
 |
 |
 |
 |
 |
Nota Técnica
|
|
| Chemostat, um software gratuito para análise exploratória de dados multivariados Chemostat: exploratory multivariate data analysis software |
|
Gilson A. HelferI,#,*;Fernanda BockI,#; Luciano MarderI,#; João C. FurtadoII,#; Adilson B. da CostaIII,#; Marco F. FerrãoIV
IDepartamento de Química e Física, Universidade de Santa Cruz do Sul, 96815-900 Santa Cruz do Sul - RS, Brasil Recebido em 09/12/2014 *e-mail: ghelfer@gmail.com The objective of this work was to develop a free access exploratory data analysis software application for academic use that is easy to install and can be handled without user-level programming due to extensive use of chemometrics and its association with applications that require purchased licenses or routines. The developed software, called Chemostat, employs Hierarchical Cluster Analysis (HCA), Principal Component Analysis (PCA), intervals Principal Component Analysis (iPCA), as well as correction methods, data transformation and outlier detection. The data can be imported from the clipboard, text files, ASCII or FT-IR Perkin-Elmer ".sp" files. It generates a variety of charts and tables that allow the analysis of results that can be exported in several formats. The main features of the software were tested using mid-infrared and near-infrared spectra in vegetable oils and digital images obtained from different types of commercial diesel. In order to validate the software results, the same sets of data were analyzed using Matlab© and the results in both applications matched in various combinations. In addition to the desktop version, the reuse of algorithms allowed an online version to be provided that offers a unique experience on the web. Both applications are available in English. INTRODUÇÃO A palavra quimiometria surgiu na década de 70 e seu desenvolvimento baseava-se na computação científica, envolvendo principalmente métodos estatísticos multivariados aplicados aos dados da química analítica. Na década de 80 a quimiometria foi organizada como uma disciplina, surgindo as primeiras publicações, associações e cursos dedicados ao tema. Porém, foi na década de 90 que ela começou a se expandir, especialmente na indústria farmacêutica. Desde então, devido à capacidade de instrumentos analíticos em adquirir grandes quantidades de dados de forma mais ágil e, associado ao aumento da capacidade de processamento dos computadores, a quimiometria se estabeleceu como uma ferramenta indispensável para mineração e análise de dados químicos.1,2 Atrelado ao avanço tecnológico e à demanda na área da pesquisa, muitos aplicativos comerciais surgiram, alguns mais flexíveis, como o Matlab® e outros nem tanto, como Pirouette® e Unscrambler®. Porém, todos tendo como requisito a compra de licenças, inviabilizando, muitas vezes, seu uso acadêmico generalizado. Outros aplicativos como o R® e Octave®, apesar de serem isentos de custos de licenciamento, necessitam, assim como o Matlab®, de algum investimento em tempo para a familiarização e interpretação de suas sintaxes. Recentemente surgiu o Chemoface®, um aplicativo gratuito tendo como requisito principal a instalação do MCR (Matlab Compiler Runtime). A vantagem do uso deste compilador é a utilização em várias plataformas como Windows®, Linux® e Mac®, no entanto, a dependência do MCR e seu suporte, a necessidade de uma grande capacidade de memória física (versão atual requer no mínimo 447 Mb) e de privilégios de administrador do sistema operacional para instalação são limitações deste aplicativo. Há ainda outros softwares da área estatística aplicada à biologia ou geografia, alguns gratuitos, outros baseados em linha de comando, entretanto desprovidos de alguns recursos específicos utilizados na quimiometria3-7 . Neste sentido, buscou-se neste trabalho desenvolver um software desktop de fácil adoção, instalação e manuseio, destinado a alunos, professores e pesquisadores, e que abrangesse, primeiramente, a análise exploratória de dados. Além disso, uma solução online básica, destinada aos dispositivos móveis, como tablets, também foi desenvolvida.
