
 |
 |
 |
 |
 |
Nota Técnica
|
|
| Considerações e implicações práticas do guia de validação e controle de qualidade analítica de fármacos em produtos para alimentação animal e medicamentos veterinários Considerations and practical implications of the guide for validation and analytical quality control of drugs in feed and veterinary drugs |
|
Susanne Rath*; Mónica Johanna Martínez-Mejia; Cláudia Hoffmann Kowalski Schröder
Departamento de Química Analítica, Instituto de Química, Universidade Estadual de Campinas, CP 6154, 13084-971 Campinas - SP, Brasil Recebido em 16/09/2014 *e-mail: susanne.rath@gmail.com Recently, the Brazilian Ministry of Agriculture, Livestock and Supply (MAPA) published a validation and analytical quality control guide called the "Guia de Validaçao e Controle de Qualidade Analítica - Fármacos em Produtos para Alimentaçao Animal e Medicamentos Veterinários", in order to guide officers and accredited laboratories in the validation of analytical methods for the quality control of veterinary medicines, drug contaminants in feed and depletion studies (residues of drugs in biological matrices). The aim of this study was to present a critical evaluation of the concepts and procedures defined in this document. The Guide was applied for the validation of chromatographic methods intended to be used for the quantification of antimicrobial and antiparasitic drugs in veterinary medicines. Methods for the determination of ivermectin, abamectin, sulfadiazine, sulfadimethoxine, sulfaquinoxaline, streptomycin, dihydrostreptomycin and florfenicol were validated using the Guide. Each validation parameter was evaluated and discussed. A total of 55 samples of veterinary drugs were analyzed. INTRODUÇÃO Medicamentos veterinários A indústria de produtos farmacêuticos veterinários corresponde ao segmento de saúde animal responsável por manter a saúde e a produtividade dos diversos rebanhos em todo o mundo, bem como por garantir a segurança e a abundância do alimento que produzem.1 O Brasil é um dos cinco maiores mercados veterinários em todo o mundo e o setor vem apresentando franco crescimento sustentado, principalmente, pelo regime de criação intensiva de aves, suínos e bovinos, que implica na maior ocorrência de infecções, ectoparasitoses e verminoses.2-4 Não menos importante é a aquicultura que vem apresentando um crescimento em todo o mundo e também no Brasil. A criação de peixes em tanque rede e o uso de medicamentos veterinários neste segmento tem sido uma prática cada vez mais comum. Além disso, o aumento das exportações de produtos veterinários, a maior fiscalização sanitária, os critérios cada vez mais exigentes para a comercialização e a maior conscientização dos criadores de animais da importância de manter os rebanhos saudáveis têm alavancado esse mercado.2 A grande demanda por esses produtos no Brasil pode ser verificada considerando-se o expressivo número de indústrias voltadas à sua produção instaladas no país, principalmente no estado de São Paulo.4 Em junho de 2014, de acordo com o Compêndio de Produtos Veterinários, publicado pelo Sindicato Nacional da Indústria de Produtos para Saúde Animal (SINDAN) com o apoio da Coordenação de Fiscalização de Produtos Veterinários (CPV) do Ministério da Agricultura, Pecuária e Abastecimento (MAPA), o número de produtos veterinários com licença vigente é de 2.696, provenientes de 108 empresas diferentes.5 Em relação aos grupos de medicamentos, o mercado veterinário está dividido em: biológicos, antimicrobianos, ecto- e endoparasiticidas, endectocidas, terapêuticos, tônicos/fortificantes, desinfetantes, dermatológicos e outros. Dentre esses, fármacos de ação ecto- e endoparasiticida e antimicrobianos somam 679 e 580 registros, respectivamente. Indubitavelmente os antimicrobianos, pelo seu amplo emprego na medicina veterinária e pelo aparecimento de resistência bacteriana, têm sido os mais discutidos, tanto por cientistas e produtores quanto por órgãos governamentais. Resíduos de ecto- e endoparasiticidas em carne proveniente de animais tratados, acima de limites considerados seguros, têm sido os responsáveis por embargos recorrentes de carne industrializada do Brasil e, portanto, têm provocado discussões entre a indústria e o MAPA. Quando os medicamentos veterinários não são utilizados conforme as boas práticas veterinárias, isto é, quando não ocorre observância das instruções de uso contidas na bula (espécie alvo, dosagem, via de administração e/ou período de carência), os mesmos podem não ser eficazes para o propósito em que são empregados. Neste caso pode ocorrer a disseminação de doenças entre os animais e/ou levar a presença de resíduos nos alimentos acima dos limites máximos de resíduos permitidos (LMR), representando não apenas uma ameaça à saúde dos animais tratados, mas também à saúde dos consumidores de produtos derivados desses animais. Ainda, pode ser um objeto de "não conformidade" em relação aos padrões de qualidade exigidos quando do comércio internacional de produtos pecuários.6 Esse cenário de falta de conformidade nos produtos veterinários é uma realidade nem sempre relatada. Em um estudo realizado na África subsaariana7 foi verificado que 42% dos produtos veterinários comercializados em Camarões e 20% dos comercializados no Senegal tinham teores de ativos abaixo do preconizado. Além disso, 4% e 2% dos medicamentos veterinários comercializados em Camarões e Senegal, respectivamente, não tinham o ativo na sua composição e 4% e 33% apresentavam sobredosagem. As não conformidades observadas no decorrer da cadeia produtiva têm sido responsáveis por embargos comerciais internacionais ocorridos nos últimos anos, como a que ocorreu no mês de maio de 2014, quando o governo brasileiro suspendeu o uso da ivermectina de longa ação no país, por terem sido encontrados resíduos do produto em carnes enlatadas exportadas para os Estados Unidos (DOU de 30 de maio de 2014).8 No que diz respeito à fiscalização de medicamentos veterinários no Brasil, existe uma grande lacuna dentro do MAPA quanto à avaliação da conformidade dos produtos que estão sendo comercializados no mercado nacional. Desde 2008 existe uma demanda para que essa fiscalização seja realizada e desde então o Laboratório Nacional Agropecuário de São Paulo (Lanagro-SP) tem preparado sua estrutura para atender essa necessidade. O objetivo final é que, juntamente com os laboratórios credenciados pelo próprio MAPA, essa rede possa realizar as análises fiscais de conformidade de teor de ativos, impurezas presentes e produtos de degradação, testes de desempenho (dureza, friabilidade, viscosidade), identificação de ativos e contaminação cruzada no caso de rações. Legislação para validação de métodos No Brasil, as legislações que regulam os produtos de uso veterinário delegam competência ao MAPA quanto às normas relativas ao registro e fiscalização destes produtos e dos estabelecimentos que os fabricam, manipulam, fracionam, comercializam, importam ou exportam.9-12 Em 2011, a Coordenação Geral de Apoio Laboratorial (CGAL/MAPA) publicou o Guia de Validação e Controle de Qualidade Analítica - Fármacos em Produtos para Alimentação Animal e Medicamentos Veterinários,13 um documento que estabelece os parâmetros a serem avaliados na validação dos procedimentos analíticos e os critérios mínimos a serem atendidos pelos laboratórios oficiais e credenciados para que o método seja considerado adequado ao fim ao qual se propõe. Até a publicação deste documento, as duas agências nacionais que disponibilizavam guias de validação para métodos analíticos eram a Agência Nacional de Vigilância Sanitária (ANVISA), por meio da Resolução RE 899 de 2003,14 cuja publicação é direcionada para fármacos de uso humano, e o Instituto Nacional de Metrologia, Normalização e Qualidade Industrial (INMETRO) por meio das orientações do DOQ-CGCRE-008.15 Já no âmbito internacional, os documentos de maior expressão são os elaborados no contexto da Conferência Internacional em Harmonização de Requerimentos para Registro de Medicamentos de Uso Humano (ICH, International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use)16 que estão harmonizados com o Capítulo 1225 da Farmacopeia Americana intitulado "Validation of Compendion Procedures", o guia "Harmonized Guidelines for Single-Laboratory Validation of Methods of Analysis",17 o guia "Validation of Chromatographic Methods" publicado pela Agência de Alimentos e Medicamentos dos Estados Unidos (FDA, Food and Drug Administration)18 e as recomendações da rede de organizações Europeias (EURACHEM) pelo guia "The Fitness for Purpose of Analytical Methods".19 Na área da medicina veterinária, o governo australiano publicou, em 2004, o Guia intitulado "Guidelines for the Validation of Analytical Methods for Active Constituent, Agricultural and Veterinary Chemical Products".20 O que se pode observar nesses documentos é que não há um procedimento padronizado sobre como avaliar os diferentes parâmetros de uma validação, bem como os critérios de aceitabilidade. Em relação ao uso de métodos normalizados, o Guia de Validação do MAPA sugere que seja realizada a verificação de desempenho desde que não seja feita alteração nos procedimentos, especificações ou técnicas analíticas descritas na farmacopeia.13 Nas versões da Farmacopeia Brasileira,21 inclusive na última versão, revisada em 2010, os medicamentos de uso exclusivo na medicina veterinária não foram contemplados. Enquanto a Farmacopeia Britânica22 tem um volume específico destinado aos medicamentos veterinários, a Farmacopeia Americana23 tem apenas monografias para alguns fármacos de uso veterinário, como por exemplo, a ivermectina. Diante do exposto acima, vê-se a importância do MAPA iniciar a colheita fiscal de medicamentos veterinários comercializados no país, seja na indústria ou no comércio, seguido da análise em laboratório oficial ou credenciado que utiliza métodos validados segundo procedimentos e critérios de aceitabilidade definidos pelo próprio MAPA, bem como um controle de qualidade eficiente na rotina analítica. A publicação do Guia de Validação e Controle de Qualidade Analítica - Fármacos em Produtos para Alimentação Animal e Medicamentos Veterinários13 foi uma iniciativa importante do MAPA no sentido de dar início ao controle da qualidade de medicamentos produzidos no País. Empregando esse guia na validação de métodos visando à determinação de fármacos em medicamentos veterinários foram encontradas dificuldades e algumas limitações, o que nos motivou a escrever esse artigo. Assim, o presente trabalho tem como objetivo aplicar e discutir os conceitos e procedimentos definidos por este guia na validação e controle de qualidade do teor de importantes antiparasitários (ivermectina e abamectina) e antimicrobianos (sulfadiazina, sulfadimetoxina sódica, sulfaquinoxalina, estreptomicina, diidroestreptomicina e florfenicol) utilizados na medicina veterinária, bem como discutir o cenário atual da conformidade dos medicamentos comercializados no país.
