
 |
 |
 |
 |
 |
Artigo
|
|
| Reação de condensação do glicerol com compostos carbonílicos. síntese, caracterização e aplicação em cristais líquidos Condensation reaction of glycerol and carbonyl compounds: synthesis, characterization, and derivatization for liquid crystals |
|
Jacqueline KautzI; Willian FeltrinI; Eric S. SalesI; Vera Lucia Eifler-LimaII; Aloir A. MerloI,*
IInstituto de Química, Universidade Federal do Rio Grande do Sul, 91501-970 Porto Alegre - RS, Brasil Recebido em 17/03/2015 *e-mail: aloir.merlo@ufrgs.br Condensation reactions of glycerol with aldehydes and ketones were performed under thermal heating and microwave irradiation regimes. Homogeneous and heterogeneous catalysts were tested in both conditions. A silica sulfated (SiO2-SO3H) heterogeneous catalyst demonstrated the best performance relative to a selectivity of >95% in favor of 5-membered ketals. For acetals, preference in favor of 5-membered or 6-membered functional groups depends on the nature of the catalyst. Homogenous catalysts favor the more stable 6-membered acetals, whereas heterogeneous catalysts favor the less stable 5-membered acetals. However, the isomer ratios in the acetalization reaction are too low, and hence the reaction cannot be used in a synthetic plan for functional materials. Ketalization processes mediated by SiO2-SO3H show a high selectivity in favor of a 5-membered ring (1,3-dioxolane). The scope of condensation was tested with different ketones. A mechanism for heterogeneous catalysis related to the selectivity in the cyclization process is presented herein. Solketal, a commercial product, was also obtained by a condensation reaction of glycerol and propanone, and showed a high selectivity in favor of 1,3-dioxolane. It was transformed to potential allylic and chiral intermediates. A mesogenic core was connected to the organic framework of glycerol to produce a monomer liquid crystal material with a stable smectic-C mesophase. INTRODUÇÃO A produção de biodiesel através da reação de transesterificação de triglicerídeos com metanol ou etanol tem produzido nos últimos anos uma quantidade enorme de glicerol como sub-produto.1 Estimativas da produção sugerem que para cada 90 m3 de óleo produzido, 10 m3 de glicerol são gerados. Processos e usos dessa enorme quantidade de glicerol são disponíveis. A incineração ou a queima em fornos industriais é um dos processos mais comuns através do qual elimina-se uma grande quantidade dos estoques de glicerol disponíveis. Contudo, existem muitas aplicações ou usos que evitam a incineração do glicerol cru como única alternativa de redução dos estoques.2 Modos alternativos estão em curso com o propósito de adicionar valor aos produtos ou processos derivados do glicerol.3 Por exemplo, o glicerol é usado em cosméticos, produtos de limpeza, farmacêuticos, entre outros. Transformações químicas do glicerol são alternativas interessantes que geram novos derivados com potencial aplicativo na indústria.4,5 A reação de condensação de álcoois com aldeídos e cetonas, denominada de acetalização ou cetalização, frequentemente empregada em síntese orgânica,6 pode ser parte da solução do problema do uso racional do glicerol e não a sua simples incineração. Essa reação é uma rota alternativa de transformação seletiva do glicerol através da proteção de dois dos três grupos hidroxilas presente no glicerol, enquanto o terceiro grupo permanece disponível para futuras transformações dentro da estratégia desejada. Dessa forma, a manipulação adequada dos três grupos hidroxilas, através das reações de proteção e desproteção em estágios de síntese distintos, leva à formação do produto alvo ou de um intermediário avançado dentro de um contexto sintético.7 Como exemplo, a preparação de material dendrítico de interesse na indústria medicinal foi descrita a partir da estratégia de proteção e desproteção dos grupos hidroxilas do glicerol.8 O método geral para a síntese de acetais ou cetais é a reação de compostos carbonílicos com álcoois na presença de um catalisador ácido. Os catalisadores ácidos comumente usados incluem os ácidos minerais, ácidos de Lewis, óxidos metálicos (Zn, Al, Zr, W, etc), bem como sílica, impregnados com ácidos minerais H2SO4, HCl e HNO3, entre outros. A incorporação de ácidos minerais em óxidos metálicos e sílica aumenta a acidez do material sólido resultante.9 Óxidos metálicos ácidos são importantes na indústria em virtude da grande área superficial do suporte sólido ácido.10 O óxido de alumínio sulfatado, Al2O3-SO4-2, por exemplo, apresenta alta estabilidade térmica e é capaz de induzir a isomerização térmica do n-butano para o iso-butano. A isomerização induzida é o resultado do poder catalítico ácido do óxido metálico revestido pelo ácido mineral.10 Os óxidos metálicos ácidos, em especial os sulfatados (sulfated metal oxide - SMO), tais como ZrO2, TiO2, SnO3, Al2O3 e SiO2, impregnados com grupos sulfatos, são ácidos sólidos muito promissores em aplicações direcionadas para a química orgânica sintética.11 A preparação da superfície sólida sulfatada - Al2O3-SO4-2 pode ser feita usando ácido sulfúrico - H2SO4 ou o ácido clorossulfônico - ClSO3H como reagentes que entregam o grupo sulfato. Esses catalisadores sólidos foram aplicados eficientemente na síntese de xantenedionas em bons rendimentos.12 Catalisadores heterogêneos derivados de sílica mesoporosa funcionalizada com grupos ácidos também têm sido desenvolvidos e empregados em muitas reações de condensação.13 A utilização de materiais sólidos porosos ácidos torna-se uma alternativa limpa e economicamente interessante para ser aplicada em síntese orgânica, com destaque para a valorização de matéria-prima disponível e abundante como o glicerol. A principal dificuldade na reação de acetalização e cetalização com o glicerol é a formação de uma mistura de quatro produtos isoméricos constituída de dois isômeros contendo o anel de 5 átomos 1,3-dioxolano e outros dois contendo o anel de 6 átomos 1,3-dioxano.14 A mistura dos 4 isômeros para aldeídos aromáticos é um fator importante a ser considerado e constitui-se numa etapa crítica para a utilização do glicerol como matéria-prima em síntese orgânica. A separação dos isômeros demanda um tempo relativamente longo, dispendiosa e resulta na perda considerável do produto alvo.15 Ao contrário da reação do acetalização do glicerol, os acetais e cetais derivados de dióis são frequentemente usados como uma forma mascarada do grupo carbonila durante as etapas de um planejamento sintético mais complexo sem o problema associado com a formação de misturas de isômeros. Nesse sentido, uma ampla variedade de métodos foi desenvolvida para as reações de proteção e desproteção de compostos carbonílicos.16 Como superar as dificuldades na síntese associada com a acetalização ou cetalização do glicerol é uma questão que não está plenamente contemplada pela comunidade química orgânica sintética. A busca por rotas alternativas em relação à transformação seletiva do glicerol nas reações de condensação com compostos carbonílicos permanece ainda como um grande desafio para os químicos e engenheiros químicos. Neste trabalho apresenta-se os resultados da reação do glicerol com compostos carbonílicos alifáticos e aromáticos utilizando aquecimento convencional (AC) e aquecimento por irradiação de micro-ondas (MO). As reações foram testadas com catalisadores homogêneos (ácidos de Brönsted) e heterogêneos (óxidos ácidos sólidos). Algumas derivatizações do solketal, o cetal mais difundido na literatura, são apresentadas na forma de intermediários sintéticos. Esses intermediários demonstram o potencial aplicativo em materiais funcionais, em particular na área de cristal líquido.