PARTE EXPERIMENTAL O software foi gerado num ambiente de desenvolvimento integrado (IDE, do inglês, "Integrated Development Environment"). A função da IDE é reunir características e ferramentas de apoio à construção de softwares com o objetivo de promover este processo de forma mais ágil. Para tanto foi utilizada a IDE Microsoft Visual Studio 2010® versão Professional, que possui um alto nível de abstração de controles e classes, decorrente do uso do pacote Microsoft.NET Framework 4.0.8 As linguagens de programação adotadas foram C# (C-Sharp) e VB (Visual Basic), e foram utilizados algoritmos e bibliotecas de terceiros, como ZedGraph para plotagens de gráficos e o Accord.NET Framework que possui inúmeros algoritmos da área da estatística, todos de código aberto. A solução online também foi desenvolvida no Visual Studio 2010 e contou com a vantagem de reutilização dos algoritmos da versão desktop.9,10 O sistema desktop é compatível com o Windows XP SP3, Windows 7, 8 e 8.1, e o sistema online para qualquer browser, sendo o mais indicado o Internet Explorer. O idioma utilizado para ambas as versões é o inglês.
RESULTADOS E DISCUSSÃO Funções da versão desktop O ChemoStat atua em dados espectrais a partir do infravermelho e na quimiometria de imagens, a partir da decomposição das camadas de cor por pixels. A tela principal do software ChemoStat possui 3 seções, ilustradas na Figura 1.
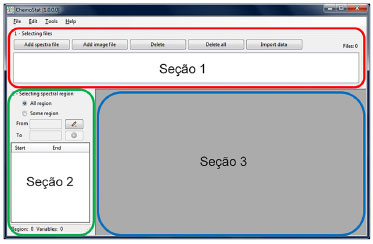 Figura 1. Tela principal do ChemoStat da versão desktop - padrão espectroscopia
Aplicação do software na análise de espectros de infravermelho A importação de dados ocorre por meio de arquivos de espectros gerados pelo espectrofotômetro de infravermelho Perkin-Elmer no formato "sp". Para outros equipamentos os dados devem ser exportados em "asc" (ASCII), além da opção da área de transferência, teclas "Control+C" (copiar) e "Control+V" (colar). Ao serem importados, os dados preenchem uma grade ou matriz de dados, ao clicar com o botão direito do mouse, é apresentado um menu com as opções de plotagem de gráficos, transformações, análises e exportação dos dados, como demonstra a Figura 2.
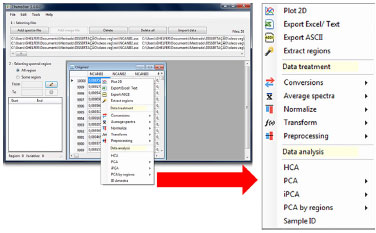 Figura 2. Tela principal padrão espectroscopia com grade de dados - detalhe do menu de operações acionado
O software possui ferramentas matemáticas de transformação e pré-processamento de sinais, como correções, suavizações e normalizações de acordo com os métodos da 1ª e 2ª derivadas, MSC (Multiple Scatter Correction), SNV (Standard Normal Variate) e método de Savitky-Golay, a partir de dados centrados na média ou escalonados, além de conversões de medidas entre absorbância e transmitância. O software também gera médias para replicatas e organiza classes de amostras por nome. Essa função, chamada de "Sample ID", permite a identificação a partir de cores pré-estabelecidas através da análise sintáxica do nome da amostra. Para que isto ocorra, basta informar o número de caracteres semelhantes entre a denominação das replicatas, conforme ilustrado na Figura 3.
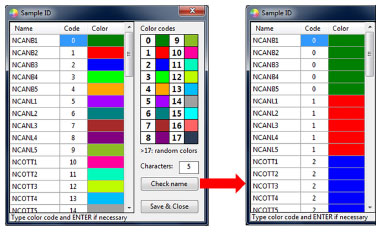 Figura 3. Tela para identificação das amostras por classe do ChemoStat versão desktop
As principais funcionalidades do software foram exploradas utilizando espectros no infravermelho médio e próximo de óleos vegetais e imagens digitais de diferentes tipos de óleo diesel, e como forma de validar os resultados do software, os mesmos conjuntos de dados foram analisados no Matlab®3, cujos resultados estão apresentados como material suplementar deste artigo. As técnicas desenvolvidas para espectroscopia foram: Análise por Agrupamento Hierárquico (HCA), Análise por Componentes Principais (PCA), Análise por Componentes Principais por Intervalos (iPCA) e detecção de amostras anômalas (outliers) pelo método T2 de Hotelling.11,12 A técnica HCA, calculada pela distância Euclidiana, foi desenvolvida com três opções de métodos de ligação: "Single-Linkage", "Complete-Linkage" e "Average-Linkage". A Figura 4 exibe o gráfico do tipo dendograma com ligação completa aplicada ao conjunto de espectros dos óleos vegetais na faixa entre 5500 e 6000 cm-1, normalizados entre os limites zero e um, corrigidos pelo método do valor normal padrão ("SNV") e, posteriormente, centrados na média.