PARTE EXPERIMENTAL Reagentes e materiais Todos os reagentes utilizados foram de grau de pureza analítica. Os solventes utilizados foram de grau HPLC e no preparo de soluções foi utilizada água purificada em sistema Milli-Q - 18,2 MΩ cm (Millipore, EUA). As soluções e solventes foram filtrados em membrana de PTFE de 0,22 µm, acopladas a filtros de seringa ou sistema de filtração (Millipore, EUA), respectivamente. Padrões dos fármacos Os padrões de abamectina B1a e B1b (ABM), 98,7%, ivermectina B1a e B1b (IVM), 97,0%, sulfadimetoxina sódica (SDM), 99,0%, e sulfaquinoxalina (SQX), 95,0%, foram adquiridos da Sigma-Aldrich (China). Florfenicol (FLF), 100%, glicose 99,0%, sulfadiazina (SDZ), 99,0%, sulfato de estreptomicina (STRE) potência 758 µg mg-1 e sulfato de diidroestreptomicina (DHS), 98,0%, foram adquiridos da Sigma-Aldrich (EUA). Instrumentação Cromatógrafo 1 (HPLC-DAD): Cromatógrafo a líquido de alta eficiência, Agilent série 1200, composto por sistema de bombeamento quaternário modelo G1311A, forno para coluna modelo G1316A e injetor automático modelo G1329A, detector espectrofotométrico por arranjo de fotodiodos (DAD), cela de 8 µL de volume, 10 nm de caminho óptico, modelo G1315D e sistema de aquisição e tratamento de dados com software ChemStation (Agilent, EUA). Cromatógrafo 2 (HPLC-PAD): Cromatógrafo de íons modular 871 Advance Bioscan, Metrohm (Suíça), composto por sistema de bombeamento 709 IC de duplo pistão com tubulações de PEEK, válvula de injeção e alça de amostragem de 20 µL, detector eletroquímico (PAD) equipado com uma célula amperométrica do tipo wall jet (volume do detector de 0,7 µL) com um eletrodo de trabalho de ouro (8,32 mm de diâmetro), um eletrodo do referência de tipo sólido (patenteado pela Metrohm) e um eletrodo auxiliar de aço inox. Cromatógrafo 3 (HPLC-DAD): Cromatógrafo a líquido de alta eficiência, Thermo Accela (Thermo, EUA), composto por sistema de bombeamento quaternário modelo Surveyor LC PLUS, injetor automático modelo Surveyor PLUS, dotado de controle de temperatura para amostras e forno de colunas integrado, detector espectrofotométrico por arranjo de fotodiodos modelo Surveyor PDA PLUS, cela de 10 µL e sistema de aquisição e tratamento de dados com software ChromQuest 5.0 (Thermo, EUA). Amostras As amostras de medicamentos veterinários foram adquiridas junto ao comércio ou de indústrias nacionais (Brasil), nos anos de 2010 a 2014. No total, foram adquiridas 55 amostras, sendo que três das amostras continham uma combinação de dois fármacos, STRE e DHS. O número de amostras e a forma farmacêutica de cada insumo ativo estão detalhados na Tabela 1, assim como a concentração nominal e o solvente para solubilização da amostra.
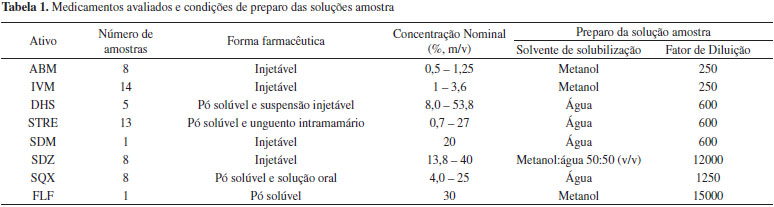
PROCEDIMENTO EXPERIMENTAL Na Tabela 2 são apresentadas as condições cromatográficas otimizadas, levando em consideração os parâmetros de conformidade do sistema, para cada um dos ativos avaliados no procedimento de validação. As sulfonamidas (SDZ, SDM e SQM)24 e o florfenicol foram analisados no cromatógrafo 1.22 Os aminoglicosídeos (STRE e DHS) e as avermectinas (ABM e IVM) nos cromatógrafos 2 e 3, respectivamente. Uma vez que os aminoglicosídeos não apresentam cromóforo, estes foram quantificados com um detector de pulso amperométrico.25
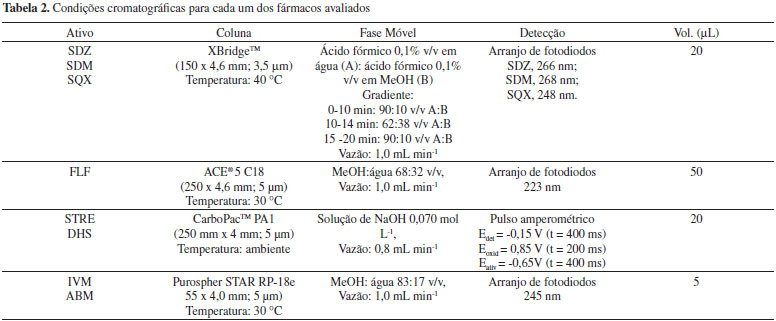
Validação dos métodos Seletividade Soluções intermediárias de cada padrão de fármaco foram preparadas na concentração de 100 µg mL-1 nos seguintes meios: 0,1 mol L-1 ácido clorídrico, 0,1 mol L-1 hidróxido de sódio, 3% v/v peróxido de hidrogênio e em água submetidas à temperatura de 60 ºC (aquecimento em banho-maria). Estas soluções, após serem previamente diluídas (avermectinas para 40 µg mL-1 e os demais fármacos para 20 µg mL-1), foram analisadas por HPLC, empregando as condições descritas na Tabela 2, após 1 hora e 24 horas do preparo das soluções. Em seguida foram analisadas soluções padrão recém-preparadas. Foi avaliada a pureza do pico cromatográfico do fármaco (no caso do detector de arranjo de fotodiodos) em três pontos: início, meio e final do pico, e a eluição de produtos de degradação que pudessem causar interferência. Para os ativos analisados usando a detecção eletroquímica (STRE e DHS) não foi possível realizar a avaliação da pureza do pico, pois o potencial de detecção é mantido constante. Linearidade Foram construídas curvas analíticas (CCAS, curvas de calibração do analito em solução) para cada fármaco usando cinco níveis de concentração (I=5), e em sextuplicata independente de preparo (J=6), seguindo o intervalo de 50 a 150% em relação ao ponto médio da curva selecionado (de 20 µg mL-1 para as sulfonamidas, aminoglicosideos e florfenicol e 40 µg mL-1 para ivermectina e abamectina). As CCAS foram ajustadas pelo método dos mínimos quadrados ordinários (MMQO), após terem sido eliminados valores extremos (outliers) identificados pelo teste de significância dos resíduos padronizados de Jacknife e ter sido comprovada a homocedasticidade da curva pelo teste de Levene.26 As equações das retas foram expressas como: y=a +b×C, na qual C é a concentração de cada fármaco em µg mL-1 e y a área do pico cromatográfico. Os valores de a e b correspondem aos coeficientes linear e angular, respectivamente. Também foram estimadas as incertezas (estimativas dos desvios padrão) de a e b e o coeficiente de regressão linear (r, linearidade). A sensibilidade do método foi obtida pelo coeficiente angular da curva analítica. Ainda, foram plotados os gráficos de resíduos para avaliar possíveis tendências. Efeito matriz de amostra As amostras de medicamentos foram previamente diluídas com solvente conforme descrito na Tabela 1 a uma concentração aproximada de 40 µg mL-1 para as avermectinas e 20 µg mL-1 para os aminoglicosídeos, FLF e sulfonamidas (com base nas informações do fabricante). Estas soluções foram analisadas por padronização externa usando a CCAS e pelo método de adição de padrão (AP). Para o método de AP as amostras contendo avermectinas foram fortificadas com os padrões analíticos nas concentrações de 0; 5,0; 10,0; 15,0 e 20,0 µg mL-1 e as demais amostras contendo florfenicol, sulfonamidas e aminoglicosídeos com 0; 2,5; 5,0; 7,5 e 10,0 µg mL-1. Todas as análises foram realizadas em sextuplicatas independentes. Os resultados foram avaliados de duas formas distintas:
