
 |
 |
 |
 |
 |
Artigo
| Novel alkaloid from Rauvolfia capixabae (Apocynaceae) |
|
Lanamar Almeida CarlosI,*; Leda MathiasII; Raimundo Braz-FilhoII; Ivo Jose Curcino VieiraII
IDepartamento de Engenharia de Alimentos, Universidade Federal de São João del-Rei, Campus Sete Lagoas, 35701-970 Sete Lagoas - MG, Brasil Recebido em 15/07/2015 *e-mail: lanamar@ufsj.edu.br A new sarpagine-type alkaloid, Na-methylrauflorine (1), was isolated from Rauvolfia capixabae together with isoreserpiline (2), Nb-oxide-isoreserpiline (3), ajmalicine (4), perakine (5) and vinorine (6) alkaloids. These compounds were characterized based on their spectral data basis, mainly one- (1H, 13C, APT) and two-dimensional (1H-1H-COSY, 1H-1H-NOESY, HMQC and HMBC) NMR, and mass spectra, also involving comparison with data from the literature. INTRODUCTION The genus Rauvolfia, family Apocynaceae, continues to be fascinating as it produces a number of indole alkaloids with novel skeletons, which are interesting from the biosynthetic point of view as well as for their medicinal aspect and spectroscopic analysis.1,2 Species of this genus have several biological activities, such as, cholinesterase inhibitors,3 and antimicrobial,4 anticonvulsant,5 anxiolytic,6 antimalarial, antipyretic,7 and antipsychotic effects,8 in addition to sedative activity.9 Rauvolfia capixabae I. Koch & Kinoshita-Gouveia,10 commonly known as "Grão-de-Gato" in Atlantic forest in the North of Espírito Santo State, appears as a tree of 6-12 m. This species has not been reported in studies on its chemical composition described in the literature. As part of the research program for the Natural Products Chemistry Group of the North Fluminense State University (UENF) on the identification of alkaloids in species of the Rauvolfia genus,4,11,12 a phytochemical investigation of the stem bark extracts of R. capixabae is described. In the present paper, we describe the isolation and characterization of a novel sarpagine-type alkaloid, Na-methylrauflorine (1), along with five known alkaloids. The known and new alkaloid structures were established on the basis of spectral data, mainly 1H and 13C (1D and 2D) NMR spectra, mass spectrometry and by comparison with literature data.
EXPERIMENTAL General procedures Optical rotation measurements were obtained on a Perkin Elmer 343 digital polarimeter. Melting points were obtained on a Microquímica MQRPF and are uncorrected. FTIR spectra were recorded on a FTIR-8300 Shimadzu spectrometer using KBr disk. EI-MS (low resolution) mass spectra were obtained on Shimadzu QP5050A mass spectrometer. Column chromatographic purifications were carried out over silica gel (70-230 mesh). Silica gel 60F254 was used in thin layer chromatography analysis. 1H and 13C NMR spectra were measured on a Jeol Eclipse 400 spectrometer, operating at 400 (1H) and 100 (13C) MHz. CDCl3 was used as solvent and TMS as internal reference. Chemical shifts are given in the δ scale (ppm) and coupling constants J in Hz. One dimensional (1D) 1H and 13C NMR spectra were acquired under standard conditions by using a direct detection 5 mm 1H/13C dual probe. Standard pulse sequences were used for two dimensional spectra by using a multinuclear inverse detection 5 mm probe with field gradient. Plant materials The stem bark of Rauvolfia capixabae I. Koch & Kinoshita-Gouveia were collected in November 2004 at Cia Vale, Linhares City, Espírito Santo State, Brazil. The specimen was identified by taxonomist Ingrid Koch. A voucher specimen (CVRD 338) is deposited at the Cia Vale herbarium, Linhares, Espírito Santo State. Extraction and isolation The stem bark (3.08 kg) was extracted with CH2Cl2 from R. capixabae I. Koch & Kinoshita-Gouveia at room temperature, furnishing 125 g of crude dichloromethane extract after solvent evaporation. A portion of the dichloromethane extract (10.1 g) was chromatographed over silica gel column with a polarity gradient of CH2Cl2/MeOH to afford fourteen fractions. Fraction 1 (58.4 mg) yielding isoreserpiline (2). The fraction 7 (1.35 g) was rechromatographed over a silica gel column with a polarity gradient of CH2Cl2/MeOH yielding thirteen fractions. Fraction 7.2 (6.8 mg) presented as an amorphous yellow solid identified as Compound (1). Fraction 7.13 (384.8 mg) was rechromatographed over a silica gel column with a polarity gradient of CH2Cl2/MeOH yielding the compounds isoreserpiline (2, 7.1 mg) and perakine (5, 166 mg). Fraction 8 (0.8 g) was rechromatographed over silica gel column with a polarity gradient of CH2Cl2/MeOH supplying eight fractions. Fraction 8.1(92.0) yielding ajmalicine (4, 92.0 mg). Fraction 8.8 (301.2) was rechromatographed over silica gel column with a polarity gradient of CH2Cl2/MeOH yielding compound 6 (199 mg). Fraction 11 (1.2 g) was rechromatographed over silica gel column with a polarity gradient of CH2Cl2/MeOH supplying seven fractions. Fraction 11.7 (217.5 mg) was rechromatographed over silica gel column with a polarity gradient of CH2Cl2/MeOH yielding compound Nb-oxide isoreserpiline (3, 13.0 mg). Na-methylrauflorine (1), yellow amorphous solid, mp 166-168 ºC; [α]D23 = + 8.06º (CHCl3, c 0.062); LREI-MS (rel. int.) 336 [M+. (100%,)] (Scheme 1); 1H and 13C NMR: see Table 1.