RESULTADOS Condensação do glicerol (1) com compostos carbonílicos aromáticos A reação de formação dos acetais de 5- e 6-átomos a partir do glicerol e de aldeídos aromáticos sob condições ácidas é descrita no Esquema 1. Neste trabalho foram selecionados três aldeídos aromáticos: benzaldeído (2a), 4-metilbenzaldeído (2b) e 4-bromobenzaldeído (2c) com o propósito de avaliar as condições reacionais que produzem os correspondentes acetais do glicerol na forma de mistura racêmica de isômeros 3-cis, 3-trans, 4-cis e 4-trans.
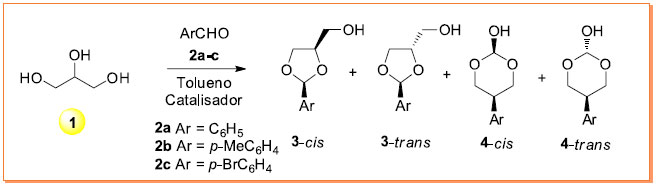 Esquema 1. Reação de condensação do glicerol (1) com aldeídos aromáticos 2a-c com a formação dos respectivos acetais racêmicos 3 e 4
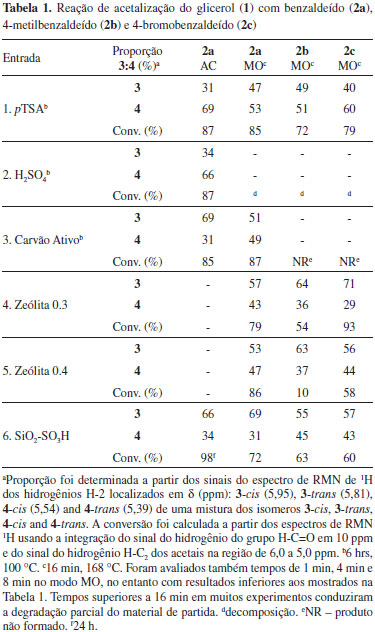
A composição da mistura dos acetais isoméricos foi determinada pela integração do sinal do hidrogênio H-C2 nos espectros de RMN de 1H. Os sinais do hidrogênio H-C2 dos isômeros 3-cis, 3-trans, 4-cis e 4-trans aparecem em 5,95, 5,81, 5,54, e 5,39 ppm, respectivamente. Para os cetais isoméricos, os espectros de RMN de 1H apresentam dois sinais característicos que pertencem ao átomo de hidrogênio H4 do anel 1,3-dioxolano e do H5 do anel 1,3-dioxano, respectivamente. Adicionalmente, os espectros de RMN de 13C apresentam sinais dos átomos de carbono C4 e C5 que ressoam em 76 ppm e 64 ppm. Esses dois sinais são atribuídos aos respectivos anéis 1,3-dioxolano e 1,3-dioxano.17 Em todos os experimentos realizados os espectros foram obtidos a partir das amostras sem purificação prévia ou tentativa de separação dos isômeros. Esse procedimento foi feito para evitar eventuais isomerizações no processo de purificação. A Tabela 1 mostra os resultados iniciais da reação de formação dos acetais 3 e 4 sob catálise ácida homogênea e heterogênea, e em duas condições reacionais distintas. As reações foram feitas sob aquecimento convencional (AC) e aquecimento por irradiação de micro-ondas (MO) em tolueno como solvente. A temperatura foi fixada em 100 ºC para AC e 168 ºC para MO. A reação modelo selecionada foi a condensação entre o glicerol (1) e o benzaldeído (2a) sob AC. Os aldeídos p-metilbenzaldeído (2b) e p-bromobenzaldeído (2c) foram testados no regime de MO. O tempo de reação no AC para os catalisadores ácidos p-toluenossulfônico (pTSA), sulfúrico (H2SO4) e carvão ativo foi de 6 h. Para catalisador sólido, a sílica modificada, SiO2-SO3H, um experimento foi realizado com o tempo de reação de 24 h. No modo MO o tempo de reação foi fixado em 16 min. Em todos os experimentos, incluindo as Tabelas 1 e 2, foi usado um excesso de glicerol em relação aos compostos carbonílicos. A proporção em massa de aldeído e catalisador foi de 100:1 e de aldeído e glicerol foi 1,1:1. Proporções diferentes de 100:1 foram também testadas, no entanto com resultados inferiores. Experimentos com o catalisador ácido sulfúrico não foram feitos em decorrência da decomposição do material de partida mesmo em tempos menores de aquecimento através do MO.
Os resultados da acetalização do benzaldeído (2a) mostrados na Tabela 1 referem-se a uma mistura de 3a e 4a sem a quantificação dos respectivos pares de isômeros cis-trans de 3a e 4a. A predominância de 4a sobre 3a é observada com catalisadores ácidos homogêneos pTSA e H2SO4 (entrada 1 e 2). No entanto, com catalisadores heterogêneos carvão ativo, zeólita-3, zeólita-4 e sílica ácida (entradas 3, 4, 5 e 6) observa-se uma inversão na proporção dos isômeros com a predominância de 3a sobre 4a.18,19 A predominância de um acetal sobre o outro é observada em ambos os regimes da reação, isto é, com AC ou MO a proporção em favor de um ou do outro se mantém. Para os experimentos de MO as condições mais ácidas (entrada 2) ou maiores tempos de reação levaram à decomposição do material de partida. A Figura 1 mostra uma expansão do espectro de RMN de 1H da mistura dos isômeros de 3a e 4a. Quatro sinais na forma de singleto são observados em 5,95, 5,81, 5,54 e 5,39 ppm, associados com o hidrogênio ligado ao átomo de carbono C2, do anel heterocíclico. Os sinais numa frequência maior estão relacionados com o anel heterociclo de 5 átomos, o 1,3-dioxolano. Para o anel de 6 átomos, 1,3-dioxano, os sinais aparecem em frequência menor no espectro de RMN de 1H. Essa atribuição está de acordo com os dados da literatura.20
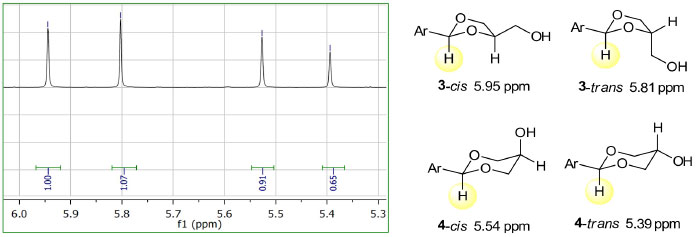 Figura 1. Expansão do espectro de RMN de 1H (300 MHz, CDCl3) da mistura de 3a e 4a (esquerda) e estruturas químicas dos isômeros 3-cis (5,95), 3-trans (5,81 ), 4-cis (5,54) e 4-trans (5,39) (direita, deslocamento químico em ppm entre parênteses)
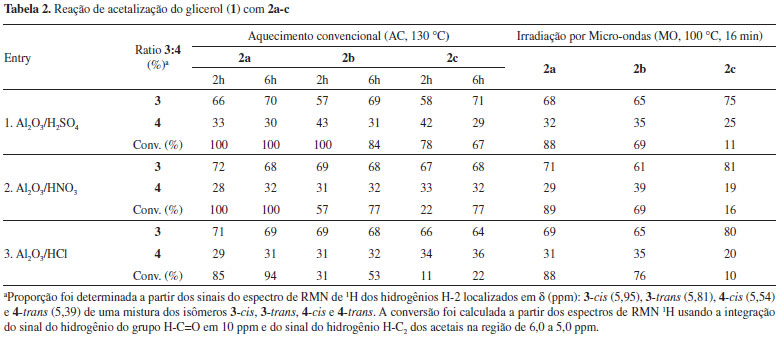