Precisão - repetitividade e reprodutibilidade intralaboratorial Os teores dos insumos farmacêuticos ativos nas amostras de medicamentos foram determinados mediante uso do método de AP conforme descrito no item anterior (efeito matriz). Para tanto, as amostras foram adicionadas do padrão analítico em quatro níveis de concentração. Para o ensaio de repetitividade cada amostra foi analisada em sextuplicata, no mesmo dia, no mesmo equipamento e pelo mesmo analista. Para o ensaio de reprodutibilidade intralaboratorial cada amostra foi analisada em mais dois dias diferentes, pelo mesmo ou outro analista e no mesmo equipamento, totalizando 18 determinações. Os resultados foram expressos como estimativas dos desvios padrão relativos. Veracidade/Recuperação Foram preparadas soluções a partir das amostras de medicamentos em quantidades equivalentes a 40 µg mL-1 para as avermectinas e 20 µg mL-1 para os demais fármacos (calculado com base no valor determinado previamente) e fortificadas com concentrações conhecidas do padrão correspondente: 5 µg mL-1 (nível baixo), 10 µg mL-1 (nível médio) e 20 µg mL-1 (nível alto) para as avermectinas e 2,5 µg mL-1 (nível baixo), 5,0 µg mL-1 (nível médio) e 7,5 µg mL-1 (nível alto) para os demais fármacos. As análises foram realizadas em sextuplicata para cada nível de fortificação. Os resultados foram comparados com o valor nominal e expressos em porcentagem de recuperação. Robustez A robustez foi avaliada por meio do Teste de Youden (ver Tabela 6, na seção de resultados) para cada ativo. As análises foram realizadas em duplicatas independentes e analisadas de forma aleatória. Para cada um dos ensaios foi usada uma solução de trabalho na concentração do ponto médio da curva analítica e foram seguidas as condições cromatográficas e de detecção previamente otimizadas e descritas na Tabela 2. A resposta avaliada foi a área do pico cromatográfico. Quantificação dos insumos farmacêuticos ativos nos medicamentos Os fármacos de todas as amostras, no total de 55, foram quantificados mediante método de AP. Três das amostras continham uma combinação de dois insumos farmacêuticos ativos, STRE e DHS. Todas as análises foram realizadas em triplicata e os valores foram expressos como o valor médio ± U (incerteza expandida) com um fator de abrangência (k) de 2. O fator de incerteza foi calculado pelo método Top Down detalhado no Guia do MAPA,27 sendo levadas em consideração as incertezas da curva analítica e da reprodutibilidade intralaboratorial.
RESULTADOS E DISCUSSÃO Neste trabalho, a validação dos métodos cromatográficos visando a determinação de ABM, IVM, SDM, SQX, SDZ, FLF, STRE e DHS em medicamentos veterinários foi realizada de acordo com as orientações do Guia de Validação e Controle de Qualidade Analítica - Fármacos em Produtos para Alimentação Animal e Medicamentos Veterinários.13 No entanto, durante a operacionalização da validação, alguns critérios e procedimentos não puderam ser seguidos na sua totalidade, como no caso da seletividade, precisão (repetitividade e reprodutibilidade intralaboratorial) e veracidade, sendo necessárias adaptações para dar prosseguimento à validação, os quais são discutidos a seguir. A determinação do fármaco em medicamentos veterinários, matérias primas, produtos para alimentação animal e matrizes de origem biológica, segundo especificado no Guia de Validação do MAPA se enquadra na categoria I (testes quali-quantitativos). Nesta categoria devem ser avaliados os seguintes parâmetros de validação do procedimento analítico: seletividade, efeito matriz, linearidade, precisão, veracidade/recuperação e robustez. Anterior ao processo de validação, os métodos cromatográficos para cada fármaco foram desenvolvidos e as condições ótimas estabelecidas estão apresentadas na Tabela 2. Para a otimização dos métodos foram levados em consideração os parâmetros de conformidade do sistema cromatográfico: resolução (Res), fator de alargamento (TF) e/ou fator de assimetria (As), número de pratos (N) e fator de retenção (k). Os valores a serem aceitos foram os recomendados nas monografias farmacopeicas para cada insumo farmacêutico ativo e, na ausência destes, foram preconizados os valores ou faixas mínimos sugeridos no Capítulo 621 da Farmacopeia Americana:23 Rs>2; 0,9 <As< 1,2; k>2 e N >2000. Ainda, anterior ao processo de validação foram realizados estudos da estabilidade das soluções estoque e de trabalho para cada fármaco (resultados não apresentados). A solução foi considerada estável quando a degradação do fármaco foi menor do que 2% em relação a uma solução recém-preparada. Todas as soluções foram usadas dentro do período de sua estabilidade, respeitando as condições de estocagem. Seletividade A seletividade do método analítico é um parâmetro importante a ser avaliado, uma vez que visa a comprovar que o procedimento analítico em uso permite determinar com confiabilidade o analito na presença da matriz (excipiente do fármaco) sem interferência de concomitantes ou presença de produtos de degradação. O teste de seletividade descrito no guia de validação do MAPA13 segue em íntegra a recomendação do ICH16, o qual prevê a avaliação deste parâmetro para fins de identificação e quantificação. Para fins de identificação o Guia13 recomenda que sejam avaliados compostos com estruturas relacionadas ao fármaco que podem estar presentes na formulação e que esses sejam escolhidos com base em evidências científicas da possível presença destes. Essa recomendação é razoável de ser atendida por laboratórios que produzem o medicamento e que conhecem o processo de síntese. No entanto, para laboratórios analíticos ou de fiscalização essa etapa é difícil de ser cumprida e praticamente não existem publicações científicas disponíveis sobre possíveis impurezas ou concomitantes em medicamentos veterinários. Para o teste de seletividade com finalidade quantitativa o Guia sugere a comparação de resultados obtidos de amostras contaminadas com quantidades apropriadas de impurezas ou excipientes de amostras não contaminadas. Ainda, indica que quando a impureza ou o padrão do produto de degradação não estiver disponível, pode-se comparar os resultados do teste das amostras contendo impurezas ou produtos de degradação com os resultados de um segundo procedimento bem caracterizado. Nesta comparação devem ser incluídas amostras armazenadas sob condições de estresse (luz, calor ou meios ácido, básico ou oxidante). Considerando que várias empresas comercializam um mesmo ativo em uma mesma forma farmacêutica, mas com diferentes excipientes (cuja composição não é revelada), fica praticamente inviável, embora tecnicamente esse procedimento seja correto, para um laboratório de fiscalização avaliar possíveis impurezas e/ou produtos de degradação para uma forma farmacêutica de cada empresa que comercializa o medicamento. Por exemplo, no caso de medicamentos contendo ivermectina existem aproximadamente 50 empresas que comercializam no país mais de seis formulações diferentes, ou seja, em torno de 300 produtos no mercado. Sendo assim, cada formulação teria que ser exposta a cinco condições de estresse, o que representaria, em uma estimativa, em torno de 1500 amostras a serem analisadas (4500 determinações em triplicata) só para a determinação deste parâmetro. Outro ponto importante a destacar é que para métodos cromatográficos é essencial que sejam avaliados os parâmetros de conformidade do sistema (system suitability). Em nenhum momento o Guia faz referência a esse item. É recomendável que a conformidade do sistema seja avaliada antes da validação ou paralelamente à seletividade. Se existir co-eluição de algum composto interferente, o procedimento do método