
 |
 |
 |
 |
 |
Artigo
|
|
| Determinação de enxofre em shampoo por espectrofotometria UV-Vis: avaliação de métodos de preparo de amostras Determination of sulfur in shampoo by UV-Vis spectrophotometry: evaluation of sample preparation methods |
|
Natalia J. Bielemann; Diogo L. R. Novo; Rodrigo M. Pereira; Julia E. Mello; Vanize C. Costa; Marcia F. Mesko*
Centro de Ciências Químicas, Farmacêuticas e de Alimentos, Universidade Federal de Pelotas, Campus Capão do Leão, 96160-000, Capão do Leão - RS, Brasil Recebido em 17/01/2017 *e-mail: marcia.mesko@pq.cnpq.br Dry ashing, dissolution in aqueous medium or wet digestion methods were evaluated for sample preparation of shampoo for the subsequent indirect determination of sulfur by UV-Vis spectrophotometry. Among the sample preparation methods evaluated, microwave-assisted wet digestion using 7 mol L-1 HNO3 was the most suitable method for this purpose. Accuracy of the proposed method was evaluated by recovery test using standard solution, and the recovery was about 96%. Moreover, for comparison of results, S (as SO42-) was also determined by ion chromatography, after microwave-assisted wet digestion using concentrated HNO3, and no significant difference was observed between the obtained results (according to the t-test for a 95% confidence level, p = 0.288). Relative standard deviations were always lower than 10% and the limit of detection was 0.007% w/v. Finally, the proposed method was applied for analysis of sixteen shampoos used to many types of hair, from different brands produced in Brazil, and the concentration of S ranged from 0.78 to 2.82% w/v. INTRODUÇAO Os shampoos têm sido amplamente utilizados como produto de beleza e de higiene pessoal visando à limpeza dos fios e do couro cabeludo.1,2 Atualmente, existe uma grande variedade desses produtos no mercado, os quais sao destinados a diferentes tipos de cabelo, e apresentam propriedades que proporcionam aos usuários diferentes resultados após a sua utilizaçao e, em alguns casos, açoes farmacológicas.3 De acordo com os últimos dados declarados no ano de 2014 pela Associaçao Brasileira da Indústria de Higiene Pessoal, Perfumaria e Cosméticos (ABIHPEC), o mercado brasileiro de produtos para cabelos faturou em torno de R$ 21,2 bilhoes, sendo o shampoo, um dos produtos mais comercializados.4 Dentre os principais constituintes dos shampoos, destaca-se o enxofre (S), uma vez que é considerado um agente terapêutico quando aplicado em diversos cosméticos. Além disso, o S apresenta açoes importantes para o cabelo como, por exemplo, queratolítica quando utilizado em elevadas concentraçoes, e queratoplástica quando aplicado em baixas concentraçoes.5 Ainda, cabe mencionar que os shampoos que apresentam S elementar e na forma de sulfacetamida podem ser eficazes no tratamento de rosácea, sarna, pitiríase versicolor, além de, geralmente, serem eficientes no tratamento de distúrbios capilares mais comuns, como a caspa, dermatite seborreica e oleosidade.6,7 O S também é adicionado aos shampoos como constituinte de surfactantes. Essas substâncias sao amplamente utilizadas na produçao de shampoos, pois auxiliam na remoçao de resíduos, bem como na oleosidade presente no cabelo.8,9 Dentre os surfactantes mais utilizados, destacam-se o lauril sulfato de sódio e o lauril éter sulfato de sódio, ambos surfactantes aniônicos que apresentam alta detergência, facilidade de produçao e menor custo.9,10 Apesar dos benefícios que o S proporciona para o cabelo, bem como para o couro cabeludo, a sua utilizaçao é contraindicada para pessoas que apresentam hipersensibilidade a este elemento, e o contato com os olhos deve ser evitado. Ademais, uma elevada concentraçao de S em shampoos pode causar aos usuários dermatite alérgica de contato, irritaçoes na pele e danos a estática dos fios, ocasionando o ressecamento do cabelo, independente da espécie química contendo S adicionada ao produto.11 Cabe ressaltar que a Food and Drug Administration (FDA) recomenda que a concentraçao de S adicionada a cosméticos que entrem em contato com o couro cabeludo nao exceda a 5%. Entretanto, quando o cosmético apresentar como principal finalidade o controle de caspas, é importante que a concentraçao de S esteja entre 2 e 5%.12 Assim, tendo em vista os benefícios e malefícios que podem advir da utilizaçao de shampoos contendo uma concentraçao inadequada de S, torna-se indispensável o controle da concentraçao deste elemento para garantir a qualidade do produto. Ainda, é importante salientar que nao foram encontrados relatos de métodos na literatura para a determinaçao de S em shampoos. Dentre as diversas técnicas analíticas que podem ser utilizadas para a determinaçao de S, pode-se destacar a espectrometria de emissao óptica com plasma indutivamente acoplado (ICP-OES, do inglês Inductively Coupled Plasma Optical Emission Spectroscopy),13 a cromatografia de íons (IC, do inglês Ion Chromatography)13 e a espectrofotometria