A preferência para um dos isômeros usando catalisador homogêneo ou heterogêneo seguiu a mesma tendência observada nos dados obtidos para o benzaldeído conforme Tabela 1. O anel 1,3-dioxolano predomina quando o catalisador é heterogêneo (entradas 2, 3, 4 e 5), enquanto que o anel 1,3-dioxano foi o majoritário para catalisador homogêneo (entrada 1). As conversões apresentaram valores intermediários. No entanto, mesmo com benzaldeídos dissubstituídos a baixa seletividade observada restringe futuras derivatizações dos acetais aromáticos na área de materiais funcionais. Foram testados diferentes tempos reacionais nos experimentos de MO. No entanto, os melhores resultados foram obtidos com tempo reacional de 16 min. Abaixo desse tempo as conversões foram menores, e em tempos superiores de 16 min verificou-se a decomposição do material de partida em alguns experimentos. Alumina é outro catalisador heterogêneo que tem sido empregado em muitos processos químicos catalíticos.21 Considerando a sua natureza química, a alumina deve ser quimicamente modificada para tornar-se em um catalisador mais ativo nas reações de condensação de álcoois e compostos carbonílicos. Uma das maneiras de aumentar as propriedades ácidas da alumina é a incorporação de ânions inorgânicos. Ânions inorgânicos derivados de ácidos minerais (ácidos de Brönsted) aumentam a acidez e a capacidade oxidativa do óxido de alumínio, provavelmente pela ativação dos grupos hidroxilas localizados na superfície no óxido de metal de Al2O3.22 Neste trabalho a alumina modificada com ácidos de Brönsted foi preparada seguindo o procedimento descrito nos trabalhos de Yang10 e Pramanik e Bhar.12 A Tabela 2 contém os dados da reação de acetalização dos aldeídos 2a, 2b e 2c catalisada pela alumina ácida preparada através da impregnação do oxido metálico Al2O3, com ácidos de Brönsted tais como H2SO4, HNO3 e HCl. As reações foram testadas com AC e com MO. As reações sob AC foram realizadas a 130 ºC e com tempos de reação de 2 e 6 h. Nas reações de MO a temperatura foi de 100 ºC em um tempo de 16 min. Em ambos os regimes de aquecimento, tempos superiores a 6 h e a 16 min não resultaram em conversões superiores e nem numa melhora da proporção entre os isômeros 3 e 4. Os dados da reação de acetalização de acordo com a Tabela 2 mostram novamente que a formação de 3 sobre 4 é favorecida em todos os experimentos realizados com o catalisador heterogêneo alumina ácida. A predominância do anel 1,3-dioxolano é favorecida na reação catalisada pelo catalisador ácido heterogêneo. A proporção de 3 sobre 4 é levemente superior em temperaturas mais elevadas (2 para 6 h), mas não muito superior com valores próximos àqueles mostrados na Tabela 1. Em todos os experimentos realizados, a conversão da reação depende da natureza do benzaldeído, sendo maior para 2a e 2b. Para o 4-bromobenzaldeído (2c) a conversão é geralmente baixa sob MO. As reações com cetonas aromáticas foram testadas sob AC na presença dos catalisadores p-TSA, sílica ácida - SiO2-SO3H e a alumina sulfatada - Al2O3/H2SO4. O tempo reacional foi de 8 h. Foram testadas as seguintes cetonas: benzofenona, acetofenona, 2-bromoacetofenona, 4-hidroxiacetofenona e a 4-octiloxiacetofenona. Os resultados de conversão para todas as cetonas listadas ficaram abaixo de 30%, com baixa seletividade em favor do isômero 1,3-dioxolano. Esse resultado está de acordo com o encontrado na literatura de reações catalisadas de formação de cetais derivados de cetonas aromáticas.11 Os dados que estão expostos nas Tabelas 1 e 2 permitem uma rápida análise da influência dos parâmetros usados nas reações de condensação de compostos carbonílicos aromáticos 2a-c com o glicerol (1). A conversão mais efetiva foi encontrada para o catalisador heterogêneo SiO2-SO3H (entrada 6, Tabela 1). A seletividade observada entre os isômeros formado 1,3-dioxolanos:1,3-dioxanos, isto é, 3:4, foi de baixa a regular, com o maior valor de 81:19 para o catalisador heterogêneo Al2O3/HNO3 (entrada 2, Tabela 2). A proporção dos isômeros 3 e 4 depende da natureza física do catalisador. Catalisadores homogêneos favorecem a formação do anel 1,3-dioxano 4, enquanto que a formação do anel 1,3-dioxolano 3 foi favorecido quando usou-se os catalisadores heterogêneos. A substituição no benzaldeído parece desfavorecer a reação de condensação tanto quanto ao aspecto da conversão quanto da seletividade para catalisadores heterogêneos. Cetonas aromáticas foram pouco reativas nas condições descritas. Os catalisadores sólidos mais efetivos em geral derivam do ácido sulfúrico. Para catalisadores homogêneos o ácido tolueno sulfônico foi o mais eficiente. Quanto ao regime das reações de AC ou MO, notou-se que as reações com AC em geral foram mais produtivas do que no regime de MO. Condensação do glicerol (1) com compostos carbonílicos alifáticos A reação de condensação de aldeídos e cetonas alifáticas também foi investigada usando o regime AC. O catalisador escolhido foi a sílica ácida SiO2-SO3H (entrada 6 da Tabela 1) em virtude da melhor conversão observada e a facilidade de separação do catalisador sólido do meio reacional. A proporção dos isômeros não foi determinante na escolha do catalisador em virtude da baixa seletividade observada em todos os experimentos. O Esquema 2 descreve a síntese de acetais e cetais de aldeídos 5a-c e cetonas 5d-f com o glicerol (1). As reações de condensação de 5a-f com 1 produzem uma mistura de isômeros cis e trans 1,3-dioxolanos 6 e 1,3-dioxanos 7, exceto para as cetonas simétricas.