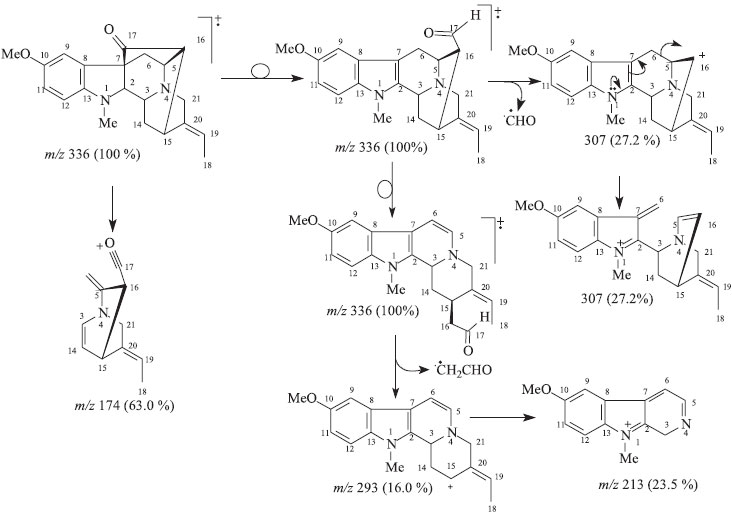 Scheme 1. Proposed fragmentation mechanisms to justify principal peaks observed in the mass spectrum (LREIMS, 70 eV) of 1
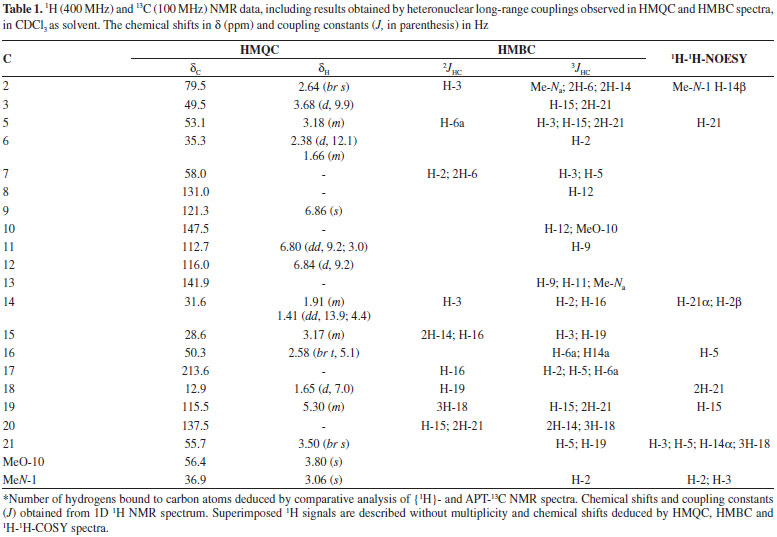
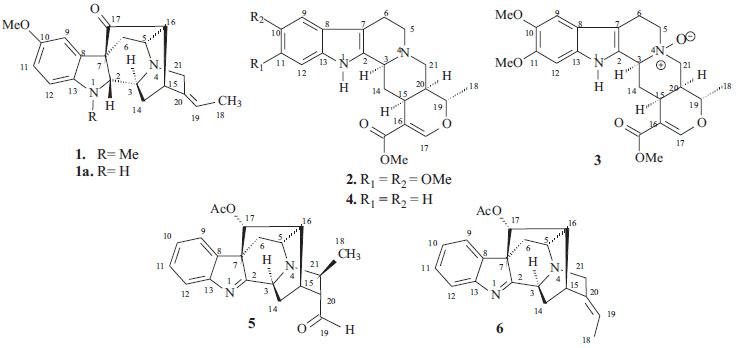 Figure 1. Compounds Isolated from R. Capixabae
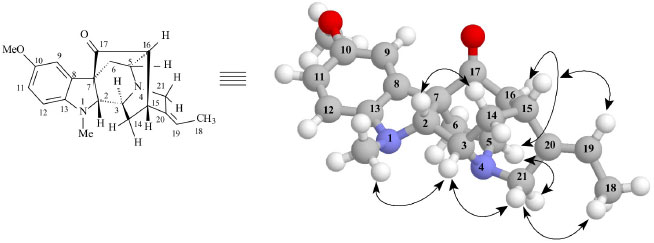 Figure 2. Select dipolar-dipolar interactions revealing spatial correlations (NOE) and relative stereochemistry for Alkaloid 1. Arrows denote NOE spatial correlations principals
RESULTS AND DISCUSSION The CH2Cl2 extracts of R. capixabae stem bark were subjected to classical chromatographic methods to yield the a new sarpagine-type alkaloid, Na-methylrauflorine (1), in addition to the known alkaloids, isoreserpiline (2),11 Nb-oxide-isoreserpiline (3),12 ajmalicine (4),13 perakine (5),14 and vinorine (6),15 that were identified in the analysis of 1H and APT-13C NMR spectra data, including 2D 1H-1H-COSY, 1H-1H-NOESY, HSQC and HMBC NMR experiments,16,17 which were also used to complete unambiguous 1H and 13C chemical shift assignments of the new alkaloid 1. The new monoterpenic indole alkaloid (1) [a]D23 = + 8.6, (CHCl3, c 0.062), was obtained as a yellow amorphous solid. Comparative analysis of {1H}- and APT-13C NMR spectra (Table 1), involving the corroboration of 1H NMR spectra (1D 1H NMR and 2D 1H-1H-COSY), allowed to recognize the presence of 21 signals corresponding to six nonhydrogenated [(C)6: one sp3, five sp2 (including one carbonyl group at δC 213.6 (C-17) and one sp2 olefinic at δC 137.5 (C-20)], nine methine [(CH)9: three sp3 (all linked to a nitrogen atom: CH-2, CH-3 and CH-5) and four sp2 including one olefinic at δC 115.5 (CH-19)], three methylene [(CH2)3, all sp3, including one linked to a nitrogen atom at δC 55.7 (CH2-21)] and three methyl [(CH3)3: including one linked to a nitrogen atom at δC 36.9/δH 3.06, one linked to an sp2 olefinic carbon atom at δC 12.9/δH 1.65 and methoxyl group (δC 56.0/δH 3.80,s)] carbon atoms, allowing to deduce the expanded molecular formula (C)5(C=O)(CH)9(CH2)3N(CH3)(NCH3)(OCH3) = C21H24N2O2 for 1. The LREI-MS (70 eV) spectrum of 1 (Scheme 1) showed of molecular peak [M+.] at m/z 336 