de Ensino no estudo de suas propriedades físicas, tais como ópticas,8 eletro-óticas9 e termocrômicas.10 Neste trabalho propomos uma interessante alternativa para disciplinas de Química Orgânica Experimental do segundo nível (II ou B, dependendo da instituição), na qual inserimos uma sequência de reações com cinco etapas. O objetivo é a preparação e caracterização de CLs derivados do nitroazobenzeno11 (Esquema 1). As sínteses abrangem reações importantes da Química Orgânica, tais como reações de substituição nucleofílica no carbono acílico (acetilação na aula 1 e hidrólise na aula 3), substituição eletrofílica aromática (nitração na aula 2 e acoplamento diazo na aula 4) e substituição nucleofílica bimolecular (O-alquilação na aula 5). Os detalhes mecanísticos das reações envolvidas são bem estabelecidos e facilmente encontrados nos diversos livros-textos de Química Orgânica, e podem ser explorados pelo professor na apresentação de cada etapa. A vantagem deste procedimento é que as três primeiras etapas da rota de síntese escolhida são bem disseminadas em manuais de laboratório de Química Orgânica.12 Os experimentos abrangem técnicas comuns em laboratórios de Síntese, tais como reações sob refluxo, destilações simples, filtrações a vácuo, reações com controle de temperatura, extrações usando funil de separação, incluindo extrações ácido-base, recristalizações e medidas de ponto de fusão, assim como a possibilidade de uso de técnicas mais sofisticadas para uma completa identificação estrutural: espectroscopia no infravermelho (IV), ressonância magnética nuclear (RMN) e cromatografia gasosa acoplada à espectrometria de massa (CG-EM). Além disso, os compostos finais preparados podem ser aproveitados em outras disciplinas, tais como em Físico-Química Experimental, no estudo das mesofases e suas propriedades termodinâmicas com uso de um equipamento DSC.
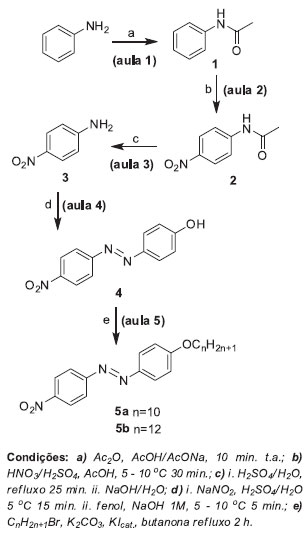 Esquema 1. Síntese multi-etapas para obtençao dos CLs derivados do nitroazobenzenos 5a-b
RESULTADOS E DISCUSSÃO A síntese multi-etapas para obtenção dos CLs 4-(alcóxi)-4'-nitrobenzenos 5a-b foi realizada de acordo com o Esquema 1. Como requisito fundamental, o aluno deverá ter cursado ou estar cursando a disciplina teórica de Química Orgânica (II ou B), sendo que as reações na rota sintética do Esquema 1 demandam o conhecimento de conceitos e mecanismos de subtituição nucleofílica no carbono acílico, substituição eletrofílica aromática (SEAr) e substituição nucleofílica bimolecular (SN2). Além da possibilidade de aprofundamento nestes conceitos teóricos envolvidos, a rota de síntese é vantajosa pelo fato de que o corante azo 4 pode ser alquilado com cadeias alifáticas de diferentes tamanhos, fornecendo CLs com n=4 até n=16.11 Assim, cada equipe poderá obter um CL diferente e discussões da relação estrutura química versus propriedades mesomórficas ficariam ainda mais interessantes. As quatro primeiras etapas desta síntese, até a obtenção do corante azo 4, foram completadas com sucesso em laboratórios de ensino de Química Orgânica por alunos de graduação dos cursos de Química, Engenharia Química e Farmácia no primeiro e segundo período de 2010 e 2012, da Universidade Federal da Paraíba. A última etapa de O-alquilação foi desenvolvida com a participação de um aluno de graduação e uma aluna de pós-graduação, coautores deste trabalho. As reações envolvidas são simples e comuns em laboratórios de Ensino, contudo, todos os roteiros foram cuidadosamente revisados, para que pudessem ser completadas no período máximo de 3 h, adequando-se assim ao tempo médio de uma aula de laboratório. Partindo da anilina, são necessárias cinco aulas, ou semanas, para completar a síntese e uma aula adicional para a caracterização da estrutura e das propriedades líquido-cristalinas dos compostos finais. O período total se enquadra perfeitamente na segunda metade da disciplina de Química Orgânica Experimental (II ou B). A Tabela 1 apresenta os rendimentos médios das equipes de alunos para cada etapa de reação. Na síntese multi-etapas, as perdas de massa devido aos processos de purificação foi o fator que mais pesou nos baixos rendimentos globais das equipes.