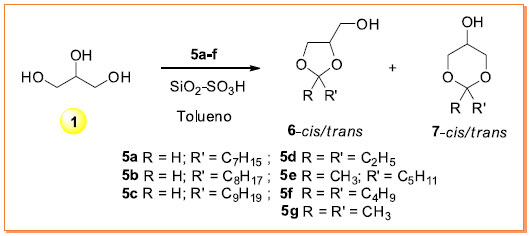 Esquema 2. Reação de ciclização do glicerol (1) com aldeídos 5a-c e cetonas 5d-f catalisadas pelo SiO2-SO3H
A reação de condensação de 1 com aldeídos alifáticos octanal (5a), nonanal (5b) e decanal (5c) foi realizada em 2 e 6 h. Os rendimentos e a seletividade da reação não se alteram com tempos de reação superiores a 6 h. A seletividade da reação em favor do isômero 1,3-dioxolano foi de baixa para moderada e está de acordo com outros trabalhos na literatura.23 Para a condensação com 3-pentanona (5d), 2-heptanona (5e), 5-nonanona (5f) e propanona (5g) a seletividade em favor do anel 1,3-dioxolano foi > 95% com o uso de sílica modifica. A conversão completa foi alcançada no tempo de reação de 8 h. O uso de sílica normal-não modificada mostrou a mesma seletividade, porém com uma conversão em 8 h abaixo de 20%. A reação de cetalização usando o catalisador heterogêneo SiO2-SO3H também foi aplicada na síntese do produto comercial solketal (6g) - que é a forma protegida do glicerol (1) com acetona (5g). A seletividade observada em favor do anel 1,3-dioxolano e a conversão para o solketal17,24 mostrou que a sílica modifica SiO2-SO3H é um excelente catalisador nas reações de condensação de compostos carbonílicos com álcoois. A conversão mostrada na Tabela 3 foi obtida da mesma forma que foi aplicada na condensação com aldeídos aromáticos (Tabela 1). Nas reações com cetonas alifáticas, a conversão foi considerada > 95% pela observação da ausência do sinal do grupo carbonila (C=O) no espectro de RMN de 13C para os cetais 5d-g.
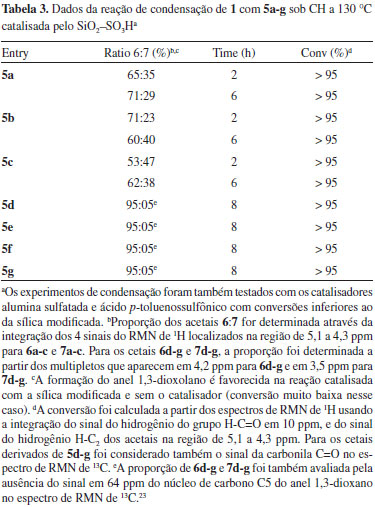
A estrutura e a proporção dos produtos 6 e 7 foram determinadas a partir dos espectros de RMN de 1H. Para os cetais 6d-f/7d-f foram também usadas as informações obtidas dos espectros de RMN de 13C. A atribuição dos sinais de ressonância para os hidrogênios dos produtos 6 e 7 nos espectros de RMN de 1H é bastante complexa devido ao acoplamento entre os núcleos vizinhos e à amostra ser composta de uma mistura de isômeros cis- e trans. A maioria dos sinais nos espectros de RMN de 1H aparecem na forma de multipletos. Do mesmo modo, a complexidade nos espectros de RMN de 13C pode ser alta em virtude da proximidade dos sinais para um mesmo átomo de carbono. Apesar da complexidade dos sinais nos espectros de RMN de 1H é possível distinguir os anéis de 1,3-dioxolanos e 1,3-dioxanos nos produtos formados. Os acetais 6a-c e 7a-c sintetizados neste trabalho apresentam um conjunto de sinais bastante complexo na região de 4,2 ppm a 3,2 ppm. No entanto, os sinais do átomo de hidrogênio ligado ao átomo de carbono C2 do anel do 1,3-dioxolano e 1,3-dioxano aparecem na forma de tripleto, na região compreendida de 5,1 ppm a 4,3 ppm. A Figura 2 descreve uma região expandida do espectro de RMN de 1H para a mistura dos isômeros de 6c e 7c. A atribuição dos sinais em maior frequência para os isômeros 1,3-dioxolanos e em menor frequência para isômeros 1,3-dioxanos foi feita com base em trabalhos da literatura de compostos semelhantes.18,19,25 A proporção dos isômeros trans-6:cis-6:cis-7:trans-7 obtidos da ciclização dos aldeídos alifáticos 5a-c com o glicerol (1) foi então determinada a partir das integrações dos respectivos sinais de acordo com a Figura 2.
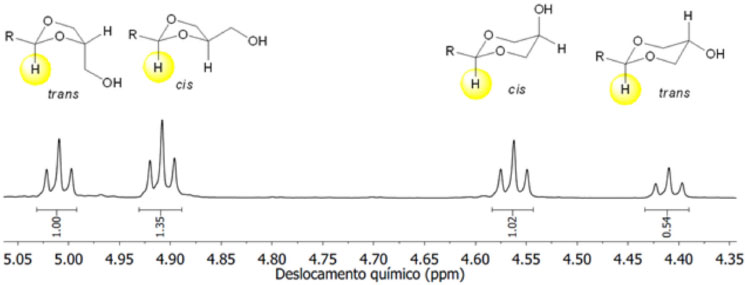 Figura 2. Expansão do espectro de RMN de 1H (CDCl3, 300 MHz) para a mistura dos isômeros cis- e trans de 6c e 7c
A determinação da proporção entre os anéis de 5 e 6 átomos produzidos na reação de cetalização foi feita com o auxílio dos dados obtidos dos espectros de RMN de 1H e de 13C. Os espectros RMN de 1H dos cetais do tipo 6 e 7 apresentam dois sinais de ressonância centrados em δ (ppm) 4,20 e 3,50 da mistura dos isômeros de 1,3-dioxolano 6 e 1,3-dioxano 7, respectivamente.17 Esses dois sinais pertencem ao átomo de hidrogênio H4 do anel 1,3-dioxolano e do H5 do anel 1,3-dioxano. A integração dos sinais que aparecem em 4,20 ppm e 3,50 ppm fornecem a proporção dos cetais de cinco e seis átomos, respectivamente. Nos experimentos realizados neste trabalho, não foi observado nos espectros de RMN de 1H o sinal em 3,50 ppm do isômero 1,3-dioxano (multipleto). Dessa forma, a ausência do sinal em 3,50 ppm permite concluir que a proporção de 6 para 7 na mistura pode ser da ordem > 95% em favor do isômero 6. Adicionalmente, espectros de RMN de 13C de cetais apresentam o sinal do átomo de carbono C4 para o 1,3-dioxolano próximo a 76 ppm, enquanto que o átomo de carbono C5 para o anel 1,3-dioxano ressoa 64 ppm. A Figura 3 apresenta a expansão dos espectros de RMN de 1H (esquerda, 300 MHz) e de 13C (direita, 100 MHz) em CDCl3, do produto 6d da reação de cetalização de 1 com 5d. A ausência do multipleto em δ = 3,50 ppm no espectro de RMN de 1H e δ = 64 ppm no espectro de RMN de 13C confirmam que a reação de ciclização de cetalização foi completamente regiosseletiva em favor do isômero 1,3-dioxolano 6d. Os sinais localizados em 4,20, 4,01 ppm são atribuídos aos núcleos de hidrogênios H4, H5'. Em 3,70 e 3,58 ppm aparecem os sinais dos H5 e CH1H1'O, respectivamente.