Daltons, allowing in conjugation with the 13C NMR spectral data to deduce the molecular formula C21H24N2O2 (1), containing eleven degrees of unsaturation and consistent with the presence of one carbonyl group and one double bond in a methoxylated pentacyclic alkaloid, compatible with the alkaloidic structure sustaining a carbonyl group at carbon atom C-17 (δC 213.6) in rauflorine skeleton (1a).4 In fact, heteronuclear long-range couplings (3JHC) of this carbon atom C-17 (δC 213.6) with H-2 (δH 2.64), H-5 (δH 3.18-3.24) and H-6a (δH 2.38) was used to confirm the presence of a function carbonyl linked at atom carbon C-17, as shown in Table 1 together with additional heteronuclear long range couplings 1H-13C-COSY-nJHC (n=2, HMQC or HSQC and HMBC, n=3) that were also used to complete unambiguous 1H and 13C chemical shift assignments. 1H NMR spectrum of 1 showed three signals corresponding to the hydrogens linked to aromatic carbons at δH 6.86 (br s, H-9), 6.84 (d, J = 9.2 Hz, H-12) and 6.80 (dd, J = 9.2 and 3.0 Hz, H-1) typical of a 1,2,4-trisubstituted benzene ring (ring A),18,19 which was confirmed by direct heteronuclear correlations (1JHC) between CH-9 (δC 121.3)/δH 6.86), CH-11 (δC 112.7/δH 6.80), C-12 (δC 116.0/δH 6.84) observed in the HMQC spectrum. The presence of a single signal at δH 3.80 was attributed to three hydrogens of a methoxyl group (1JHC δC 56.0/δH 3.80), in agreement with a methoxyl group in the indole nucleus.18,19 The single signal at δH 3.06 (three hydrogen atoms) suggested the presence of a N-Me group that was confirmed in the HMQC by the correlation 1JHC Me-Na (δC 37.0/δH 3.06) and compatible with a substituted indole nucleus and N-methylated.20 The location of the methoxyl and Me-Na groups in the indole moiety was confirmed by heteronuclear long-range couplings observed in the HMBC spectrum by correlations 3JHC between by C-8 (δC 131.1) and H-12 (δH 6.84), C-10 (δC 147.5), H-12 (δH 6.84) and MeO-10 (δH 3.80), C-13 (δC 141.9), H-9 (δH 6.86), H-11 (δH 6.80) and Me-Na (δH 3.06), CH-11 (δC 112.7) and H-9 (δH 6.86), and CH-2 (δC 79.5) and Me-Na (δH 3.06), shown in Table 1. The presence of a methyl group bonded to a sp2 methine carbon was recognized