na regiao do ultravioleta e visível (UV-Vis, do inglês, Ultraviolet and Visible Spectrophotometry).14,15 No entanto, embora sejam técnicas bastante sensíveis para a determinaçao de S, a ICP-OES e a IC apresentam elevado custo de aquisiçao e manutençao quando comparadas com a espectrofotometria UV-Vis. Nesse sentido, tendo em vista o baixo custo operacional, robustez, rapidez de análise, confiabilidade nos resultados e fácil manuseio, a espectrofotometria UV-Vis pode ser uma interessante alternativa para a determinaçao de S em shampoo.14,15 A determinaçao de S por espectrofotometria UV-Vis é comumente realizada através de uma análise turbidimétrica.16 Contudo, para que isso seja possível, faz-se necessário que a amostra seja preparada de maneira adequada, pois a presença de compostos oriundos da matriz da amostra pode dificultar a formaçao do precipitado ou conferir turbidez a soluçao, culminando em resultados errôneos. Nesse contexto, é importante ressaltar que, apesar de ser uma amostra líquida, o shampoo apresenta grande quantidade de ingredientes com longas cadeias carbônicas, os quais podem interferir na etapa de determinaçao. Portanto, a realizaçao de uma etapa de preparo de amostras, visando disponibilizar os analitos a partir da matriz da amostra para uma soluçao preferencialmente livre de interferências e adequada a técnica de determinaçao, é essencial para o êxito da análise.5,17 Embora a dissoluçao do shampoo em meio aquoso permita que os constituintes estejam presentes na soluçao, interferências relacionadas à matriz da amostra e a possibilidade do analito nao estar disponível para ser detectado podem comprometer a exatidao dos resultados. Diante disso, dentre os métodos comumente utilizados para o preparo de amostras de shampoo, pode-se destacar a decomposiçao por via seca em forno mufla18 e a decomposiçao por via úmida com aquecimento convencional19 ou assistida por radiaçao micro-ondas.20,21 No entanto, é importante mencionar que esses métodos de preparo de amostras ainda nao foram utilizados para a posterior determinaçao de S em shampoo. A decomposiçao por via seca em forno mufla apresenta diversas vantagens, principalmente, devido a decomposiçao completa da matéria orgânica, bem como pela possibilidade de reduzir os limites de detecçao do método, uma vez que é realizada em sistema aberto, permitindo a decomposiçao de elevadas massas de amostra. No entanto, o procedimento necessita demasiado tempo de execuçao, além de ser bastante suscetível a contaminaçoes e perdas de elementos por volatilizaçao e/ou adsorçao nas paredes do recipiente utilizado durante o processo.22,23 Por outro lado, a decomposiçao por via úmida promove a oxidaçao da matéria orgânica da amostra através do uso de um ácido oxidante, de uma mistura de ácidos oxidantes ou de uma mistura contendo um ácido oxidante e peróxido de hidrogênio. Contudo, quando realizada em sistemas abertos a temperatura de decomposiçao é limitada ao ponto de ebuliçao dos reagentes. Nesse sentido, geralmente é requerido um elevado tempo de decomposiçao, bem como uma elevada quantidade de reagentes para a total oxidaçao da matéria orgânica, principalmente daqueles que apresentam temperaturas de ebuliçao mais elevadas como, por exemplo, o H2SO4.22-24 Assim, a utilizaçao de sistemas de decomposiçao com frascos fechados associado ao aquecimento com radiaçao micro-ondas tem sido uma tendência para o preparo de amostras que apresentam composiçao predominantemente orgânica, bem como para amostras de shampoo.6 Apesar de permitir o uso de massas de amostras menores quando comparado aos métodos com fracos abertos, a utilizaçao de frascos fechados possibilita o uso de menores quantidades de reagentes sem comprometer a eficiência de decomposiçao. Isso está associado a reduçao na perda dos reagentes por volatilizaçao, por se tratar de um sistema fechado, bem como pela possibilidade de realizar a decomposiçao das amostras sob elevadas pressoes, o que resulta em um aumento no ponto de ebuliçao dos reagentes e, consequentemente, da eficiência de oxidaçao da matéria orgânica. Além disso, a possibilidade de trabalhar com sistemas de decomposiçao com frascos fechados minimiza a ocorrência de perdas e contaminaçoes durante o preparo das amostras.25,26 Diante disso, esse trabalho teve como objetivo avaliar a decomposiçao por via seca em forno mufla, bem como a dissoluçao em meio aquoso e a decomposiçao por via úmida, ambas em sistemas com frascos abertos ou fechados, com aquecimento convencional ou assistido por radiaçao micro-ondas, visando o preparo de amostras de shampoo para a subsequente determinaçao indireta de S por espectrofotometria UV-Vis. A exatidao do método considerado mais adequado foi avaliada através de ensaios de recuperaçao, bem como pela comparaçao dos resultados com os obtidos após a determinaçao por IC. Por fim, foi realizada a determinaçao de S em shampoos de variados tipos e marcas utilizando o método proposto.