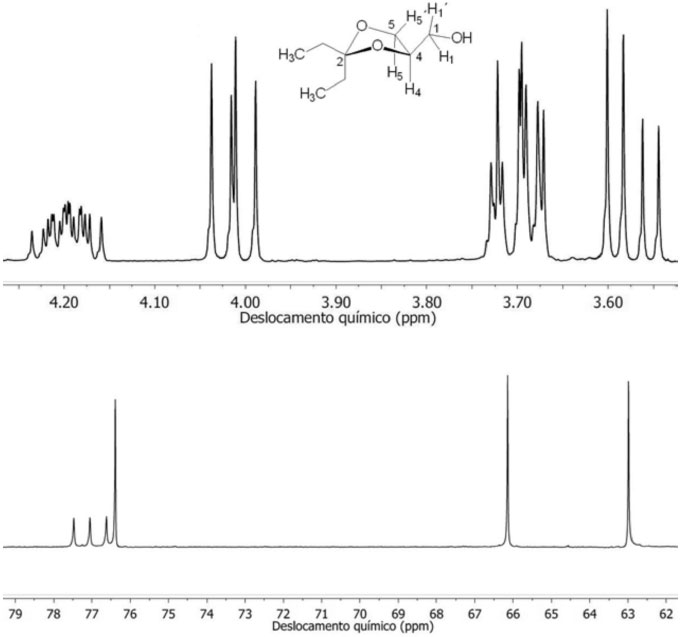 Figura 3. Expansão dos espectros de RMN de 1H (acima, 300 MHz) e de 13C (abaixo, 100 MHz) em CDCl3, do produto 6d da reação de cetalização de 1 com 5d
Os resultados da seletividade nas reações de ciclização obtidos neste trabalho estão de acordo com a tendência geral de predominância dos acetais ou cetais cíclicos de 5 átomos sobre os de 6 átomos na presença de catalisador heterogêneo. Com catalisadores homogêneos predomina o isômero mais estável de 6 átomos. Assim, com catalisadores homogêneos o produto favorecido é o termodinâmico, enquanto que com catalisadores heterogêneos a predominância é do produto cinético.26 A reação de formação dos acetais e cetais é uma reação reversível a qual acontece em duas etapas chaves. A etapa inicial é a formação do hemiacetal seguido da remoção de água. A etapa lenta é a formação do carbocátion a partir do hemiacetal protonado. Entretanto, a protonação do hemiacetal é também uma etapa importante que influencia na velocidade global do mecanismo. Meio fortemente ácido aumenta a eficiência da reação também em razão do aumento da velocidade de protonação do hemiacetal. Fatores de natureza estérica e eletrônica são importantes e influenciam na reatividade dessas reações.11,18 O Esquema 3 descreve o mecanismo da reação de condensação da propanona (5g) com o glicerol (1) sob catálise ácida.
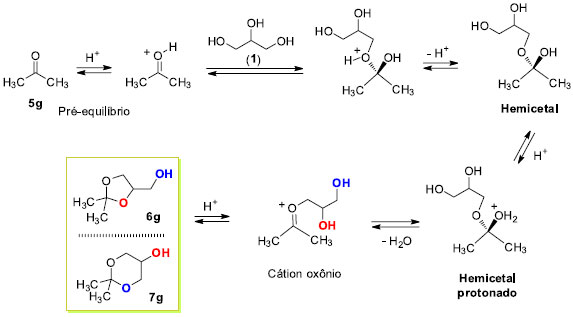 Esquema 3. Mecanismo geral de formação do cetal da propanona (5g) via catálise ácida
O pré-equilibrio inicial ativa a reação da propanona (5g) com o glicerol (1) pelo ataque preferencial da hidroxila primária. A desprotonação leva à formação do hemicetal. A eliminação de água do hemicetal produz o correspondente carbocátion, que se estabiliza na forma de cátion oxônio. Uma vez protonado, o hemicetal vai a produto através dele próprio ou do cátion oxônio. A ciclização pode acontecer de duas maneiras (i) ciclização pelo ataque da hidroxila secundária gerando o anel 1,3-dioxolano - controle cinético; ou (ii) ciclização pelo ataque da hidroxila primária levando à formação do anel 1,3-dioxolano - controle termodinâmico. A proposta neste trabalho, que procura explicar a formação preferencial do anel 1,3-dioxolano com catalisadores heterogêneos, parte do pressuposto que a reação é composta por uma série de equilíbrios ácido-base conforme Esquema 3. A superfície ácida do catalisador oferece os sítios ácidos de Brönsted, a qual disponibiliza uma concentração apropriada do cátion H+ para as etapas de transferências protônicas. Entretanto, o intermediário hemicetal também pode se complexar com o catalisador sólido através dos grupos hidroxilas livres residuais do glicerol. Assim, não estão sendo considerados outros equilíbrios ácido-base importantes no Esquema 3. A Figura 4 descreve as possíveis interações ácido-base entre o catalisador sólido heterogêneo SiO2-SO3H e o hemicetal. As duas formas de complexação mostradas apresentam uma hidroxila primária e uma secundária interagindo com o grupo hidrossulfato. A superfície modificada da sílica é também mostrada com a ligação do átomo de silício ao grupo SO3H. Modelos similares são encontrados na literatura com outros catalisadores ácidos sólidos.11,27 A interação da hidroxila primária (azul) é por natureza mais intensa do que a secundária (vermelha) e as conformações possíveis da cadeia carbônica do glicerol assumem os arranjos ou conformações descritos na forma W e Foice.28,29 A reação de ciclização acontece pelo ataque nucleofílico das hidroxilas coordenadas ao suporte ácido sólido. As estabilidades relativas dos produtos parecem não ser o fator preponderante na proporção dos isômeros 1,3-dioxolano e 1,3-dioxano. A forma em W facilita o ataque da hidroxila secundária ao carbono que carrega o grupo de saída (H2O) ou ao átomo de carbono catiônico, enquanto que na forma Foice é a hidroxila primária que atua no centro reativo de carbono. A forma W leva à formação do produto cinético e a forma Foice gera o produto termodinâmico. As velocidades de ciclização são diferentes em virtude do efeito de complexação do catalisador sólido ácido e do arranjo da cadeia do hemicetal na superfície do catalisador.