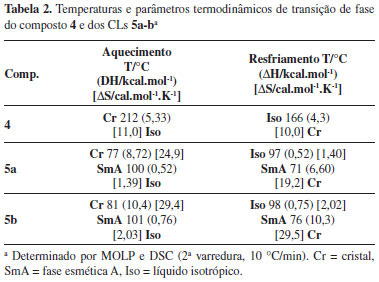
Os CLs 5a-b foram preparados pela sequência de reações: a) a proteção do grupo amina por uma reação de acetilação da anilina com anidrido acético, em meio tamponado de ácido acético/acetato de sódio, na preparação da acetanilida (1). Esta reação é rápida e eficiente. A acetanilida obtida tem elevado grau de pureza, não necessitando recristalização, desde que a anilina empregada seja previamente destilada. A destilação da anilina pode ser realizada pelos próprios alunos no inicio da aula, em paralelo com a síntese da acetanilida. b) a nitração da acetanilida para obtenção da p-nitroacetanilida (2). Aqui a reação forma majoritariamente o produto de substituição para em relação ao orto (64:36 por CG), sendo este último eliminado na recristalização com etanol, devido a sua maior solubilidade neste solvente. O emprego de técnicas de cromatografia gasosa (CG ou CG-EM) para a caracterização da proporção dos regioisômeros orto e para no produto nitrado é uma alternativa interessante para esta aula. A proposta é que os próprios alunos façam as análises, injetando alíquotas do produto bruto, do produto recristalizado e da água-mãe da recristalização (vide Material Suplementar). c) a hidrólise da p-nitroacetanilida para fornecer o intermediário-chave p-nitroanilina (3). Para uma pureza adequada e evitar perdas na recristalização, foi observada a necessidade de uma filtração, usando papel pregueado, da solução ácida do sal de amínio intermediário, antes de precipitar a p-nitroanilina com adição da base. d) diazotização da p-nitroanilina seguida de reação de acoplamento diazo com fenolato de sódio, fornecendo o corante azo 4, de coloração vermelho-alaranjado. Esta etapa é a mais crítica de toda a sequência, havendo bastante perda no rendimento, principalmente relacionada à dificuldade do controle da temperatura, que não deve exceder 10 ºC. Os alunos de graduação obtiveram no máximo 21% de rendimento nesta etapa. A aluna de pós-graduação, com mais experiência em síntese orgânica, conseguiu obter 47% de rendimento na síntese do corante azo 4, embora tenha empregado vidraria e equipamentos mais adequados para pequena escala, não disponíveis no laboratório de Ensino onde foi realizado este trabalho. De qualquer forma, as quantidades obtidas pelos alunos de graduação (em média 450 mg por equipe, quando partindo de 3,6 g de anilina) são mais que suficientes para serem usadas na última etapa. e) O-alquilação do corante azo, empregando método de eterificação de Willianson com 1-bromodecano ou 1-bromododecano, carbonato de potássio como base e butanona como solvente.13 Esta reação em meio heterogêneo (cetona e carbonato) tem a desvantagem de necessitar de agitação constante, contudo, mostrou-se mais eficiente quando comparado ao método empregando meio homogêneo (metanol e hidróxido de potássio). O refluxo foi realizado usando uma chapa de aquecimento com agitação magnética, de forma muito eficaz. O fato de ser uma reação de SN2 faz com que o solvente polar aprótico seja mais eficiente, aumentando a velocidade da reação. Ainda assim, o tempo necessário de refluxo para a total conversão no produto, sem uso de catalisador, foi de 30 h. A conversão foi acompanhada por cromatografia em camada delgada (SiO2-gel, eluente hexano:acetato de etila 4:1). Para adequar ao tempo de uma aula experimental, decidimos por 2 h de refluxo. Ainda que todo o fenol não tenha sido consumido durante este período, ele foi facilmente removido do meio, por extração com solução aquosa de NaOH 10%, em um funil de separação. O produto 5a foi obtido com rendimento de apenas 28%, usando-se este protocolo, todavia suficiente para caracterização estrutural e de sua mesofase. O rendimento foi aprimorado significativamente com uso de iodeto de potássio como catalisador. Embora sem a conversão total em 2 h de refluxo, o composto 5a foi isolado com 77% de rendimento, após o tratamento com base. Com relação ao solvente empregado, a butanona, por possuir uma temperatura maior de refluxo (p.e. 80 