PARTE EXPERIMENTAL Amostras Os shampoos utilizados nesse trabalho foram adquiridos no comércio local da cidade de Pelotas/RS e sao destinados a variados tipos de cabelos (lisos, ondulados, cacheados e tingidos) e finalidades (controle de oleosidade, para cabelos secos, reduçao de frizz ou caspas). As amostras utilizadas foram provenientes de diferentes marcas da indústria brasileira. Além disso, vale mencionar que dentre as dezesseis amostras utilizadas, duas sao comercializadas como produtos isentos de S na forma de SO42-. As otimizaçoes iniciais foram realizadas utilizando um shampoo destinado a cabelos normais e oleosos, o qual foi selecionado aleatoriamente, e as outras amostras foram, posteriormente, analisadas utilizando o método proposto. Instrumentaçao Para a realizaçao da decomposiçao do shampoo por via seca, um forno tipo mufla (2000G, Tekpluz, Brasil) foi utilizado, enquanto, a solubilizaçao das cinzas foi realizada com o auxílio de um agitador magnético (C-MAG HS 7, IKAr, Alemanha). Os procedimentos de dissoluçao em meio aquoso e decomposiçao por via úmida em frasco aberto com aquecimento convencional foram realizados em um bloco digestor (MA4025, Marconi, Brasil) equipado com oito frascos de vidro de borossilicato (50 x 250 mm). Por sua vez, a dissoluçao em meio aquoso e a decomposiçao por via úmida assistida por radiaçao micro-ondas em frascos fechados foram realizadas em um forno de micro-ondas (Multiwave 3000r, Anton Paar, Austria), equipado com oito frascos de politetrafluoretileno modificado quimicamente (PTFE-TFMr) com volume máximo de 100 mL e, temperatura e pressao de trabalho em torno de 260 ºC e 60 bar, respectivamente. Enxofre foi determinado como SO42- utilizando um espectrofotômetro (IL-592, Kasuaki, Brasil) que opera na faixa UV-Vis de 190 a 1100 nm e apresenta banda de passagem de 2 nm. Ainda, para comparaçao dos resultados, S também foi determinado como SO42- utilizando um cromatógrafo de íons (861 IC Advanced Compact, Metrohm, Suiça) equipado com sistema de supressao química - Metrohm Suppressor Module (MSM) - e detecçao por condutividade. Para tanto, foi utilizada uma coluna de troca aniônica com grupos amônio quaternário suportados em poli(álcool vinílico), modelo Metrosep A Supp 5 (250 x 4 mm d.i., 5 µm de diâmetro de partícula, Metrohm), e uma coluna-guarda de mesmo material de preenchimento da coluna de troca iônica, modelo Metrosep A Supp 4/5 Guard (5 x 4 mm d.i., 5 µm de diâmetro de partícula, Metrohm). A alça de amostragem e a vazao do eluente utilizados neste trabalho foram de 25 µL e 0,7 mL min-1, respectivamente. Reagentes e soluçoes Todos os reagentes utilizados neste trabalho foram de grau analítico (Merck, Alemanha) e as soluçoes foram preparadas com água ultrapura (18,3 MΩ cm) obtida através de um sistema de ultrapurificaçao (MEGA UP, MegaPurity, Coreia do Sul). Agua ultrapura também foi utilizada para a lavagem de materiais e vidrarias após a descontaminaçao utilizando ácido nítrico 10% (v/v) ou concentrado. Por sua vez, o HNO3 65% (m/m) utilizado nos métodos de decomposiçao por via úmida foi previamente purificado em um sistema de destilaçao abaixo do ponto de ebuliçao - em torno de 65 °C (Milestone, Model Duopor, Itália). Para a calibraçao dos equipamentos e para a realizaçao dos ensaios de recuperaçao, uma soluçao estoque de SO42- (10000 mg L-1) foi preparada a partir da dissoluçao do sal de sulfato de sódio em água. Para a análise por espectrofotometria UV-Vis uma curva de calibraçao de 5 a 30 mg L-1 de SO42- foi preparada a partir da diluiçao da soluçao estoque de SO42- (1000 mg L-1) e da adiçao de cloreto de bário 10% (m/v) e de ácido clorídrico 