necessita ser modificado no sentido de permitir a separação do analito dos demais compostos. No presente trabalho a seletividade foi avaliada em termo do sinal proveniente da análise da amostra processada e do padrão analítico de interesse. A identidade foi confirmada pela pureza do pico cromatográfico em três pontos do pico. Ainda, em termos de quantificação foi avaliada a resolução do pico do analito com os produtos de degradação formados após exposição dos padrões de ABM, IVM, SDM, SQX, SDZ e FLF a condições de estresse (0,1 mol L-1 ácido clorídrico, 0,1 mol L-1 hidróxido de sódio, 3% v/v peróxido de hidrogênio e soluções aquosas submetidas à temperatura de 60 ºC). A análise dos cromatogramas (HPLC-DAD) permitiu concluir que, embora em alguns meios fosse verificada a formação de produtos de degradação dos padrões, não houve alteração do espectro de absorção no decorrer do pico cromatográfico do fármaco em nenhuma das condições de estresse e nos tempos estudados e também não houve co-eluição de compostos degradados. Ainda, foi realizada uma comparação entre os espectros do padrão e das amostras comerciais nas condições ambientais e não se observou diferenças nos espectros provenientes da matriz.24,25 Estes resultados comprovam que os métodos se comportam de forma seletiva frente às condições e parâmetros cromatográficos previamente otimizados. Para a STRE e DHS não foi possível avaliar a pureza do pico pelo detector empregado ter sido um amperométrico. Para esse método foi avaliada apenas a coeluição de produtos de degradação e/ou do excipiente.25 Uma vez que existem detectores que não permitem a avaliação da pureza do pico, como detectores tipo eletroquímicos ou de espalhamento da luz, é importante a inclusão no Guia de procedimentos e critérios de avaliação da seletividade também para esses detectores. Curva de calibração, linearidade, sensibilidade e faixa de trabalho para fins quantitativos, o método analítico deve gerar resultados que sejam diretamente proporcionais à concentração do analito em solução. Para os métodos cromatográficos o sinal avaliado de modo geral é a área do pico cromatográfico. Para garantir uma curva homocedástica, a relação deve ser linear dentro de um determinado intervalo de concentração, sendo que a faixa de trabalho deve ser selecionada neste intervalo com base na resposta do detector para o analito em questão. Se concentrações muito baixas forem empregadas há um aumento associado da incerteza e se concentrações muito altas forem usadas, pode ocorrer uma sobrecarga na coluna cromatográfica e, em consequência, ocasionar alargamento dos picos e/ou picos assimétricos (cauda frontal). Como a faixa de trabalho deve contemplar a faixa de concentração esperada para o analito na amostra é importante considerar que a amostra se situe no meio da CCAS e que o primeiro e o último ponto não fiquem muito distantes para evitar curvas heterocedásticas. Enquanto a faixa recomendada no Guia da ANVISA14 é de 80 a 120% do teor do analito alvo na amostra (ensaios quantitativos), no Guia Harmonizado de Validação de Métodos Laboratoriais de Análise, publicado pela União Internacional de Química Pura e Aplicada IUPAC17 é de 50 a 150%. Por outro lado, o Guia do MAPA não faz recomendação alguma para esse parâmetro. Neste trabalho foi seguida a recomendação da IUPAC. Para construção da CCAS o Guia do MAPA requer que sejam contemplados cinco níveis de concentração (I=5) em triplicata de preparo (J=3) e duplicata de injeção (L=2), o que leva a um total de obtenção de 30 cromatogramas para cada ativo. Para avaliar a linearidade dos métodos, geralmente os Guias avaliam o coeficiente de correlação linear (r) da CCAS que deve ser superior a 0,99. No entanto, o Guia de Validação do MAPA faz referência a um tratamento estatístico mais detalhado e rigoroso antes da determinação da linearidade, que consiste em avaliar estatisticamente a presença de possíveis valores extremos ou outliers (avaliado pelo teste de resíduos padronizados Jacknife) e verificar se os dados seguem ou não uma distribuição homocedástica (avaliada pelo teste de Levene).26,27 Estas recomendações foram seguidas para cada uma das curvas dos diferentes fármacos e os resultados estão apresentados na Tabela 3.
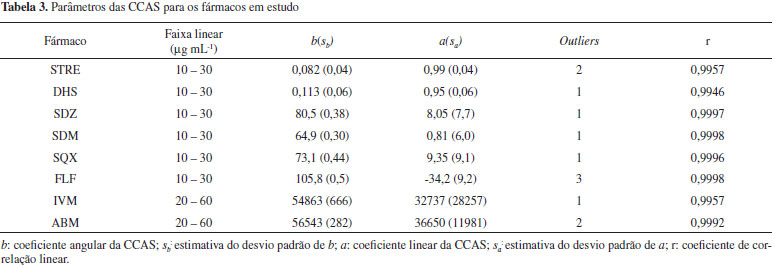
Os desvios padrão de repetitividade das áreas em cada nível de concentração da CCAS para todos os fármacos avaliados foram estatisticamente iguais (α=0,05), confirmando a homocedasticidade das curvas e, portanto, a equação da reta foi estabelecida pelo MMQO. Pela experiência adquirida, constata-se que se as CCAS respeitarem o intervalo de 50% a 150% da concentração do ativo, não haveria necessidade da realização deste teste, pois as CCAS tendem a ser homocedásticas neste intervalo restrito de concentração. A avaliação dos valores extremos pelo teste de resíduos padronizados Jacknife revelou a presença de pelo menos um valor extremo em cada CCAS (Tabela 3). Embora estas tendências fossem observadas também por meio da inspeção visual dos gráficos dos resíduos da regressão, este teste confirmou a exclusão do outlier. Ainda, pela inspeção visual dos gráficos de resíduos observou-se que todos os resíduos estiveram aleatoriamente distribuídos ao redor do eixo "x". Além disso, não foram evidenciados heterocedasticidade (formato cônico) ou desvios de linearidade (forma em "U"). Efeito matriz de amostras Apesar da relevância deste parâmetro, nem todos os protocolos de validação de métodos discorrem sobre experimentos para avaliação dos efeitos de matriz quando se trata da análise de fármacos em medicamentos. Isso se justifica, porque os excipientes geralmente não interferem na determinação ou são insolúveis e separados antes da quantificação. O Guia de Validação do MAPA13 articula que a avaliação do efeito matriz é um estudo de seletividade que objetiva averiguar possíveis interferências causadas pelas substâncias que compõem a matriz amostral, e ainda propõe que este estudo seja delineado com o preparo de curvas analíticas no solvente e curvas analíticas na matriz branca. No caso da indisponibilidade da matriz branca, o Guia preconiza que o efeito matriz possa ser avaliado pela determinação do analito por padronização externa (CCAS) e pelo método de AP. Ainda, recomenda que o procedimento de AP contenha no mínimo cinco níveis de concentração e que a análise completa seja realizada em sextuplicata independente. Para o tratamento estatístico dos dados é sugerido aplicar o teste de homogeneidade de variâncias (teste F) para cada nível i de fortificação (comparação dos resultados da CCAS com AP). Uma vez verificado que a matriz não tem efeito significativo sobre a variância em um nível de significância de 0,05, então devem ser comparadas as médias para cada nível i de fortificação pelo teste t de Student. Finalmente, o Guia propõe a aceitação da não existência do efeito matriz somente quando em todos os níveis de concentração não for detectado um efeito significativo. Esse tratamento estatístico recomendado