by a double signal at δH 1.65 (d, J = 7.0 Hz, 3H-18) and the multiplet at δH 5.29-5.32 (m, H-19), characterizing a C=CH-CH3 moiety corresponding to CH-19 and CH3-18, respectively, present in several alkaloids. This moiety was further confirmed by long-range correlations revealed by the HMBC spectrum through correlations observed between the CH3-18 (δC 12.9) and H-19 (δH 5.29-5.32, 2JHC), CH-19 (δC 115.5), 3H-18 (δH 1.65, 2JHC) and C-20 (δC 137.5) and 3H-18 (δH 1.65, 3JHC).18,19,21 The presence of a carbonyl carbon at C-17 was confirmed by the heteronuclear correlations observed in the HMBC spectrum, which revealed long-range correlations between C-17 (δC 213.6) and H-16 (δH 2.58, 3JHC), H-2 (δH 2.64, 3JHC), H-5 (δH 3.22, 3JHC) and H-6a (δH 1.66, 3JHC).18,19,21 The other correlations are shown in Table 1. All these data allowed the definition of a sapargine-type skeleton18,19,21 and propose the Structure 1 for the alkaloid isolated. The comparison of the 1H and 13C NMR spectral data of 1 with values described in the literature for the rauflorine alkaloid (1a)18 allowed to verify that the significant difference between these two alkaloids can be justified by presence of the methyl group linked to N-atom of the alkaloid 1. The relative stereochemistry proposed for alkaloid 1 was based on biogenic argument,22 by comparison with data described in the literature for sapargine-type alkaloids skeleton18-21 and results revealed by dipolar-dipolar interactions observed in a 1H-1H-NOESY experiment (Table 1).
CONCLUSION From dichloromethane extract of R. capixabae stem bark a new sarpagine-type alkaloid was isolated, Na-methylrauflorine (1), in addition to known alkaloids, isoreserpiline (2), Nb-oxide-isoreserpiline (3), ajmalicine (4), perakine (5) and vinorine (6).
SUPPLEMENTARY MATERIAL The LREIMS, 1H NMR, 13C NMR -APT, 1H-1H COSY NMR, HMQC, HMBC and NOESY spectra for compound 1 are freely available at http://quimicanova.sbq.org.br in PDF file.
ACKNOWLEDGEMENTS The authors are grateful to Fundação Carlos Chagas Filho de Amparo à Pesquisa do Rio de Janeiro (FAPERJ), Consenho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) for scholarships and financial support.