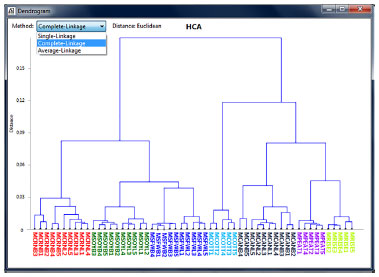 Figura 4. Gráfico do tipo dendrograma HCA - ligação completa do ChemoStat versão desktop
O software ChemoStat possibilita a análise de componentes principais (PCA) nas opções: "Meancenter", para centrar os dados na média; "Autoscale" para autoescalar os dados; ou "None", para nenhum pré-processamento, ou seja, para quando algum dos pré-processamentos já tenha sido realizado em etapas anteriores à grade de dados. A Figura 5 exibe o gráfico de scores a partir da análise dos componentes principais (PCA) aplicada no mesmo conjunto de dados de óleos vegetais utilizados na análise de agrupamento hierárquico.
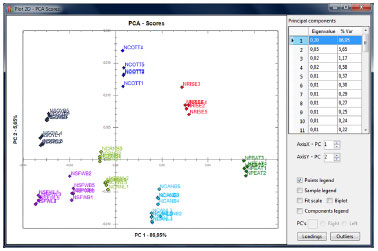 Figura 5. Gráfico de scores PCA do ChemoStat versão desktop
A interface do gráfico PCA (scores) apresenta ainda opções "Loadings" (Figura 6), "Biplot", que permite a visualização dos gráficos de scores e loadings num mesmo plano (Figura 7) e "Outliers", que exibe dados relativos às amostras anômalas calculadas através do método multivariado T2 de Hotelling, demonstrado na Figura 8, todas aplicadas sobre a mesma análise executada nos óleos vegetais.
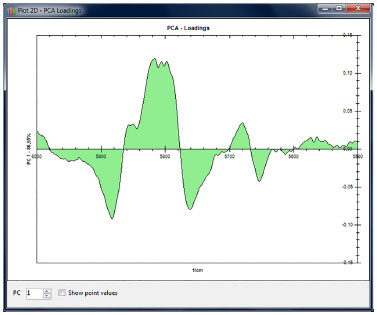 Figura 6. Gráficos de loadings PCA do ChemoStat versão desktop
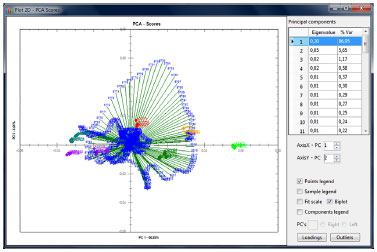 Figura 7. Gráfico Biplot (scores x loadings) do ChemoStat versão desktop
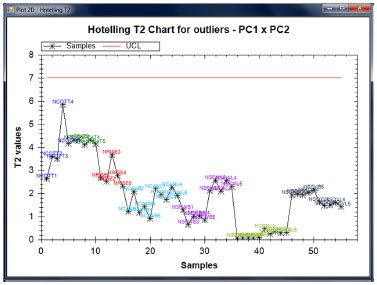 Figura 8. Gráficos de Outliers a partir do T2 de Hotelling do ChemoStat versão desktop
Além disso, permite também a análise dos componentes principais por intervalos (iPCA), muito utilizada para seleção de variáveis espectrais. Nessa opção é solicitado ao usuário o número de intervalos pelos quais deve ser dividido o espectro para geração dos scores (PCA).13 O resultado dessa operação é ilustrado na Figura 9, onde foram escolhidos 6 componentes principais, de modo que as alturas das barras representam, em forma percentual, a variância contida em cada componente principal para cada intervalo. A linha traçada horizontalmente representa a variância de cada uma das componentes principais da análise de PCA para toda a informação do espectro.