0,5 mol L-1, os quais foram utilizados para promover a formaçao de precipitado. Por outro lado, para a análise por IC foi preparada uma curva de 0,1 a 1,0 mg L-1 de SO42- a partir da diluiçao da soluçao estoque de SO42-. O eluente utilizado para a determinaçao de SO42- por IC [Na2CO3 (3,2 mmol L-1)/NaHCO3 (1,0 mmol L-1)] foi preparado pela dissoluçao dos seus respectivos sais em água. Uma soluçao de ácido sulfúrico na concentraçao de 200 mmol L-1 e água foram utilizadas como soluçoes regeneradoras do sistema de supressao química. Para avaliar a acidez dos digeridos, foi realizada uma titulaçao ácido/base utilizando hidróxido de sódio 0,09474 mol L-1 previamente padronizado com biftalato de potássio. Decomposiçao de shampoo assistida por radiaçao micro-ondas e determinaçao indireta de enxofre por cromatografia de íons A decomposiçao por via úmida assistida por radiaçao micro-ondas em frasco fechado, utilizando HNO3 14 mol L-1 como soluçao digestora, foi utilizada como método de referência previamente a determinaçao por IC. Para isso, 0,5 mL de shampoo, o qual foi escolhido aleatoriamente, foi transferido para os frascos de reaçao, aos quais foram adicionados 6 mL de HNO3 14 mol L-1. Posteriormente, os frascos foram fechados, fixados ao rotor e submetidos ao seguinte programa de irradiaçao com micro-ondas: i) rampa de 10 min até 1000 W; ii) 1000 W por 10 min; iii) 0 W por 20 min (etapa de resfriamento). Finalizadas as etapas de aquecimento e resfriamento, as soluçoes obtidas foram transferidas para baloes volumétricos de 25 mL e o volume foi aferido com água. Por fim, S foi determinado na forma de SO42- por IC, e os resultados obtidos por este método foram utilizados para fins de comparaçao com os obtidos utilizando os demais métodos avaliados. A exatidao do método usado como referência e do método proposto foi avaliada através de ensaios de recuperaçao, os quais foram realizados a partir da adiçao de 900 µL de uma soluçao estoque contendo 10000 mg L-1 de SO42- a amostra, previamente a etapa de decomposiçao. Além disso, os digeridos obtidos pelo método proposto também foram analisados por IC para a comparaçao dos resultados. Decomposiçao de shampoo por via seca utilizando um forno tipo mufla Inicialmente, cerca de 2 mL de shampoo foram transferidos para cadinhos de porcelana, os quais foram submetidos ao seguinte programa de aquecimento em um forno tipo mufla: i) 30 min a 90 ºC; ii) 60 min a 500 ºC. Posteriormente, as cinzas foram solubilizadas com 500 µL de HNO3 14 mol L-1 e 5 mL de água. Por fim, as soluçoes foram filtradas e transferidas para um balao volumétrico de 25 mL e avolumadas com água ultrapura. Essas condiçoes foram selecionadas com base no trabalho desenvolvido por Salvador et al.,6 que descreve um método para determinaçao de selênio, zinco e cádmio em shampoo antiscaspas, por espectrometria atômica. Preparo de amostra de shampoo utilizando aquecimento convencional em frasco aberto Inicialmente, para a realizaçao da dissoluçao em meio aquoso utilizando aquecimento convencional em sistema aberto, cerca de 0,5 mL de shampoo foi transferido para frascos do bloco digestor sendo, posteriormente, adicionados 20 mL de água ultrapura. Para a decomposiçao por via úmida, 0,5 mL de shampoo foi transferido para os frascos, no entanto, 20 mL de HNO3 em diferentes concentraçoes (4, 7 ou 14 mol L-1) foram adicionados nos frascos de decomposiçao. Em ambos os procedimentos, as amostras foram submetidas a aquecimento a 80 ºC por 2 h e as soluçoes obtidas foram transferidas para baloes volumétricos de 25 mL e aferidas com água. Preparo de amostra de shampoo utilizando aquecimento com radiaçao