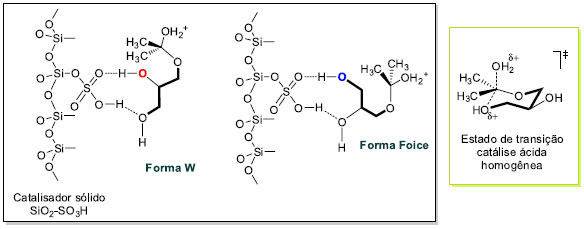 Figura 4. Descrição do modo de complexação do hemicetal com sílica ácida - SiO2-SO3H com a formação do produto cinético a partir da forma W e do produto termodinâmico da forma Foice, e estado de transição de seis átomos na catálise ácida homogênea
A etapa de formação dos produtos cinético e termodinâmico pode acontecer a partir do hemicetal protonado ou do cátion oxônio derivado da eliminação de água. A aproximação do nucleófilo ao centro reativo é favorecido na forma W em virtude da melhor disposição dos orbitais no estado de transição de 5 átomos.30 Na reação de catálise ácida homogênea as questões dos modos de complexação do hemicetal na forma W ou em Foice são mais difíceis de serem analisadas em virtude da grandeza do ácido de Lewis (H+) presente em solução. Nessas circunstâncias, o produto majoritário formado tem origem num estado de transição de seis átomos. Síntese de derivados do solketal 6g A preparação de derivados orgânicos do glicerol foi feita a partir do solketal 6g conforme Esquema de síntese 4. Apesar de ser um produto comercial, o solketal foi preparado em excelentes rendimentos e seletividade da reação de acetalização do glicerol com propanona conforme descrito nos parágrafos anteriores. Foram selecionadas duas reações de transformação do solketal 6g nos correspondentes produtos alquilados 9 e 11. Sob condições de catálise de transferência de fase, usando o brometo de tetra n-butilamônio (TBAB) como catalisador em meio básico,31 foram obtidos os intermediários 9 e 11 com rendimentos não optimizados de 60% e 50%, respectivamente.
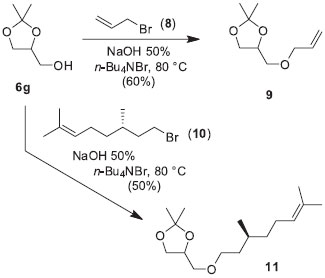 Esquema 4. Síntese de precursores 9 e 11 de materiais funcionais
O derivado 9 contém a porção alílica, enquanto que 11 apresenta o grupo quiral citronelila. Ambos são intermediários valiosos na preparação de materiais funcionais. A unidade alílica permite o ingresso em materiais cuja cadeia polimérica carrega ligações de éter. A presença de um substituinte quiral em 11 nos dá acesso à preparação de novos cristais líquidos com mesofases nemática e esméticas quirais além da possibilidade de encontra a fase azul (blue phase). Os compostos 9 e 11 representam uma alternativa concreta de uso do glicerol (1) em reações regiosseletivas. Assim, o solketal (6g) permite a manipulação correta da reatividade de dois grupos hidroxilas do glicerol. O grupo isopropilideno acetal em 9 ou 11 abre a possibilidade de transformação de 6g em diferentes compostos de interesse com posterior hidrólise ou abertura do anel 1,3-dioxolano. Nesse sentido, a hidrólise em meio ácido (ácido acético glacial 80%, 3 h, 40 ºC) forneceu os dióis 12 e 13 em 92% e 75% de rendimento, respectivamente.
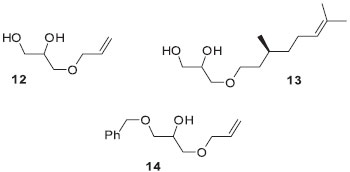
Estabelecida a primeira reação de alquilação, seguida da hidrólise do isopropilideno acetal como em 12 e 13, pode-se agora explorar de maneira eficiente as diferenças de reatividades dos grupos hidroxilas livres remanescentes do glicerol. O excesso diastereoisomérico de 11 e 13 não foram obtidos em virtude da baixa resolução dos picos observado no cromatograma usando a coluna quiral ciclodextrina. Os compostos 13 e 14 são assim apresentados como intermediários sintéticos não simétricos importantes derivados do glicerol. Esses intermediários serão explorados em trabalhos futuros na área de cristais líquidos. Por exemplo, a síntese de 14 foi feita usando o procedimento descrito no Esquema 4 em 78% de rendimento. Síntese de monômero líquido-cristalino a partir de 14 A transformação de derivados do glicerol em materiais funcionais é uma das formas apropriadas da sua utilização como fonte de matéria prima. No Esquema de síntese 5 é descrita a síntese do monômero alílico 16 contendo três unidades moleculares distintas: uma unidade polimerizável e duas unidades que podem ser desenhadas e sintetizadas com fins específicos. A funcionalização ou a incorporação de unidades que respondam a estímulos externos torna os derivados de 14 muito interessantes com respeito a potenciais aplicabilidades. A seleção do brometo 15 como reagente alquilante visou a transformação de 14 em um monômero com propriedade líquido-cristalina. Nesse sentido, é a exploração das propriedades de cristal líquido do monômero 16 que será destacada no contexto de materiais funcionais.32
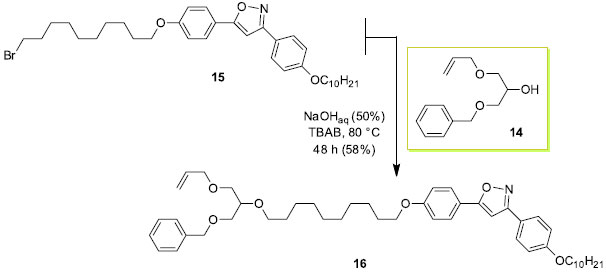 Esquema 5. Monômero alílico líquido-cristalino 16 derivado do glicerol 1,3-dissubstituído 14
O comportamento térmico observado para o 16 estimula a preparação de novos monômeros com propriedades líquido-cristalinas. Dessa forma, novos materiais poliméricos podem ser sintetizados com alguma propriedade de interesse, tais como, fotoluminescência, geleificação, ferroeletricidade e de óptica não-linear. A polimerização de monômeros do tipo 16 permitirá a obtenção de novos polímeros de cadeia lateral em que a cadeia polimérica é construída com base na ligação éter presente no glicerol e não de ésteres derivados do ácido acrílico ou metacrílico.33 Essa diferença de conexão química da cadeia polimérica com os grupos laterais imprime no material final uma maior estabilidade térmica e fotoquímica. Esses materiais serão objetos de estudos futuros. A preparação de 16 foi feita a partir da reação de alquilação do glicerol alílico 1,3-dissubstituído não simétrico 14 com o correspondente brometo 15.34 A condição de reação de síntese de 16 seguiu o método de alquilação conforme trabalho descrito na referência 25. A reação foi feita em meio básico na presença TBAB. O composto 16 é um sólido branco acinzentado obtido em 58% de rendimento a partir de sucessivas recristalizações em etanol. O sólido obtido foi redissolvido em diclorometano e a solução foi filtrada passando por uma camada de celite para remover partículas sólidas remanescentes, tais como fibras de papel, pó, etc. Estabelecida a síntese do monômero 16 iniciou-se a investigação das propriedades funcionais desse monômero. A análise do comportamento mesomórfico de 16 foi feita a partir dos dados de microscopia óptica de luz polarizada (MOL) e de calorimetria diferencial de varredura (DSC). As temperaturas de transição (ºC) foram adquiridas via DSC com uma taxa de aquecimento e resfriamento de 10 ºC min-1. A caracterização das mesofases e as texturas observadas foram obtidas e analisadas utilizando microscópio de luz polarizada. A análise de microscopia foi feita no resfriamento a partir da fase líquida isotrópica. A amostra foi aquecida até o ponto de clareamento, que corresponde à transição da mesofase para a fase líquida. A seguir, iniciou-se o processo de resfriamento da amostra. Isso se faz necessário para a eliminação completa de qualquer informação estrutural oriunda da mesofase ou da fase cristalina. Dessa forma é possível obter uma textura que está livre das informações ou impressões deixadas pela cristalina ou de mesofases na fase de interesse. Na transição da fase cristalina para a mesofase SmC foi considerada a temperatura Tonset e na transição SmC foi considerada a temperatura de transição do pico em 95 ºC. A Figura 5 apresenta o termograma do monômero alílico 16 com taxa de aquecimento de 10 ºC min-1. A amostra 16 no aquecimento apresenta as seguintes transições de fase: Cristal → SmC → I. No resfriamento são observadas as mesmas transições em ordem reversa I → SmC → Cristal. Em 72,0 ºC a amostra entra na mesofase SmC e, em 95,0 ºC é a temperatura de transição da mesofase SmC para a fase isotrópica (líquida). No resfriamento, a mesofase SmC reaparece em 94 ºC e cristaliza a 68 ºC. As entalpias de transição calculadas para as transições Cristal → SmC e SmC → I correspondem aos valores de 8,7 kcal mol-1 e 0,40 kcal mol-1, respectivamente. Os valores obtidos estão de acordo com a natureza da transição. A transição da fase cristalina para a fase esméctica C apresenta uma entalpia muito elevada enquanto que a transição da mesofase SmC para a fase isotrópica tem um valor muito abaixo em virtude das diferenças dos graus de ordem das diferentes fases em análise.