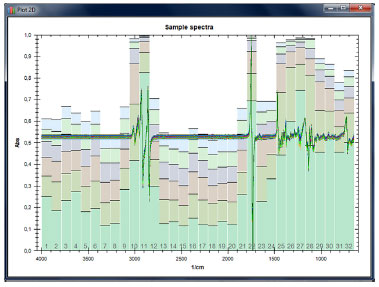 Figura 9. Variaçao percentual das componentes principais divididas em 32 intervalos aplicados nos espectros de óleos vegetais (FT-MIR)
A partir dessa análise, gráficos de scores e loadings são efetuados para cada intervalo de região espectral. Os resultados de todas as análises podem ser exportados para extensão ".xls", ".txt" e ".asc". Já os gráficos, possuem opção de exportação nos formatos de figuras ".bmp", ".png" e ".jpg". Aplicação do software na análise de imagens Na quimiometria de imagens, podem ser analisados dados do histograma R, G e B, ou de cada componente de cor R, G, B, R relativo, B relativo, G relativo, H, S, V, I e L. A importação destes ocorre por meio de arquivos de imagens nos formatos "bmp", "jpg" e "png", além da opção da área de transferência, teclas "Control+C" (copiar) e "Control+V" (colar), conforme ilustra a Figura 10.
 Figura 10. Tela principal padrão imagens - detalhe do menu de operações acionado
As técnicas desenvolvidas para quimiometria de imagens foram: Análise por Agrupamento Hierárquico (HCA), Análise por Componentes Principais (PCA), detecção de amostras anômalas (outliers) pelo método T2 de Hotelling e visualização do histograma. Da mesma forma como nos resultados dos espectros de infravermelho, os resultados das análises de imagens podem ser exportados para extensão ".xls", ".txt" e ".asc". Já os gráficos, possuem opção de exportação nos formatos de figuras ".bmp", ".png" e ".jpg". Funções da versão web (online) O acesso ao software se dá pelos endereços "http://www.chemostat.com.br" e "http://www.chemostat.net", cuja tela de entrada apresenta a opção de registro de usuário (link "here"), e para usuários já registrados, as opções de login (e-mail de entrada), "password" (senha de entrada), "can't access your account" (recuperação de senha), para entrada no sistema.
 Figura 11. Detalhe do acesso via login do ChemoStat versão web
Assim que acessado o sistema, aparecerá a tela principal com comandos básicos para importação de dados via planilha de dados/texto, tratamento de dados, identificação das amostras, técnicas PCA, iPCA e HCA. O sistema permite a entrada de dados via planilhas ou texto utilizando o recurso da área de transferência, teclas "Control+C" (copiar) e "Control+V" (colar), ou via importação de arquivos de espectros gerados pelo espectrofotômetro de infravermelho Perkin-Elmer nos formatos "sp" e "asc" (ASCII), ilustrado na Figura 12.
 Figura 12. Detalhe da entrada de dados do ChemoStat versão web
Além disso, para dados de espectroscopia, podem ser aplicados os tratamentos de sinais pela marcação nas caixas checagem, detalhado na Figura 13. A coluna de marcação "Run order" significa a ordem em que serão aplicados os tratamentos, caso mais de um seja selecionado. Os tratamentos "Moving average", "1st derivative" e "2nd derivative" necessitam que seja informada a quantidade de pontos ("Points"), ou número de ondas, nos quais será aplicado o algoritmo. O sistema ainda permite a identificação a partir de cores pré-estabelecidas através da análise sintáxica do nome da amostra. Para que isto ocorra, deve ser marcada a caixa de checagem "Classify by number of sample characters" e informado o número de caracteres que são semelhantes entre as replicatas, sendo que o valor zero o sistema calcula de forma automática.