micro-ondas em frasco fechado A dissoluçao em meio aquoso e a decomposiçao por via úmida utilizando HNO3 (1, 4, 7 ou 14 mol L-1) foram realizadas em sistema com frascos fechados assistido por radiaçao micro-ondas. Em um primeiro momento, cerca de 0,5 mL de shampoo foi transferido para frascos de reaçao, aos quais foram adicionados 6 mL de água ultrapura ou de HNO3 nas concentraçoes avaliadas. Posteriormente, os frascos foram fechados, fixados ao rotor e submetidos ao programa de irradiaçao com micro-ondas conforme descrito no item "Decomposiçao de shampoo assistida por radiaçao micro-ondas e determinaçao indireta de enxofre por cromatografia de íons". Após finalizado o programa, as soluçoes obtidas foram transferidas para baloes volumétricos de 25 mL e o volume foi aferido com água. Determinaçao indireta de S em shampoo por espectrofotometria UV-Vis Para a determinaçao de S por espectrofotometria UV-Vis através de análise turbidimétrica, as soluçoes provenientes dos diversos métodos de preparo de amostras foram diluídas em um balao volumétrico de 25 mL, sendo adicionados 500 µL da amostra obtida após dissoluçao ou decomposiçao, 2 mL de HCl 0,5 mol L-1 e 1 mL de BaCl2 10% (m/v). Posteriormente, as soluçoes foram homogeneizadas e a determinaçao de S como SO42- foi realizada no comprimento de onda de 420 nm. Essas condiçoes foram utilizadas com base no trabalho desenvolvido por Tabatai.16
RESULTADOS E DISCUSSAO Determinaçao indireta de S em shampoo por cromatografia de íons após decomposiçao assistida por radiaçao micro-ondas Com o intuito de obter um valor que pudesse ser utilizado para comparaçao dos resultados, tendo em vista que nao existem métodos oficiais ou relatos na literatura para a determinaçao de S em shampoo, a amostra S1 foi decomposta em um sistema com frasco fechado assistido por radiaçao micro-ondas com 6 mL de HNO3 14 mol L-1, conforme descrito na seçao "Parte experimental" e, posteriormente, os digeridos foram analisados por IC para a determinaçao de S na forma de SO42-. Esse método foi utilizado como referência, visto que proporciona condiçoes adequadas para a eficiente decomposiçao da matéria orgânica, além de apresentar menores riscos de perda por volatilizaçao e contaminaçao quando comparado aos métodos convencionais de digestao.26 A determinaçao de S foi realizada por IC, pois essa técnica apresenta sensibilidade adequada, além de ser consolidada para esse tipo de análise e amplamente utilizada para determinaçao de S na forma de SO42- em diferentes tipos de amostra.27-29 Todavia, cabe mencionar que a determinaçao de S por IC nesses digeridos somente foi possível devido à elevada concentraçao deste analito na amostra avaliada. Isso permitiu a realizaçao de diluiçoes de até 2000 vezes, minimizando os efeitos da elevada acidez residual dos digeridos e também da elevada concentraçao de íons nitrato, os quais poderiam inviabilizar a determinaçao de S por IC.13 A exatidao do método foi avaliada através da realizaçao de ensaios de recuperaçao utilizando soluçao padrao, e recuperaçoes obtidas foram em torno de 98 ± 9%. Assim, a concentraçao de S obtida para a amostra S1 (1,04 ± 0,10% m/v) foi adotada como referência para a avaliaçao dos demais métodos. Avaliaçao de métodos de preparo de amostra visando a posterior determinaçao de S em shampoo Após a obtençao de um valor de referência para a concentraçao de S na amostra de shampoo, os métodos de preparo de amostras descritos na seçao "Parte experimental" foram avaliados. Os resultados obtidos para S por espectrofotometria UV-Vis e o aspecto dos digeridos após a avaliaçao dos métodos de preparo de amostras estao descritos na Tabela 1.