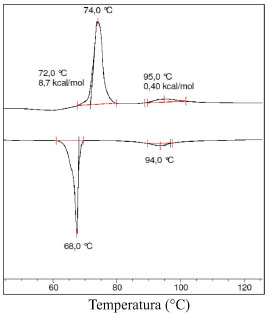 Figura 5. Termograma do monômero alílico 16 a 10 ºC min-1. Tonset foi a temperatura considerada na transição cristal Cr → SmC
A Figura 6 mostra uma sequência de microfotografias obtidas de uma amostra de 3 no ciclo de resfriamento. A amostra está confinada entre duas lâminas de vidro e a linha mais escura vertical é a borda da amostra. Na Figura 5(a) é observada a formação das esfirulitas da fase cristalina, enquanto que em 5(b), 5(c) e 5(d) a mesofase SmC é observada com a textura Schlieren. A variação da cor nas microfotografias na mesofase SmC é uma consequência do arranjo molecular inclinado das moléculas na forma de bastão. Ao entrar na mesofase SmC a partir do estado isotrópico, as moléculas se organizam em camadas com uma inclinação molecular que aumenta com a diminuição da temperatura. A inclinação atinge valores máximos próximos a 25º no resfriamento. À medida que a temperatura diminui a inclinação molecular aumenta e, em consequência, a amostra torna-se mais birrefringente e mais colorida. A coloração muda de cinza (Figura 5d, 90 ºC) para amarelado em (Figura 5b, 75 ºC).
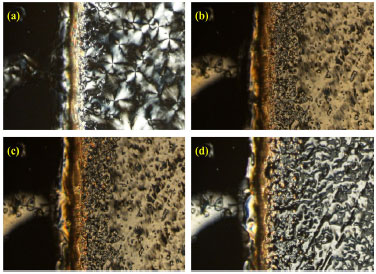 Figura 6. Sequência de fotografias da fase cristalina a 65 ºC (a) e da mesofase SmC a 75 ºC (b), 82 ºC (c) e 90 ºC (d) para o monômero 16
CONCLUSÃO A reação de condensação catalisada por ácidos do glicerol com aldeídos e cetonas formou os respectivos compostos cíclicos isoméricos 1,3-dioxolanos e 1,3-dioxanos. Foram testados catalisadores homogêneos (H2SO4 e pTSA) e ácidos heterogêneos (alumina, carvão ativo e sílica) em dois regimes de aquecimento - convencional (AC) e irradiação via micro-ondas (MO). Com catálise homogênea a seletividade foi moderada em favor do anel 1,3-dioxano. Sob catálise heterogênea, os melhores resultados foram com SiO2 - SO3H no regime AC com a seletividade em favor do 1,3-dioxolano. A proporção dos isômeros foi determinada por RMN de H1. A maior seletividade foi encontrada na cetalização de cetonas alifáticas em > 95% em favor do anel 1,3-dioxolano. Um mecanismo foi proposto para a catálise heterogênea com base no modo de complexação do hemicetal. Intermediários importantes do glicerol foram sintetizados com potencial aplicativo na área de materiais funcionais. Um deles foi transformado em um monômero liquido-cristalino com mesofase sméctica C (SmC). Os resultados preliminares demonstraram que o glicerol funcionalizado é um modo alternativo de acesso a materiais funcionais que serão explorados em trabalhos futuros na área de polímeros quirais e de cristais líquidos.
MATERIAL SUPLEMENTAR Descrição experimental, dados espectroscópicos, espectros, figuras, etc estão disponíveis em http://quimicanova.sbq.org.br/ em pdf e com acesso livre.
AGRADECIMENTOS Os autores agradecem às agências de fomento CNPq e Fapergs (Edital PqG 02/2014) pelo suporte financeiro e à CAPES. Aos professores E. V. Benvenutti e D. Russowsky pelas análises dos catalisadores sólidos e da síntese da sílica ácida.