 Figura 13. Detalhe das opções de pré-processamento de dados do ChemoStat versão web
O método de PCA para "Scores" é executado através dos botões "Autoscale", cujos dados serão previamente autoescalados; "Meancenter", centrados na média; e "None", para nenhum pré-processamento. Em todos os casos, quando pressionados, uma nova janela abrirá com o gráfico de scores conforme os campos "PC's" preenchidos. O método de PCA para "Loadings" ocorre de forma semelhante à opção de "Scores", no que diz respeito aos botões "Autoscale", "Meancenter" e "None". A tela padrão foi configurada para PC1, podendo ser alterada manualmente. Já o método de HCA, calculado pela distância euclidiana, é executado através dos botões "Single-Linkage", "Complete-Linkage" e "Average-Linkage", denominado pelo método de ligação. Em todos os casos, quando pressionado o botão, uma nova janela abrirá com o respectivo dendrograma. A Figura 14 ilustra o detalhe da página web com os botões de análise multivariada.
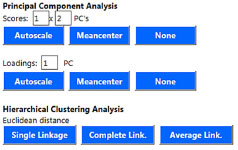 Figura 14. Detalhe das opções de métodos de análise de dados do ChemoStat versão web
Na versão web a única forma de saída dos resultados é a partir de gráficos, que podem ser salvos no formato de figura ".png".
CONCLUSÃO O desenvolvimento deste trabalho permitiu criar um software gratuito contemplando os métodos de análise de agrupamento hierárquico (HCA), análise de componentes principais (PCA), análise de componentes principais por intervalos (iPCA), assim como técnicas de correção, transformação dos dados e detecção de amostras anômalas, com as seguintes características:
Todos os dados das amostras utilizadas foram também analisados no Matlab® e os resultados em ambas as ferramentas coincidiram nas mais diversas combinações. Além da versão desktop, o reuso dos algoritmos permitiu disponibilizar uma versão online com alguns recursos básicos de tratamento de dados além dos métodos de análise de agrupamento hierárquico (HCA) e análise de componentes principais (PCA).
MATERIAL SUPLEMENTAR Os resultados obtidos com o Matlab® encontram-se disponíveis em formato pdf, com acesso livre, a partir do website da revista Química Nova (http://quimicanova.sbq.org.br/).
AGRADECIMENTOS Os autores agradem à Capes, pelo apoio financeiro, e à UNISC, em especial ao Programa de Pós-Graduação em Sistemas e Processos Industrais.
REFERÊNCIAS 1. Brereton, R. G.; Chemometrics for Pattern Recognition, 1th ed., Wiley: Chichester, 2009. 2. Brereton, R. G.; Applied Chemometrics for Scientists, 1th ed., Wiley: Chichester, 2007. 3. Matlab® ; The Mathworks, Inc.; USA, 2012. 4. Infometrix Inc.; Pirouette User Guide; Version 4.5, USA, 2011. 5. Camo.; Unscrambler Software Inc.; USA, 2006. 6. Nunes, C. A.; Freitas, M.; Pinheiro, A.; Bastos, S; J. Braz. Chem. Soc. 2012, 23, 11. 7. Jarvis, R. M.; Broadhurst, D.; Johnson, H.; O'boyle, N.; Goodacre, R.; Bioinformatics 2006, 22, 20. 8. Microsoft Visual Studio 2010® . Microsoft Corporation: Redmond, USA, 2010. 9. http://sourceforge.net/projects/zedgraph/, acessada em Novembro 2013. 10. http://accord. googlecode.com, acessada em Julho 2013. 11. Wehrens, R.; Chemometrics with R: Multivariate data analysis in the natural sciences and life sciences, 1th ed., Springer:Berlin , 2011. 12. http://www.sccg.sk/~haladova/principal_components.pdf/, acessada Setembro 2013. 13. Leardi, R.; Norgaard, L.; J. Chemometrics 2004, 18, 486. DOI: http://dx.doi.org/10.1002/cem.893
# Programa de Pós-Graduação em Sistemas e Processos Industriais |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access