pelo Guia é bastante trabalhoso e foi verificado neste trabalho que os resultados das variâncias das amostras não-matrizadas e matrizadas nem sempre são iguais para todos os níveis i de fortificação, o que se justifica pelo método de AP levar naturalmente a uma maior dispersão dos resultados do que a CCAS. Neste caso, antes da aplicação do teste t teria que ser estimado o número de graus de liberdade. De modo geral as primeiras fortificações (concentrações menores) levaram a diferenças significativas e as últimas fortificações não. Nesta situação, segundo o Guia, a conclusão seria da presença de efeito matriz, que nem sempre foi confirmada quando o resultado final da análise do medicamento foi avaliado. De fato, a resposta que se busca é avaliar se a matriz pode afetar o resultado final da análise e, portanto, é muito mais razoável comparar os resultados de concentração obtidos na análise de uma amostra pelo método de AP e pela CCAS ou avaliar estatisticamente a inclinação das duas curvas. Realizar o teste estatístico a cada nível i de fortificação é desnecessário para o objetivo a que se propõe. Portanto, neste trabalho, os resultados de quantificação das amostras de medicamentos obtidos pelo método de AP foram comparados com os resultados obtidos pela CCAS. A comparação foi realizada de duas formas: comparando as inclinações das duas curvas matrizada e não-matrizada e comparando o resultado final de concentração do fármaco nas amostras quantificadas por cada um dos métodos (CCAS e AP). Para o medicamento contendo florfenicol, de acordo com o teste F as variâncias não puderam ser consideradas estatisticamente iguais (α=0,05), portanto, foi empregado o teste t para duas médias presumindo variâncias diferentes. Para os aminoglicosideos e as avermectinas , uma vez confirmada a homogeneidade das variâncias foi aplicado o teste t para variâncias iguais. Mediante teste t, foi confirmado que as médias para esses três grupos farmacológicos são estatisticamente iguais nas formulações avaliadas, indicando que não há efeito matriz e, portanto, a determinação destes ativos poderá ser feita usando a CCAS. No caso das sulfonamidas, a avaliação estatística mostrou a presença do efeito matriz, sendo que sua quantificação deverá ser realizada pelo método de adição padrão. Precisão A precisão deve ser avaliada no contexto de repetitividade e precisão intermediária, também denominada de reprodutibilidade intralaboratorial. É importante ressaltar que o Guia do MAPA13 descreve de forma específica os delineamentos para a avaliação deste parâmetro, inclusive traz informação no caso do laboratório não dispor de amostras brancas. No caso de medicamentos veterinários, os laboratórios de fiscalização ou de controle de qualidade podem não ter acesso às matrizes brancas e, neste sentido, a precisão deve ser avaliada mediante análise de amostras pelo método de adição de padrão. Por esse motivo, neste trabalho, a precisão foi determinada pela análise de amostras empregando o método de AP, conforme detalhado na parte experimental. Os resultados (Tabela 4) foram expressos como estimativas do desvio padrão relativo (DPR).
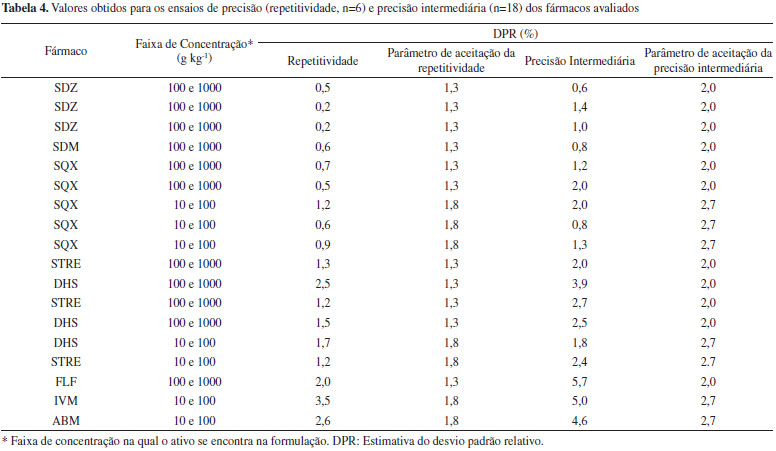
O Guia de Validação do MAPA13 preconiza que em condições de repetitividade o desvio padrão relativo não deve exceder 2/3 do valor previsto pela Equação de Horwitz. No caso da precisão intermediária, o Guia determina que o parâmetro de aceitação seja o próprio valor definido pela Equação de Horwitz. Esses valores estão expressos na Tabela 4 como parâmetro de aceitação da repetitividade e da precisão intermediária, respectivamente. Das 18 amostras analisadas, cinco e seis não se enquadraram nos valores preconizados para a repetitividade e precisão intermediária, respectivamente. Os fármacos foram os aminoglicosídeos, determinados por HPLC com detector eletroquímico, as avermectinas que estão em concentração na faixa de 10 a 100 g L-1 e o florfenicol. De modo geral a detecção eletroquímica leva a uma dispersão maior dos resultados do que um detector de arranjo por fotodiodos. O uso de glicose como padrão interno melhorou os resultados em termos de precisão, mas mesmo assim os resultados não se enquadraram na sua íntegra dentro dos parâmetros de aceitação preconizados pelo Guia do MAPA. Verifica-se também que para a faixa de concentração de 10 a 100 g kg-1 o critério de aceitação nem sempre conseguiu ser atingido e um valor mais apropriado seria considerar aceitável uma variação de 5%. Deve se considerar que o resultado obtido pelo método de AP foi gerado a partir de 30 determinações independentes. Se a quantificação fosse realizada na matriz branca segundo o Guia, o resultado seria gerado a partir de 3 níveis de concentração em 6 réplicas independentes (6 determinações para cada nível). Deste modo, a incerteza do resultado obtido pelo método de AP é maior do que aquela usando a matriz branca, uma vez que mais operações analíticas estão envolvidas, o que se reflete na precisão do conjunto de dados. Ainda, o Guia recomenda que na ausência de matriz branca, a precisão seja calculada para cada nível de fortificação realizado na execução do método de AP e que a precisão em cada nível deva atender o critério de aceitação preconizado. Seria recomendado e mais prático avaliar a precisão do resultado final da análise, que de fato é o valor a ser considerado no cálculo da incerteza da medição e deve ser expresso no resultado final. De acordo com o Guia para realizar o teste de precisão (repetitividade e precisão intermediária), não tendo a matriz branca, são necessárias, no mínimo, 90 determinações para uma única formulação. Considerando que cada corrida cromatográfica leva em torno de 10 minutos, totalizam 15 horas de dedicação do equipamento apenas para avaliar esse parâmetro. Veracidade/Recuperação O termo veracidade do método é empregado para avaliar a concordância entre a média de um número suficientemente grande de resultados de um ensaio e o valor aceito convencionalmente como verdadeiro.13 Em outros guias, como o DOQ-CGCRE-008 do INMETRO,28 esse parâmetro é denominado exatidão. Segundo o Guia do MAPA, a veracidade deve ser avaliada mediante análise de material de referência certificado (6 replicatas) e, na indisponibilidade deste, por intermédio da análise de matriz branca fortificada (3 níveis de fortificação). Novamente, laboratórios de fiscalização ou de controle de qualidade nem sempre possuem a amostra branca, que seria o excipiente, assim como também não existe disponível MRC para medicamentos veterinários. Portanto, o Guia do MAPA não apresenta uma alternativa de procedimento a ser seguido para avaliação da veracidade no caso que não se disponha de MRC ou da matriz branca. Para tanto, neste trabalho foi escolhido avaliar a veracidade dos métodos a partir da fortificação das amostras em três níveis de concentração (baixo, médio e alto) e comparação do resultado com o valor nominal. Os valores foram expressos como porcentagem de recuperação e estão apresentados na Tabela 5.