ºC), foi mais eficiente que a acetona (p.e. 57 ºC), embora esta última possa ser empregada. Naturalmente, o uso de acetona como solvente forneceu um menor rendimento do produto (35% em 2 h de refluxo com KI), devido ao tempo mais prolongado, necessário para uma maior conversão. Outro solvente, que pode ser empregado, é a N,N-dimetilformamida (DMF),11,14 não testado em nossos experimentos. Os compostos obtidos nas sínteses foram caracterizados a cada etapa, usando técnicas simples de medida do ponto de fusão e comparação com dados da literatura e algumas vezes com a formação de derivados.12 O uso nas aulas práticas de técnicas modernas de análise, tais como Infravermelho (IV) e Ressonância Magnética Nuclear (RMN), são fundamentais para a formação do químico. Neste contexto, a presente síntese multietapas produz compostos muito interessantes para a aprendizagem de interpretação de espectros, uma vez que os compostos vão crescendo em complexidade estrutural na medida em que as sínteses avançam. Os CLs 5a e 5b obtidos para este trabalho foram caracterizados por IV, CG-EM e RMN de 1H. As propriedades líquido-cristalinas exibidas pelos compostos 5a-b são apresentadas na Tabela 2.
A caracterização das mesofases, exibidas pelos CLs sintetizados, pode ser feita por diversas técnicas, desde a simples observação com um equipamento de ponto de fusão, até o uso de microscópio ótico de luz polarizada e de calorimetria diferencial de varredura (DSC). Mesmo sem o uso de equipamento sofisticado, foi possível caracterizar as transições de fase, com a amostra em um tubo capilar, sob o aquecimento em um aparelho de medida de fusão (Figura 1S no material suplementar deste artigo). Contudo, a transição Cr-SmA foi mais evidente e melhor relatada pelo observador do que a transição SmA-Iso, devido à coloração avermelhada típica de composto azo, que apenas fica mais translúcida quando vai ao líquido isotrópico. Os compostos exibiram mesofase SmA, confirmada pela textura focal cônica,15 observada por MOLP no resfriamento do líquido isotrópico (Figura 2). As temperaturas de transição para a fase SmA ficaram um pouco abaixo dos valores relatados para estes compostos na literatura. Mesmo as poucas referências existentes,11 contendo dados térmicos destes compostos, são discrepantes no que diz respeito às temperaturas de entrada na fase SmA. Contudo, as transições de SmA-Iso foram praticamente idênticas às relatadas. As transições de fusão para a SmA, observadas pelo método simples com tubo capilar, ficaram em média 4 ºC acima dos valores obtidos por DSC.
 Figura 2. Fotomicrografia da textura focal cônica exibida pela SmA do composto 5b a 93 °C, obtidas por MOLP (ampliaçao 66 x)
O termograma obtido por DSC do composto 5b é apresentado na Figura 3a. Ambos compostos deram termogramas muito didáticos, com picos bem definidos para as transições de fase. Para efeito comparativo de um CL que funde para uma mesofase, com a de um composto sólido, que funde diretamente para a fase líquida, o termograma do composto 4 é exibido na Figura 3b. As entalpias de transição foram calculadas (Tabela 2) e ficou evidente que as energias envolvidas na fusão do estado sólido para a fase líquido-cristalina são significativamente maiores do que as energias no processo de entrada no estado líquido. Este resultado é interessante ponto para discussão nas aulas, pois reflete a similaridade estrutural da fase CL com a fase líquida. Assim, o estudo do comportamento térmico desta intrigante classe de materiais por DSC é uma excelente alternativa para aulas experimentais de Físico-Química, onde os CLs preparados no laboratório de Ensino de Orgânica seriam utilizados. Além da propriedade mesomórfica, também existe a possibilidade de fotoisomerização da dupla N=N do grupo azo, levando ao estereoisômero Z, com a perda da estrutura anisotrópica que sustenta a mesofase.16 Deste modo, experimentos de fotoisomerização destes compostos são possíveis em aulas de Físico-Química, ampliando a relevância da síntese multietapas que propomos neste trabalho.