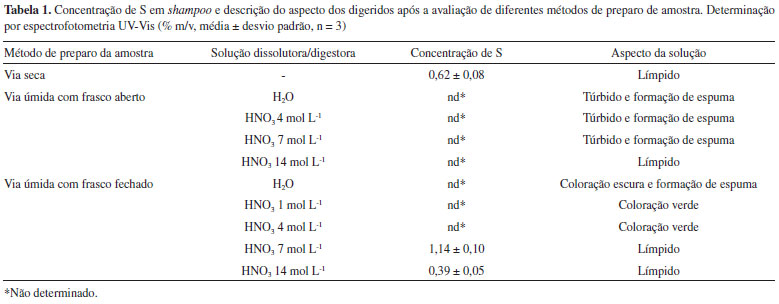
Como pode ser observado na Tabela 1, a maioria das soluçoes obtidas após o uso de diferentes métodos de preparo de amostra nao foram compatíveis para a determinaçao de S em shampoo por espectrofotometria UV-Vis, tendo em vista que estas soluçoes apresentaram coloraçao, turbidez e/ou acidez nos digeridos inadequados a análise. Com relaçao a presença de coloraçao e turbidez na amostra, é importante salientar que estas podem influenciar na medida turbidimétrica, culminando em resultados inexatos, tendo em vista que somente a turbidez associada a formaçao do BaSO4 deve ser determinada. Por outro lado, a elevada acidez dos digeridos após a etapa de preparo da amostra pode aumentar a dissociaçao do precipitado de BaSO4, o que também pode influenciar na exatidao dos resultados.30 Ainda, com base nos resultados obtidos, pode-se observar que quando realizado o preparo da amostra de shampoo através da decomposiçao por via seca utilizando forno tipo mufla, a soluçao resultante apresentou um aspecto límpido e adequado para a análise turbidimétrica (Figura 1A). No entanto, quando realizada a determinaçao de S por espectrofotometria UV-Vis a concentraçao obtida (0,62 ± 0,08% m/v) foi bastante inferior quando comparada com o valor de referência (1,04 ± 0,10% m/v). Provavelmente, a diferença observada pode estar associada a erros causados pela decomposiçao incompleta da amostra, uma vez que foi observada a presença de fuligem no cadinho após a solubilizaçao das cinzas. Além disso, dependendo da composiçao da amostra, espécies voláteis de S, como, por exemplo, SO2 e SO3, podem ser formadas durante o aquecimento e combustao da amostra, sendo perdidas por volatilizaçao devido à utilizaçao de recipientes abertos e elevadas temperaturas.13
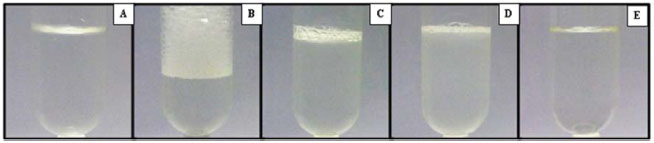 Figura 1. Aspectos das soluçoes obtidas após (A) decomposiçao do shampoo por via seca com a dissoluçao das cinzas utilizando 500 µL HNO3 14 mol L-1 e 5 mL de água, (B) dissoluçao em meio aquoso e decomposiçao por via úmida em frasco aberto com aquecimento convencional utilizando (C) HNO3 4 mol L-1, (D) HNO3 7 mol L-1 e (E) HNO3 14 mol L-1
Quando realizada a dissoluçao do shampoo em meio aquoso utilizando frasco aberto com aquecimento convencional, as soluçoes apresentaram formaçao de espuma e turbidez, o que pode influenciar na determinaçao turbidimétrica de S por espectrofotometria UV-Vis, como mencionado anteriormente (Figura 1B). Além disso, as soluçoes obtidas após a decomposiçao por via úmida com aquecimento convencional em frasco aberto utilizando HNO3 nas concentraçoes de 4 ou 7 mol L-1, também apresentaram aspecto inadequado devido a presença de turbidez e espuma, o que pode estar relacionado com a decomposiçao incompleta da matriz da amostra após a etapa de decomposiçao, nao sendo possível realizar a determinaçao de S por análise turbidimétrica (Figura 1C e 1D). Por outro lado, quando utilizado HNO3 na concentraçao de 14 mol L-1, os digeridos apresentaram um aspecto límpido, conforme pode ser verificado na Figura 1E. Entretanto, após a adiçao do reagente precipitante (BaCl2) nao foi observada a formaçao do precipitado, pois a presença de elevada concentraçao de íons H+ em soluçao pode favorecer a dissociaçao do BaSO4, aumentando a sua solubilidade em meio aquoso.30 Nessa condiçao, a concentraçao de íons H+ remanescentes em soluçao após o preparo da amostra utilizando a digestao por via úmida em sistema aberto com HNO3 14 mol L-1 foi bastante elevada (cerca de 11,2 mol L-1), o que provavelmente favoreceu esse processo. Com relaçao aos resultados obtidos utilizando a dissoluçao do shampoo em meio aquoso em frasco fechado assistido por radiaçao micro-ondas, as soluçoes também apresentaram coloraçao e espuma devido à dissoluçao de compostos presentes na matriz da amostra (Figura 2A). Além disso, as soluçoes obtidas após a decomposiçao por via úmida em frasco fechado assistido por radiaçao micro-ondas utilizando HNO3 nas concentraçoes de 1 e 4 mol L-1, também apresentaram aspecto inadequado para a análise turbidimétrica devido à presença de coloraçao esverdeada, o que pode estar relacionado a decomposiçao incompleta da matriz da amostra após a etapa de decomposiçao (Figura 2 B e C). Por outro lado, quando utilizado HNO3 na concentraçao de 7 e 14 mol L-1, os digeridos apresentaram um aspecto límpido, conforme pode ser verificado nas Figuras 2 D e E.