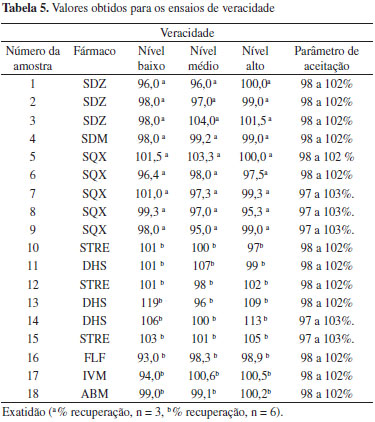
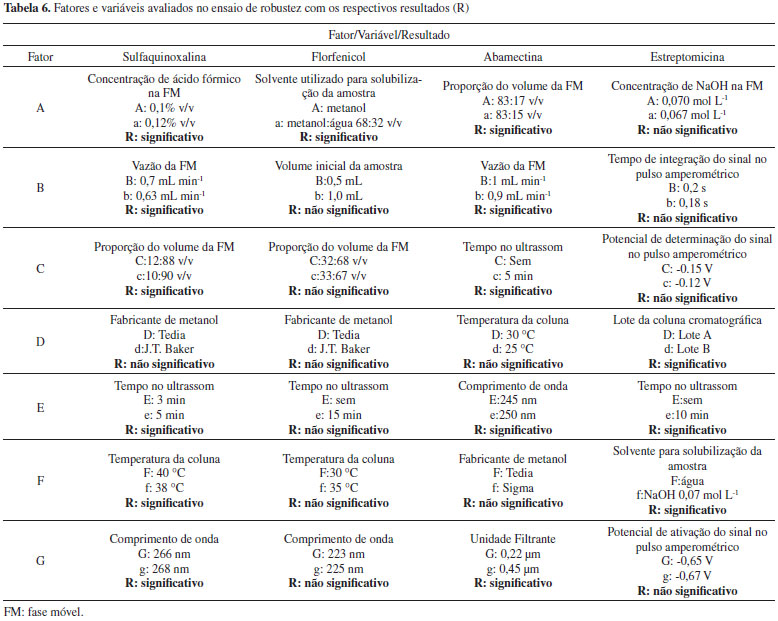
Segundo recomendações do Guia de Validação do MAPA,13 o critério de aceitação de veracidade/recuperação para o teor do fármaco nas amostras é dependente da concentração do mesmo na formulação e previsto pela Equação de Horwitz. Segundo este critério, apenas 4 das 18 amostras analisadas se enquadram nos três níveis de fortificação simultaneamente. As demais amostras encontram-se fora da faixa de recuperação aceita, em pelo menos um dos níveis de fortificação avaliados. Se considerarmos uma faixa aceitável de recuperação de 95% a 105%, apenas duas amostras ficariam fora da faixa aceitável, que são duas amostras de aminoglicosideos (DHS) analisadas por HPLC com detector eletroquímico. Robustez A definição do termo robustez e o procedimento a ser empregado não são harmonizados entre os guias de validação de métodos e nem sempre é mandatória a sua avaliação. Segundo o Guia do MAPA "o estudo da robustez de um procedimento analítico procura avaliar o quão sensível o resultado analítico é às variações nas condições experimentais do procedimento analítico."13 Segundo a Comunidade Europeia29 a robustez (robustness) é a susceptibilidade de um método de ensaio às alterações das condições experimentais, as quais podem ser expressas como tipos de amostras, analitos, condições de armazenamento, condições ambientais ou de preparo de amostra em que o método pode ser aplicado tal como apresentado ou com pequenas alterações específicas. O Guia de Validação do MAPA13 recomenda que o teste de robustez seja realizado utilizando a abordagem clássica, variando um fator de cada vez, ou utilizando a abordagem do planejamento fatorial completo ou fracionário. Contudo, o Guia não propõe quais parâmetros podem ou devem ser avaliados nos testes de robustez de métodos analíticos e nenhuma menção é dada referente a testes estatísticos para avaliar os resultados. Já o Guia do INMETRO28 recomenda o Teste de Youden para determinar a robustez do método. Neste trabalho a avaliação da robustez foi realizada para quatro fármacos (um representante para cada classe de antimicrobiano e um antiparasitário) e foi escolhido o teste de Youden,30 visto que neste experimento é possível examinar, simultaneamente, os efeitos de alterações em sete diferentes fatores do método. Na Tabela 6 são mostrados os fatores e as variáveis selecionados para cada ativo, os quais foram variados levemente em dois níveis (tratamentos) e os resultados (significância dos efeitos) após tratamento estatístico. Foram elencados fatores que poderiam causar alterações no resultado.
A significância dos efeitos foi interpretada graficamente e estatisticamente. A análise dos resultados foi feita mediante a construção do gráfico de Pareto (Figura 1), no qual os efeitos padronizados são calculados mediante o quociente do efeito de cada fator e o erro associado. Para considerar a significância estatística destes fatores, o valor do efeito padronizado foi comparado com o valor de tcrítico tabelado (α=0,05) para oito graus de liberdade (linha vermelha da Figura 1). O resultado obtido para os diferentes fármacos avaliados revela a importância deste parâmetro de validação, pois apesar do método ser devidamente desenvolvido e validado, nem sempre ele pode ser considerado robusto.
 Figura 1. Gráfico de pareto normalizado para um planejamento fatorial fracionário com oito experimentos (N = 8) e sete variáveis
O Guia do MAPA13 descreve que caso seja verificado que o método não é influenciado ou fracamente influenciado por pequenas variações dos fatores, o procedimento analítico é classificado como "procedimento analítico robusto e adequado para análises de rotina". No entanto, se o método é fortemente influenciado, o procedimento pode ser classificado como: (i) "procedimento analítico não robusto e inadequado para análise de rotina", (ii) "procedimento analítico de uso restrito" ou (iii) "procedimento analítico não robusto e inadequado para análise de rotina na faixa de variação estudada dos fatores de influência". Cabe destacar que essa definição é um tanto quanto subjetiva, tendo em vista que fica difícil definir o que seria fracamente ou fortemente influenciado. Para o florfenicol verificou-se que apenas o Fator A - solvente para solubilização da amostra (comparando metanol com metanol:água, 68:32 v/v,) possui um efeito significativo e necessita ser rigorosamente controlado, portanto, poderia se enquadrar o método em "procedimento analítico robusto e adequado para análises de rotina". No caso da abamectina, cinco dos sete fatores foram significativos, sendo os de maior significância a variação no comprimento de onda e na vazão da fase móvel. Para a estreptomicina três dos sete fatores foram significativos: solubilização da amostra na fase móvel e tempo no ultrassom (de 0 a 10 min) e lote da coluna de separação cromatográfica (Fator D). Cabe destacar que, embora já seja conhecido que a STRE é instável em meio alcalino, o fator "F" foi elencado por este ser a fase móvel do sistema. Já no caso da sulfaquinoxalina, seis dos sete fatores foram significativos, sendo o de maior significância a vazão da fase móvel. Pela avaliação dos gráficos de Pareto podem-se classificar os métodos para ABM, SQX e STRE como "procedimentos analíticos de uso restrito". Quantificação dos antimicrobianos em amostras comercialmente disponíveis de medicamentos de uso veterinário Os métodos validados foram empregados na determinação dos oito diferentes fármacos (ABM, IVM, SDZ, SDM, SQX, FLF, STRE e DHS) em cinco tipos de formulações (injetável, suspensão, solução oral, pó solúvel e unguento mamário), totalizando a análise de 55 medicamentos, todos comercializados no mercado nacional. Na Tabela 7 são apresentados 58 resultados sendo que três dos medicamentos tinham dois fármacos (STRE e DHS). Uma vez que os medicamentos eram provenientes de empresas diferentes, mesmo se tratando do mesmos ativos e formulação, poderia haver diferenças na composição do excipiente e haver efeito matriz e, portanto, as quantificações foram realizadas todas pelo método de adição de padrão. Os resultados foram expressos como valor médio da triplicata de análise ± incerteza expandida (U), empregando um fator de abrangência (k) de 2. A incerteza foi calculada pelo método de Top-Down, sendo consideradas as incertezas da curva analítica e da reprodutibilidade intralaboratorial. Não foi considerada a incerteza de amostragem, uma vez que não se tinha esse parâmetro, e da recuperação, uma vez que os resultados eram provenientes de uma curva de adição de padrão. Os resultados das concentrações do ativo em cada amostra, suas respectivas incertezas, tipo de formulação, concentração nominal, limites farmacopeicos e região de aceitação estão reportados na Tabela 7.