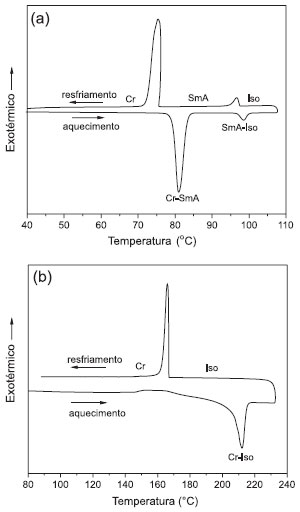 Figura 3. Termogramas obtidos por análises de DSC dos compostos (a) CL 5b e (b) sólido 4, (2ª varredura, 10 °C/min)
PARTE EXPERIMENTAL Síntese Multi-etapas Aula 1: Síntese da acetanilida (1) Em um béquer de 100 mL, foram adicionados 3,5 mL (0,04 mol) de anilina, 1,1 g (0,01 mol) de acetato de sódio e 4,0 mL (0,07 mol) de ácido acético. Sob agitação constante, anidrido acético (5,0 mL, 0,05 mol) foi adicionado, em pequenas porções. Após 10 min, a mistura resultante foi vertida sobre 120 mL de água gelada. Os cristais formados foram coletados por filtração a vácuo e colocados em dessecador até a aula seguinte. Forneceu 3,4 g de um sólido branco (Rend. = 66 %). P.f.: 111-113º C. (lit. 114-115 ºC).17 Uma pequena amostra da acetanilida foi recristalizada em água, fornecendo um material mais cristalino (p.f. 113-114 ºC). Aula 2: Síntese da p-nitroacetanilida (2) Em um béquer de 50 mL, foram adicionados 3,34 g (0,025 mol) de acetanilida, 4,6 mL (0,08 mol) de ácido acético glacial e 9,6 mL de H2SO4. A solução resultante foi resfriada entre 0 e 5 ºC, em banho de gelo. Separadamente, em um tubo de ensaio, foi preparada uma solução nitrante, composta de 2,2 mL (0,05 mol) de HNO3 e 1,4 mL de H2SO4. Esta solução foi resfriada entre 0 e 5 ºC e, a seguir, adicionada lentamente sobre a solução ácida de acetanilida, com agitação constante e de maneira que a temperatura no meio reacional nunca ultrapassou os 10 ºC. Após adição, a mistura reacional foi deixada em repouso por 30 min à temperatura ambiente. Depois foi vertida sobre uma mistura de gelo/água (70 mL), e o precipitado amarelo foi filtrado, sob vácuo. O produto bruto foi recristalizado em etanol, fornecendo 3,16 g de cristais amarelos (Rend. = 62 %). Os cristais permaneceram em dessecador até a aula seguinte. P.f.: 214-215 ºC. (lit. 215 ºC ).18 Aula 3: Síntese da p-nitroanilina (3) Para um balão de fundo redondo de 50 mL, equipado com condensador, foram adicionados 3,10 g (0,017 mol) de p-nitroacetanilida e 16 mL de uma solução aquosa 70% de H2SO4. A mistura resultante foi refluxada por 25 min. Após esfriar, o meio reacional é vertido sobre 130 mL de água fria em um béquer. A solução avermelhada foi filtrada com papel pregueado para eliminar impurezas sólidas presentes. Para precipitar a p-nitroanilina, o pH do meio foi ajustado a 13 com adição cuidadosa de solução de NaOH 6 M. Os cristais foram coletados por filtração a vácuo, lavando-os com água fria. O sólido amarelo escuro foi deixado em dessecador até a aula seguinte. Forneceu 1,80 g, (Rend. = 76 %). P.f.: 147-149 ºC. (lit. 146-147 ºC).19 Aula 4: Síntese do p-(4-nitrobenzenoazo)-fenol (4) Em um béquer de 100 mL, foram adicionados 1,38 g (0,01 mol) de p-nitroanilina (3) 2 mL (0,036 mol) de H2SO4 e 10 mL de água. A solução resultante foi resfriada abaixo de 5 ºC com agitação constante. Uma solução gelada de NaNO2 (0,69 g, 0,01 mol) em 2 mL de água foi adicionada lentamente, de maneira que a temperatura do meio reacional não ultrapassasse 10 ºC, durante 10 min. A suspensão resultante foi adicionada lentamente sobre uma solução de 0,94 g (0,01 mol) de fenol em 5 mL de NaOH(aq.) 1 M, mantida a 5 ºC em um béquer de 100 mL. A suspensão avermelhada resultante foi agitada constantemente por 5 min, sob banho de gelo, sendo os cristais coletados por filtração sob vácuo. Forneceu 1,14 g (47%) de um sólido