REFERÊNCIAS 1. Melero, J. A.; Vicente, G.; Paniagua, M.; Morales, G.; Munoz, P.; Bioresour. Technol. 2012, 103, 142; Suarez, P. A. Z.; Meneghetti, S. M. P.; Quim. Nova 2007, 30, 2068. DOI: http://dx.doi.org/10.1016/j.biortech.2011.09.105 PMID: 22018752 2. Zhou, C. H. C.; Beltramini, J. N.; Fan, Y. X.; Lu, G. Q. M.; Chem. Soc. Rev. 2008, 37, 527; Gonçalves, V. L. C.; Pinto, B. P.; Silva, J. C.; Mota, C. J. A.; Catal. Today 2008, 133-135, 673. DOI: http://dx.doi.org/10.1039/B707343G PMID: 18224262 3. Aguirre, I.; García, I.; Requies, J.; Barrio, V. L.; Guemez, M. B.; Cambra, J. F.; Arias. P. L.; Biomass Bioenergy 2011, 35, 3636; Nichele, T. Z.; Fávero, C.; Monteiro, A. L.; Catal. Commun. 2009, 10, 693. DOI: http://dx.doi.org/10.1016/j.biombioe.2011.05.008 4. Behr, A.; Eilting, J.; Irawadi, K.; Leschinski, J.; Lindner, F.; Green Chem. 2008, 10, 1. DOI: http://dx.doi.org/10.1039/b718911g 5. Zhou, C.-H.; Beltramini, J. N.; Fan, Y.-X.; Lu, G. Q.; Chem. Soc. Rev. 2008, 37, 527. DOI: http://dx.doi.org/10.1039/B707343G PMID: 18224262 6. Clerici, A.; Pastori, N.; Porta, O.; Tetrahedron 1998, 54, 15679; da Silva, G. P.; Contiero, J.; Ávila Neto, P. M.; de Lima, C. J. B.; Quim. Nova 2014, 37, 527. DOI: http://dx.doi.org/10.1016/S0040-4020(98)00982-X 7. Crotti, C.; Farnetti, E.; Guidolin, N.; Green Chem. 2010, 12, 2225. DOI: http://dx.doi.org/10.1039/c0gc00096e 8. Meyers, S. R.; Juhn, F. S.; Griset, A. P.; Luman, N. R.; Grinstaff, M. W.; J. Am. Chem. Soc. 2008, 130, 14444. DOI: http://dx.doi.org/10.1021/ja806912a PMID: 18842041 9. Thomas, B.; Prathapan, S.; Sugunan. S.; Microporous Mesoporous Mater. 2005, 80, 65; Ramos, A. L. D.; Tanase, S.; Rothenberg, G.; Quim. Nova, 2014, 37, 123. DOI: http://dx.doi.org/10.1016/j.micromeso.2004.12.001 10. Yang, T-S.; Chang, T-H.; Yeh, C-T.; J. Mol. Catal. A: Chem. 1997, 115, 339. DOI: http://dx.doi.org/10.1016/S1381-1169(96)00283-X 11. Lin, C.-H.; Lin, S. D.; Lin, T.-P.; Huang, Y-J.; Appl. Catal. A 2003, 240, 253. DOI: http://dx.doi.org/10.1016/S0926-860X(02)00453-2 12. Pramanik, A.; Bhar, S.; Catal. Commun. 2012, 20, 17. DOI: http://dx.doi.org/10.1016/j.catcom.2011.12.036 13. Diaz, I.; Marquez-Alvarez, C.; Mohino, F.; Prez-Pariente, K.; Sastre, E.; J. Catal. 2000, 193, 283; Shylesh, S.; Sharma, S.; Mirajkar, S. P.; Singh, A. P.; J. Mol. Catal. A: Chem. 2004, 212, 219. DOI: http://dx.doi.org/10.1006/jcat.2000.2898 14. Silva, P. H. R.; Gonçalves, V. L. C.; Mota, C. J. A.; Bioresour. Technol. 2010, 101, 6225. DOI: http://dx.doi.org/10.1016/j.biortech.2010.02.101 PMID: 20304633 15. Deutsch, J.; Lieske, H.; Martin, A.; J. Catal. 2007, 245, 428. DOI: http://dx.doi.org/10.1016/j.jcat.2006.11.006 16. Kocienski, P. J.; Protecting Groups, Georg Thieme Verlag: Stuttgart, 1994. 17. Fadnavis, N. W.; Reddipalli, G. S.; Ramakrishna, G.; Mishra, M. K.; Sheelu, G.; Synthesis 2009, 557. 18. Carlsen, P. H. J.; Sorbye, K.; Ulven, T., Aasbo, K.; Acta Chem. Scand. 1996, 50, 185. DOI: http://dx.doi.org/10.3891/acta.chem.scand.50-0185 19. Pawar, R. R.; Jadhav, S. V.; Bajaj, H. C.; Chem. Eng. J. 2014, 235, 61. DOI: http://dx.doi.org/10.1016/j.cej.2013.09.018 20. Piasecki, A.; Sokołowski, A.; Burczyk, B.; Gancarz, R.; Kotlewska, U.; J. Colloid Interface Sci. 1997, 192, 74. DOI: http://dx.doi.org/10.1006/jcis.1997.4967 PMID: 9268545 21. Cheng, Z.-X.; Ponec, V.; Catal. Lett. 1994, 27, 113; Ferreira, M. L.; Rueda, E. H.; J. Mol. Catal. A: Chem. 2002, 178, 147. DOI: http://dx.doi.org/10.1007/BF00806984 22. Cheng, Z.-X.; Ponec, V.; J. Catal. 1994, 143, 607; Cheng, Z.-X.; Ponec, V.; Appl. Catal., A 1994, 118, 127; Peri, J. B.; Phys. Chem. 1965, 69, 220; Knözinger, H.; Kaerlein, C. P.; J. Catal. 1972, 25, 436; Pryzystajko, W.; Fiedorow, R.; Lana, I. G. D.; Appl. Catal. 1985, 15, 265. DOI: http://dx.doi.org/10.1006/jcat.1994.1247 23. Ruiz, V. R.; Velty, A.; Santos, L. L.; Leyva-Pérez, A.; Sabater, M. J.; Iborra, S.; Corma, A.; J. Catal. 2010, 271, 351. DOI: http://dx.doi.org/10.1016/j.jcat.2010.02.023 24. Deutsch, J.; Martin, A.; Lieske, H.; J. Catal. 2007, 245, 428. DOI: http://dx.doi.org/10.1016/j.jcat.2006.11.006 25. Piasecki, A.; Sokołowski, A.; Burczyk, B.; Gancarz, R.; Kotlewska, U.; Langmuir 1997, 13, 1434; Juaristi, E.; Antúnez, S.; Tetrahedron 1992, 48, 5941; Inch, T. D.; Williams, N.; J. Chem. Soc. C 1970, 263; Eliel, E.; Banks, H. D.; J. Am. Chem. Soc. 1972, 94, 171. DOI: http://dx.doi.org/10.1021/la960207z 26. da Silva, C. X. A.; Gonçalves, V. L. C.; Mota, C. J. A.; Green Chem. 2009, 11, 38; Chandrasekhar, S.; Chem. Soc. Rev. 1987, 16, 313. DOI: http://dx.doi.org/10.1039/B813564A 27. Su, F.; Guo, Y.; Green Chem. 2014, 16, 2934. DOI: http://dx.doi.org/10.1039/c3gc42333f 28. Lehn, J. M.; Wipff, G.; Tetrahedron Lett. 1984, 21, 159; Mark, V; Tetrahedron Lett. 1974, 15, 299. DOI: http://dx.doi.org/10.1016/S0040-4039(00)71401-8 29. Merlo, A. A.; Reações pericíclicas. Uma sinfonia de moléculas e elétrons, Editora da UFRGS: Porto Alegre, 2012, p. 471. 30. Baldwin, J. E.; J. Chem. Soc., Chem. Commun. 1976, 734; Baldwin, J. E.; Lusch, M. J.; Tetrahedron 1982, 38, 2939. 31. Dumoulin, F.; Lafont, D.; Boullanger, P.; Mackenzie, G.; Mehl, G. H.; Goodby, J. W.; J. Am. Chem. Soc. 2002, 124, 13737. DOI: http://dx.doi.org/10.1021/ja020396+ PMID: 12431104 32. Duarte, R. C.; Ongaratto, R.; Piovesan, L. A.; Lima, V. R.; Soldi, V.; Merlo, A, A.; Montes D'Oca, M. G.; Tetrahedron Lett. 2012, 53, 2454. DOI: http://dx.doi.org/10.1016/j.tetlet.2012.03.015 33. Passo, J. A.; Merlo, A. A.; Eccher, J.; Bechtold, I. H.; Kelly, S. M.; Química Nova 2012, 35, 1527. DOI: http://dx.doi.org/10.1590/S0100-40422012000800007 34. Sales, E. S.; Dissertação de Mestrado, Universidade Federal do Rio Grande do Sul, Brasil, 2015. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access