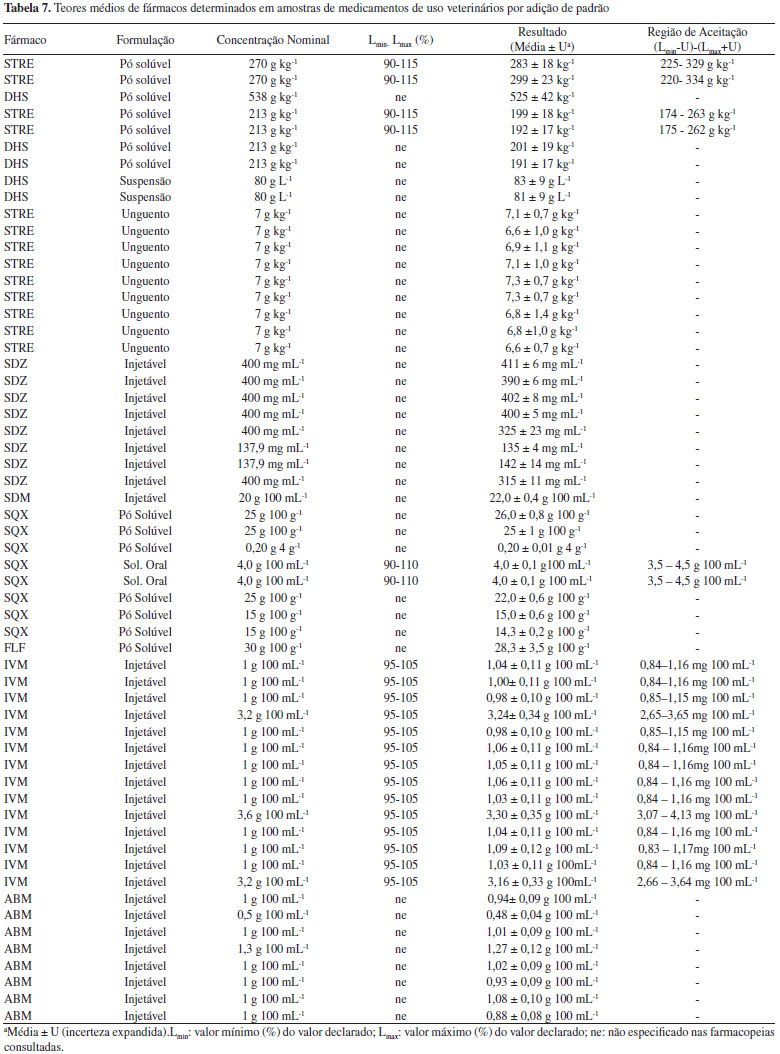
Para a interpretação dos resultados de medição, o Manual de Garantia da Qualidade Analítica do MAPA27 recomenda o estabelecimento de regras de decisão contemplando bandas de segurança, a fim de evitar a probabilidade de falsa rejeição (falsa não conformidade, falso não conforme, falso positivo) ou a probabilidade de falsa aceitação (falsa conformidade ou falso negativo). Assim sendo, o resultado de quantificação obtido é comparado com uma faixa ou intervalo. O estabelecimento destas faixas leva em consideração os valores de referência estabelecidos nas farmacopeias (mínimo e máximo) e a incerteza expandida (U) com fator de abrangência (k) de 2 calculada para cada formulação. Cabe destacar que a Farmacopeia Brasileira21 não contempla monografias específicas para medicamentos de uso veterinário. Dos fármacos avaliados apenas a estreptomicina na formulação em solução injetável e em pó estéril tem monografia descrita na Farmacopeia Brasileira, indicando os limites de aceitação de no mínimo 90,0% e, no máximo 115,0% da quantidade declarada no rótulo do medicamento, sendo essa mesma faixa especificada pela Farmacopeia Americana.23 No entanto, a resolução da ANVISA RDC nº 37 de 200931 estabelece que na ausência de monografia oficial do fármaco, formas farmacêuticas, correlatos e métodos gerais inscritos na Farmacopeia Brasileira poderão ser adotados os intervalo estabelecidos em outras monografias oficiais. Sendo assim, neste trabalho foram também consultadas as Farmacopeias Americana e Britânica. Nestas Farmacopeias foram encontradas monografias para sulfaquinoxalina solução oral23 e ivermectina formulação injetável.22,23 Para os demais medicamentos (35 ao total) não existem, até o presente, monografias disponíveis nestas farmacopeias e, portanto, não foi possível avaliar se o fármaco no medicamento está em conformidade ou não com o teor declarado. Para a SDZ, embora não se tenha monografia disponível, observou-se discrepância entre o valor determinado (325 mg mL-1 e 315 mg mL-1) e o nominal (400 mg mL-1) para duas amostras injetáveis, o que leva a indicar uma não conformidade. As amostras analisadas que tem monografias descritas (20 no total), contendo os fármacos estreptomicina, sulfaquinoxalina e ivermectina tiveram seus resultados dentro da região de aceitação e, portanto, podem ser declaradas conformes. Vale a pena observar que a região de aceitação inclui a faixa especificada descrita na monografia e a incerteza expandida do método o que leva a uma ampliação considerável da faixa de aceitação final, em torno de ±20%.
CONCLUSÕES E PERSPECTIVAS A publicação do Guia de Validação do MAPA para medicamentos veterinários foi sem dúvida um grande avanço no sentido de harmonizar, em nível nacional, um procedimento de validação de métodos destinados para o controle e qualidade de medicamentos veterinários. Esse Guia, no entanto, é muito abrangente e visa além do estabelecimento de parâmetros de desempenho que devem ser atendidos em procedimentos analíticos destinados ao controle de qualidade de medicamentos veterinários, determinação de fármacos como contaminantes em ração, assim como estudos de depleção de resíduos em matrizes de origem animal. Apesar de a ideia de elaborar um guia geral e abrangente ser interessante, na prática introduz dificuldades e questionamentos. Os objetivos de um método para a determinação de resíduos de fármacos em matrizes biológicas são diferentes do que os de um método a ser empregado no controle e qualidade de insumos farmacêuticos ativos em medicamentos. Neste sentido, fica evidente a dificuldade em estabelecer um procedimento único para finalidades tão distintas no que tange a validação dos métodos. Embora o Guia descreva os parâmetros necessários a ser avaliados para cada finalidade, o mesmo não detalha o procedimento a ser adotado para cada caso. No decorrer da realização dos experimentos conforme descrito neste trabalho identificaram-se algumas dificuldades de ordem prática, seja pela falta de clareza na descrição dos procedimentos, seja pelos critérios rígidos de aceitação exigidos. O Guia recomenda um número excessivo de repetições na avaliação dos diferentes parâmetros, o que torna o processo de validação altamente dispendioso, tanto no que se refere ao trabalho laboratorial como ao custo do processo como um todo., , Pelos resultados obtidos neste trabalho foi verificado que seria possível reduzir o número de análises sem comprometer a qualidade dos resultados finais. O tratamento estatístico a ser seguido é bastante trabalhoso e requer um número excessivo de planilhas. Em alguns parâmetros o tratamento estatístico poderia ser reduzido ou simplificado. Um ponto importante a destacar é a não recomendação da avaliação dos parâmetros cromatográficos do sistema (system suitability) pelo Guia, que podem levar a falhas na otimização do método, principalmente no que tange à seletividade, que é um parâmetro de validação. Seria interessante incluir no Guia uma recomendação geral para os parâmetros: resolução, fator de retenção, número de pratos e assimetria que poderiam ser usados na ausência de especificações descritas nas monografias. Ainda, a falta de monografias para medicamentos veterinários nas farmacopeias com descrição das faixas de tolerância do insumo farmacêutico ativo nos medicamentos não permite a tomada de decisão com respaldo legal quanto a declarar uma amostra conforme ou não conforme. E mesmo para os medicamentos que têm limites mínimos e máximos estabelecidos nas monografias, a região de aceitação se torna bastante ampla quando a incerteza expandida é considerada no cálculo, permitindo uma ação fiscal somente quando alterações bastante significativas na formulação forem diagnosticadas.