vermelho-alaranjado. P.f. 202 - 204 ºC (lit. 207 ºC).20 IV (KBr pastilha) νmax/cm-1: 3405 (O-H), 1507, 1337. Aula 5: Procedimento geral para os 4-(alquilóxi)-4'-nitroazobenzenos (5a-b) Para um balão de fundo redondo de 100 mL, equipado com condensador, foram transferidos p-(4-nitrobenzenoazo)-fenol (4) (0,320 g, 1,30 mmol), butanona (5 mL), K2CO3 (0,720 g, 5,2 mmol), KI (0,02 g, 0,13 mmol), e o respectivo bromoalcano (1,56 mmol). A mistura reacional foi aquecida à temperatura de refluxo por 2 h, sob agitação constante. Após esfriar à temperatura ambiente, o conteúdo do balão foi vertido sobre 40 mL de água. A solução resultante foi transferida para um funil de separação, adicionando-se 20 mL de acetato de etila. A fase orgânica foi separada, lavada com solução aquosa 10% de NaOH (2 x 15 mL), água (1 x 15 mL) e, por fim, com solução saturada de NaCl (1 x 15 mL). Os solventes foram evaporados em rotaevaporador. O produto bruto foi macerado em hexano (5 mL) para retirar o excesso do bromoalcano, precipitando um sólido alaranjado. O sólido foi filtrado e lavado com hexano, sendo recristalizado em etanol 95%. 4-(Deciloxi)-4'-nitroazobenzeno (5a). IV (KBr pastilha) νmax/cm-1: 2920, 2860, 1585, 1507, 1337, 1243, 1145. RMN 1H (CDCl3) δ/ppm: 8,36 (d, J = 8,9 Hz, 2 H, Ar-H), 8,01-7,94 (m, 4 H, Ar-H), 7,03 (d, J = 8,9 Hz, 2 H, Ar-H), 4,07 (t, J = 6,5 Hz, 2 H, OCH2), 1,90-1,76 (m, 2 H, CH2), 1,49-1,27 (m, 14 H, CH2), 0,88 (t, J = 6,4 Hz, 3 H, CH3). CG-EM m/z (%): 383 (M+, 32), 261 (18), 243 (14), 121 (89), 107 (98), 93 (68), 83 (50), 69 (64), 57 (98), 43 (100). 4-(Dodeciloxi)-4'-nitroazobenzeno (5b). IV (KBr pastilha) νmax/cm-1: 2920, 2860, 1600, 1530, 1345, 1250, 1150. CG-EM m/z (%): 411 (M+, 23), 289 (7), 243 (9), 121 (68), 107 (57), 93 (45), 83 (34), 69 (75), 57 (98), 43 (100).
CONCLUSÕES Os experimentos envolvendo a síntese e caracterização dos CLs derivados do nitroazobenzeno são adequados para a segunda metade do semestre da disciplina de Química Orgânica Experimental (II ou B), tanto no número de aulas como na aprendizagem dos temas abordados. Os compostos alvos foram preparados a partir da anilina em uma sequência de reações com cinco etapas, cada uma, sendo realizada em uma aula de no máximo 3 h. Todas as reações empregaram métodos simples, cujos fundamentos teóricos estão presentes nos programas das disciplinas de Química Orgânica. Esta abordagem é uma ferramenta interessante para a introdução da Química de Materiais Orgânicos Avançados nas aulas de graduação, uma alternativa aos já bem estabelecidos exemplos de síntese de compostos naturais e sintéticos bioativos. As fases líquido-cristalinas puderam ser estudadas pelo uso de um aparato simples de ponto de fusão, bem como usando técnicas mais sofisticadas de microscopia ótica de luz polarizada (MOLP) e calorimetria diferencial de varredura (DSC). As equipes de alunos obtiveram um bom desempenho na parte sintética e seus resultados reunidos podem ser explorados pelo professor nas discussões envolvendo as relações estrutura-propriedade mesomórfica: o efeito de variar o comprimento da cadeia alifática terminal sobre o comportamento térmico do material, com base no que já existe descrito na literatura.11 Além disso, os compostos obtidos nos laboratórios de Ensino de Química Orgânica podem ser usados nas aulas de Físico-Química Experimental, no estudo dos parâmetros termodinâmicos de suas transições de fase.