REFERENCES 1. Roberts, M. F.; Wink, M.; Alkaloids: Biochemistry, Ecology and Medicinal Applications; Plenum Press: New York, 1998. 2. Bruneton, J.; Pharmacognosy, Phytochemistry, Medicinal Plants, Lavoisier: Londres, 1995. 3. Tappayuthpijarn, P.; Jansom, C.; Jansom, V.; Ingkaninun, K.; Planta Med. 2010, 76, 1209. 4. Carlos, L. A.; Silva, A. K. A.; Vieira, I. J. C.; Mathias, L.; Braz-Filho, R.; Samarão, S. S.; Vieira-da-Motta, O.; Braz. J. Microbiol. 2010, 41, 612. DOI: http://dx.doi.org/10.1590/S1517-83822010000300011 5. Quintans-Junior, L. J.; Siqueira, J. S.; Melo, M. S.; Silva D. A.; Morais, L. C. S. L.; Souza, M. F. V.; Almeida, R. N.; Rev. Bras. Farmacogn. 2010, 20, 54. DOI: http://dx.doi.org/10.1590/S0102-695X2010000100012 6. Netto, S. M.; Warela R. W. B.; Fechine, M. F.; Queiroga, M. N.; Quintans-Júnior, L.; J. Rev. Bras. Farmacogn. 2009, 19, 888. DOI: http://dx.doi.org/10.1590/S0102-695X2009000600017 7. Amole, O. O.; Onabanjo, A. O.; Agbaje, E. O.; Arch. Pharmacol. 1998, 358, 497. 8. Obembe, A.; Sokomba, E. N.; Sijuwola, O. A.; Olorunfemi, P. O.; Alemika, T. O. E.; Isichei, C. O.; Ogunkeye, O. O.; Bangudu, A. B.; Phytother. Res. 1994, 8, 218. DOI: http://dx.doi.org/10.1002/ptr.2650080406 9. Madawala, P. G.; Arambewela, L. S. R.; Premakumara, G. A. S.; Ratnasooria, W. D.; J. Ethnopharmacol. 1994, 42, 63. DOI: http://dx.doi.org/10.1016/0378-8741(94)90024-8 PMID: 8046945 10. Koch, I.; Kinoshita, L. S.; Bittrich, V.; Novon 2007, 17, 462. DOI: http://dx.doi.org/10.3417/1055-3177(2007)17[462:TNIRAR]2.0.CO;2 11. Cancelieri, N. M.; Vieira, I. J. C.; Mathias, L.; Schripsema, J.; Braz-Filho, R.; Tetrahedron Lett. 2002, 43, 1783. DOI: http://dx.doi.org/10.1016/S0040-4039(02)00151-X 12. Cancelieri, N. M.; Vieira, I. J. C.; Mathias, L.; Braz-Filho, R.; Magn. Reson. Chem. 2003, 41, 287. DOI: http://dx.doi.org/10.1002/mrc.1168 13. Lounasmaa, M.; Tolvanem, A.; Heterocycles 1986, 24, 3229. DOI: http://dx.doi.org/10.3987/R-1986-11-3229 14. Rahman, A.-U.; Sultana, A.; Nighat, F.; Bhatti, K.; Kuruku, S.; Kartal, M.; Phytochemistry 1995, 38, 1057. DOI: http://dx.doi.org/10.1016/0031-9422(94)00638-A 15. Kato, L.; Braga, R. M.; Kock, I.; Kinoshita, L. S.; Phytochemistry, 2002, 60, 315. DOI: http://dx.doi.org/10.1016/S0031-9422(02)00122-X PMID: 12031452 16. Claridge, T. D. W.; High-Resolution NMR Techniques in Organic Chemistry, Pergamon: Amsterdam, 1999. 17. Friebolin, H.; Basic One and Two-Dimensional NMR Spectroscopy, 2nd ed., VCH: New York, 1993. 18. Jokela, R.; Lounasmaa, M. A.; Heterocycles 1996, 43, 1015. DOI: http://dx.doi.org/10.3987/COM-96-7401 19. Chatterjee, A.; Ghosh, A. K.; Hagaman, E. W.; J. Org. Chem. 1982, 47, 1732. DOI: http://dx.doi.org/10.1021/jo00348a026 20. Azoug, M.; Loucaci, A.; Richard, B.; Nuzillard, J.; Moreti, C.; Hanrot, Z.; Men-Oliver, L.; Phytochemistry 1995, 39, 1223. DOI: http://dx.doi.org/10.1016/0031-9422(95)00050-H 21. Braga, R. M.; Reis, F. A. M.; Phytochemistry 1987, 26, 833. DOI: http://dx.doi.org/10.1016/S0031-9422(00)84797-4 22. Danieli, B.; Palmisano, G.; In The Alkaloids; Brossi, A., ed.; Academic Press: New York, 1986. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access