AGRADECIMENTOS Os autores agradecem ao Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq/MAPA/SDA 064/2008 e Projeto Sagres 457417/2012-9) e a Fundação de Amparo à Pesquisa do Estado de São Paulo (Proc. 2009/10443-1 e Proc. 2013/09543-7) pelos auxílios recebidos. C. H. K. Schröder agradece ao CNPq pela bolsa de pesquisa concedida (Proc. 350404/2014-3).
REFERÊNCIAS 1. Omote, H. S. G.; Sluszz, T.; Prospecçao de Mercado visando P&D para Medicamentos Veterinários para Bovinocultura no Brasil. Anais SIMTEC - ISSN: 2318-3403, Aracaju, 2013, 1, 444. DOI: http://dx.doi.org/10.7198/S2318-34032013001038 2. Capanema, L. X. L.; Velasco, L. O. M.; Souza, J. O. B.; Noguti, M. B.; Panorama da indústria farmacêutica veterinária. BNDES Setorial, Rio de Janeiro, 2007, 25, 157. 3. Sindicato Nacional da Indústria de Produtos para Saúde Animal, SINDAN, http://www.sindan.org.br/sd/base.aspx?controle=8, acessado em Setembro 2014. 4. da Costa, F. M.; Netto, A. D. P.; Quim. Nova 2012, 35, 616. DOI: http://dx.doi.org/10.1590/S0100-40422012000300031 5. Coordenaçao de fiscalizaçao de produtos veterinários (CPV); Relatório de produtos com licença vigente. http://www.cpvs.com.br/cpvs/>, acessado em Junho 2014. 6. Marshall, B. M.; Levy, S. B.; Clin. Microbiol. Rev. 2011, 24, 718. DOI: http://dx.doi.org/10.1128/CMR.00002-11 PMID: 21976606 7. Teko-Agbo, A.; Messomo Ndjana, F.; Walbadet, L.; Akoda, K.; Niang, EL.H.; Abiola, F.A.; Quality of veterinary medicinal products in circulation in Cameron and Senegal. OIE Conference on Veterinary Medicinal Products in Africa, Dakar, 2008. 8. Diário Oficial da Uniao de 30 de Maio de 2014, Seçao 1, Página 55. Disponível em <http://pesquisa.in.gov.br/imprensa/jsp/visualiza/index.jsp?jornal=1&pagina=55&data=30/05/2014>, acessado em Junho 2014. 9. Ministério da Agricultura, Pecuária e Abastecimento (MAPA); Decreto-Lei nº 467, de 13/02/1969. Dispoe Sobre a Fiscalizaçao de Produtos de Uso Veterinário, Dos Estabelecimentos que os Fabriquem e da Outras Providencias. Publicada no Diário Oficial da Uniao de 14/02/1969, Seçao 1. http://extranet.agricultura.gov.br/sislegis-consulta/>, acessado em Julho 2014. 10. Ministério da Agricultura, Pecuária e Abastecimento (MAPA); Decreto nº 1662, de 06/10/1995. Aprova o Regulamento de fiscalizaçao de produtos de uso veterinário e dos estabelecimentos que os fabriquem e comerciem, e dá outras providências. Publicada no Diário Oficial da Uniao de 09/10/1995. http://extranet.agricultura.gov.br/sislegis-consulta/>, acessado em Junho 2014. 11. Ministério da Agricultura, Pecuária e Abastecimento (MAPA); Decreto nº 301, de 19/04/1996. Aprova as normas complementares anexas, elaboradas pela Secretaria de Defesa Agropecuária, a serem observadas pelos estabelecimentos que fabriquem e ou comerciem produtos de uso veterinário.Publicado no Diário Oficial da Uniao de 25/04/1996, Seçao 1. http://extranet.agricultura.gov.br/sislegis-consulta/>, acessado em Junho 2014. 12. Ministério da Agricultura, Pecuária e Abastecimento (MAPA); Decreto nº 5053, de 22/04/2004. Aprova o Regulamento de Fiscalizaçao de Produtos de Uso Veterinário e dos Estabelecimentos que os Fabriquem ou Comerciem, e dá outras providências. Publicado no Diário Oficial da Uniao de 23/04/2004, Seçao 1. http://extranet.agricultura.gov.br/sislegis-consulta/>, acessado em Junho 2014. 13. Ministério da Agricultura, Pecuária e Abastecimento (MAPA); Guia de Validaçao e Controle da Qualidade Analítica. Fármacos em Produtos para Alimentaçao Animal e Medicamentos Veterinários, Secretaria de Defesa Agropecuária. MAPA/ACS. Brasília, 2011. 14. Agência Nacional de Vigilância Sanitária (ANVISA); Guia para Validaçao de Métodos Analíticos e Bioanalíticos, RE nº 899, de 29/05/2003. http://portal.anvisa.gov.br/wps/content/Anvisa+Portal/Anvisa/Inicio/Medicamentos/Assunto+de+Interesse/Equivalencia+farmaceutica/Legislacoes/>, acessado em Junho 2014. 15. Instituto Nacional de Metrologia, Normalizaçao e Qualidade Industrial (INMETRO). Orientaçoes sobre Validaçao de Métodos de Ensaios Químicos, DOQ-CGCRE-008, 2ª revisao, 2007. 16. International Conference on Harmonization (ICH); Validation of Analytical Procedures: Text and methodology Q2 (R1), 2005. 17. Thompson, M.; Ellison, S. L. R.; Wood, R.; Pure Appl. Chem. 2002, 74, 835. 18. Center for Drug Evaluation and Research, Food and Drug Administration (CDER-FDA); Reviewer Guidance: Validation of Chromatographic Methods, 1994. 19. Eurachem; The Fitness for Purpose of Analytical Methods. A Laboratory Guide to Method Validation and Related Topics, 1998. http://www.eurachem.org/images/stories/Guides/pdf/valid.pdf, acessado em Junho 2014. 20. Australian Pesticides and Veterinary Medicines Authority (APMVA). Guidelines for the Validation of Analytical Methods for Active Constituent, Agricultural and Veterinary Chemical Products, 2004. http://www.apvma.gov.au/, acessado em Junho 2014. 21. Farmacopeia Brasileira, 5ª ed., v 2. Agência Nacional de Vigilância Sanitária: Brasília, 2010. 22. British Pharmacopoeia (Veterinary); Her Majesty's Stationery Office, London, 2013. 23. United States Pharmacopeia (USP) Convention; US Pharmacopeia 37, Rockville, 2014. 24. Doretto, K. M.; Tese de doutorado, Universidade Estadual de Campinas, Brasil, 2013. 25. Martinez-Mejia, M. J.; Dissertaçao de Mestrado, Universidade Estadual de Campinas, Brasil, 2013. 26. de Souza, S. V. C.; Junqueira, R. G. A.; Anal. Chim. Acta 2005, 552, 25. DOI: http://dx.doi.org/10.1016/j.aca.2005.07.043 27. Ministério da Agricultura, Pecuária e Abastecimento (MAPA); Manual de Garantia da Qualidade Analítica - Resíduos e Contaminantes em Alimentos. MAPA/ACS, Brasília, 2011. 28. Instituto Nacional de Metrologia, Normalizaçao e Qualidade Industrial (INMETRO); Orientaçoes sobre Validaçao de Métodos de Ensaios Químicos, DOQ-CGCRE-008, 4ª revisao, 2011. 29. European Commission; Official Journal of the European Communities, 17/08/2002, L221/8-36. 30. Youden, W. J.; Steiner, E. H.; Statistical Manual of AOAC, Association of Official Analytical Chemistry. Washington: AOAC, 1975. 31. Agência Nacional de Vigilância Sanitária (ANVISA); Resoluçao nº 37, de 06/07/2009. Trata sobre a Admissibilidade das Farmacopeias Estrangeiras. Publicada no Diário Oficial da Uniao de 08/07/2009, Seçao 1. http://portal.anvisa.gov.br/wps/wcm/connect/e9d6f98048e524df8f859f466b74119d/RDC_37_2009_Trata%2Bda%2Badmissibilidade%2Bdas%2BFarmacop%C3%A9ias%2Bestrangeiras..pdf?MOD=AJPERES, acessado em Agosto 2014. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access