 Figura 2. Aspectos das soluçoes obtidas após a (A) dissoluçao em meio aquoso e decomposiçao por via úmida em frasco fechado assistida por radiaçao mi cro-ondas utilizando (B) HNO3 1 mol L-1, (C) HNO3 4 mol L-1, (D) HNO3 7 mol L-1 e (E) HNO3 14 mol L-1
Embora a soluçao obtida após a digestao em sistema fechado utilizando HNO3 na concentraçao de 14 mol L-1 tenha apresentado um aspecto límpido (Figura 2E), o resultado obtido após a determinaçao de S por espectrofotometria UV-Vis (0,39 ± 0,05% m/v) apresentou diferença significativa quando comparado com o valor adotado como referência (1,04 ± 0,10% m/v). Isso possivelmente está associado a solubilizaçao parcial do precipitado devido a concentraçao de íons H+ remanescentes em soluçao após o preparo da amostra, que foi de 1,98 mol L-1. Visando contornar esse efeito, sucessivas diluiçoes foram realizadas previamente a etapa de determinaçao. Contudo, nao houve a formaçao de precipitado, provavelmente, em virtude da baixa concentraçao do analito em soluçao após realizar sucessivas diluiçoes. Por outro lado, é importante mencionar que os digeridos obtidos após a decomposiçao por via úmida assistida por radiaçao micro-ondas em frasco fechado utilizando HNO3 7 mol L-1, além de terem apresentado aspecto límpido (Figura 2D), viabilizaram a obtençao de resultados por espectrofotometria UV-Vis (1,14 ± 0,10% m/v) concordantes com o valor adotado como referência (1,04 ± 0,10% m/v). Vale ressaltar que a concentraçao de íons H+ remanescente em soluçao após o preparo da amostra nessa condiçao foi inferior (1,06 mol L-1) às observadas nas condiçoes em que nao houve a formaçao de precipitado (decomposiçao por via úmida com aquecimento convencional em sistema aberto utilizando 14 mol L-1), bem como na condiçao em que o resultado nao foi concordante com o valor adotado como referência (decomposiçao por via úmida assistida por radiaçao micro-ondas em frasco fechado utilizando 14 mol L-1). Assim, diante desses resultados, a decomposiçao por via úmida assistida por radiaçao micro-ondas em frasco fechado utilizando HNO3 na concentraçao de 7 mol L-1 foi selecionada para os estudos subsequentes. Nesse contexto, é importante enfatizar que o método selecionado proporciona a obtençao de digeridos compatíveis com a técnica de determinaçao, além de envolver a utilizaçao de frascos fechados, o que minimiza a ocorrência de contaminaçoes e de perdas por volatilizaçao. Avaliaçao da exatidao do método proposto Para avaliaçao da exatidao das condiçoes selecionadas foram realizados ensaios de recuperaçao, nos quais foram obtidas recuperaçoes em torno de 96 ± 7%. Desta forma, os resultados demonstram que, assim como o método de referência, a decomposiçao por via úmida assistida por radiaçao micro-ondas em sistema fechado utilizando ácido 7 mol L-1 e subsequente determinaçao de S em shampoo por espectrofotometria UV-Vis apresenta exatidao adequada. Adicionalmente, os digeridos obtidos utilizando o método de preparo de amostra proposto foram analisados por cromatografia de íons, e a concentraçao de S determinada nao apresentou diferença significativa (de acordo com o test-t Student ao nível de confiança de 95%, p = 0.288) quando comparado com o valor obtido por espectrofotometria UV-Vis. Além disso, a concentraçao de S obtida (1,14 ± 0,10% m/v) foi concordante em 110% quando comparada com o valor de referência (1,04 ± 0,10% m/v). Cabe mencionar que o desvio padrao relativo (RSD, do inglês Relative Standard Deviation) do método proposto foi inferior a 10%, enquanto que o limite de detecçao (LOD do inglês Limit of Detection) foi de 0,007% m/v, estando este de acordo para atender aos limites recomendados para S pela FDA. O LOD do método foi calculado de acordo com as recomendaçoes do Instituto Nacional de Metrologia, Qualidade e Tecnologia (INMETRO).31 Determinaçao de S em shampoo Como descrito anteriormente, o método de decomposiçao por via úmida assistida por radiaçao micro-ondas em frasco fechado utilizando HNO3 na concentraçao de 7 mol L-1 foi selecionado para a decomposiçao de variados tipos de shampoos visando à determinaçao de S por espectrofotometria UV-Vis. Os resultados obtidos sao apresentados na Tabela 2.