MATERIAL SUPLEMENTAR No material suplementar, disponível gratuitamente em arquivo PDF em http://quimicanova.sbq.org.br, encontram-se fotografias do capilar contendo amostra do composto 5b, obtidas do interior do aparelho de fusão, fotomicrografias coloridas da fase SmA obtidas por MOLP e cromatogramas obtidos por CG dos regioisômeros orto e para nitroacetanilida (2) presentes no produto antes e depois da recristalização.
AGRADECIMENTOS Aos alunos que cursaram a disciplina de Química Orgânica Experimental II da UFPB no primeiro e segundo semestre de 2010 e 2012. Ao técnico de laboratório de Ensino, C. S. Diniz. Ao CNPq pelo auxílio financeiro. M. G. B. Cabral agradece à CAPES a bolsa concedida.
REFERÊNCIAS 1. Collings, P.J.; Hird, M.; Introduction to Liquid Crystals, 1st ed., Taylor & Francis Ltd.: London, 1997. 2. Brown, G. H.; J. Chem. Educ. 1983, 60, 900. 3. Goodby, J. In Handbook of Liquid Crystals. Part II; Demus, D; Goodby, J.; Gray, G. W.; Spiess, H.-W.; Vill, V. Goodby, J., eds.; Weinheim: Wiley-VCH, 1998, chap. 1. 4. Lehn, J.-M.; Ball, P. Em Neoquímica. A química moderna e suas aplicações; Hall, N., ed.; Bookman: Porto Alegre, 2004, chap. 12; Munn, B. Em Neoquímica. A química moderna e suas aplicações; Hall, N. e colaboradores eds.; Bookman: Porto Alegre, 2004, chap. 14. 5. Bechtold, I. H.; Revista Brasileira de Ensino de Física 2005, 27, 333; Merlo, A. A.; Gallardo, H.; Taylor, T. R.; Quim. Nova 2001, 24, 354. 6. Ault, A.; Techniques and Experiments for Organic Chemistry, 6th ed., University Science Books: Sausalito, CA, 1998, pp 428-430; Patch, G.; Hope, G. A.; J. Chem. Educ. 1985, 62, 454. 7. Jensen, J.; Grundy, S. C.; Bretz, S. L.; Hartley, C. S.; J. Chem. Educ. 2011, 88, 1133. 8. Van Hecke, G. R.; Karukstis, K. K.; Li, H.; Hendargo, H. C.; Cosand, A. J.; Fox, M. M.; J. Chem. Educ. 2005, 82, 1349. 9. Waclawik, E. R.; Ford, M. J.; Hale, P. S.; Shapter, J. G.; Voelcker, N. H.; J. Chem. Educ. 2004, 81, 854. 10. Ely, F.; Hamanaka, M. H. M. O.; Mammana, A. P.; Quim. Nova 2007, 30, 1776. 11. Goldmann, D.; Janietz, D.; Schmidt, C.; Wedorff, J. H.; Liq. Cryst. 1998, 25, 711; Karasz, F. E.; Imrie, C. T.; Attard, G. S.; Macromolecules 1993, 26, 545; Ho, M. -S.; Fung, B. M.; Bayle, J. -P.; Mol. Cryst. Liq. Cryst. 1993, 225, 383. 12. Mohrig, J. R.; Morrill, T. C.; Hammond, C. N.; Neckers, D. C.; Experimental Organic Chemistry; Freeman: New York, NY, 1997; 456-467. 13. Srivastava, R.M.; Neves Filho, R. A. W.; Schneider, R.; Vieira, A. A.; Gallardo, H.; Liq. Cryst. 2008, 35, 737. 14. Saravanan, C.; Kannan, P.; Polym. Degrad. Stabil. 2009, 94, 1001. 15. Demus, D.; Richter, L.; Textures of Liquid Crystals. VCH: Weinheim, 1978; Dierking, I. Textures of Liquid Crystals. Wiley-VCH: Weinheim, 2003. 16. Westphal, E.; Bechtold, I. H.; Gallardo, H.; Macromolecules 2010, 43, 1319. 17. Narahari, S. R.; Reguri, B. R.; Mukkanti, K.; Tetrahedron Lett. 2011, 52, 4888. 18. Ramana, M. M. V.; Malik, S. S.; Parihar, J. A.; Tetrahedron Lett. 2004, 45, 8681. 19. Theodoridis, G.; Manfredi, M. C.; Krebs, J. D.; Tetrahedron Lett. 1990, 31, 6141. 20. Rahman, M. L.; Tschierske, C.; Yusoff, M.; Silong, S.; Tetrahedron Lett. 2005, 46, 2303. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access