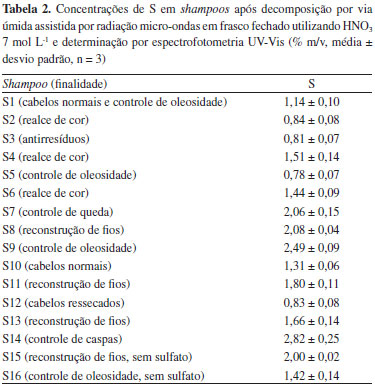
Com base nos resultados, foi possível observar que as concentraçoes de S para os shampoos avaliados variaram de 0,78 a 2,82% m/v, portanto, todas as amostras apresentaram uma concentraçao de S menor que 5%, conforme recomendado pela FDA. O shampoo S5, que tem como principal funçao o controle da oleosidade, foi o que apresentou a menor concentraçao de S (0,78% m/v) o que, provavelmente, deve estar relacionado com a utilizaçao majoritária de surfactantes isentos de SO42- em sua composiçao, bem como de outros compostos isentos de S. É importante destacar que os shampoos S9 (destinado ao controle de oleosidade) e S14 (destinado ao controle de caspas) foram os que apresentaram as maiores concentraçoes de S (2,49% e 2,82% m/v, respectivamente). Acredita-se que a concentraçao obtida para a amostra S9 esteja relacionada a adiçao de compostos contendo S com a finalidade de remover a oleosidade, como os surfactantes lauril sulfato de sódio e lauril éter sulfato de sódio que constam no rótulo do produto. A maior concentraçao de S foi obtida para o shampoo S14 que tem como finalidade o controle de caspas, e esse resultado pode estar relacionado com a recomendaçao da FDA, a qual indica uma faixa de concentraçao de S neste tipo de produto de 2 a 5% para a eficiente eliminaçao de caspas. É importante destacar que os shampoos comercializados como livre de sulfato (S15 e S16) apresentaram concentraçoes consideráveis de S, as quais foram de 2,00% e 1,42% m/v, respectivamente, o que possivelmente está relacionado a adiçao de outros compostos a base de S aos shampoos. Além disso, shampoos com a mesma açao principal, como o S2 e S6 (realce de cor), apresentaram concentraçoes de S que diferiram significativamente, bem como o S5 e 59 para o controle de oleosidade, S8, S11 e S13 para reconstruçao de fios e S1 e S10 para cabelos normais. Esses resultados evidenciam a necessidade do desenvolvimento de um método adequado para a determinaçao da concentraçao de S em shampoos, o que foi alcançado através do desenvolvimento desse trabalho, bem como de métodos que viabilizem a determinaçao das espécies de S adicionadas nesse tipo de produto.
CONCLUSAO De acordo com os resultados obtidos foi possível verificar que a decomposiçao por via úmida assistida por radiaçao micro-ondas em sistema fechado utilizando HNO3 7 mol L-1 e posterior determinaçao indireta de S por espectrofotometria UV-Vis se mostrou um método adequado para o controle de qualidade de shampoos. Além disso, o método proposto nao necessitou do uso HNO3 concentrado, reduzindo, assim, os riscos ao analista durante a análise, assim como o consumo de reagentes e a geraçao de resíduos. Com a utilizaçao desse método foi possível digerir 8 amostras em 40 minutos (20 min de decomposiçao + 20 min de resfriamento do sistema) e ainda utilizar uma técnica de determinaçao rápida, precisa e exata, de fácil manuseio e de baixo custo. Também é importante mencionar que o LOD foi adequado a faixa de concentraçao de S nas amostras avaliadas, e que apesar das variaçoes observadas na concentraçao de S, todas as amostras estao de acordo com o limite recomendado pela FDA. Nesse sentido, o método proposto pode ser uma alternativa promissora para a aplicaçao no controle de qualidade de shampoo, tendo em vista a importância de controlar a concentraçao de S nesse tipo de amostra, as características do método proposto e a carência de métodos que atendam a essa finalidade.
AGRADECIMENTOS Os autores agradecem à FAPERGS, a CAPES e ao CNPq pelo apoio financeiro para o desenvolvimento deste trabalho.
REFERENCIAS 1. Gossens, J.; Beleza, um conjunto em harmonia, 1st ed., Harbra: Sao Paulo, 2004. 2. Gomes, A. L.; O Uso da tecnologia Cosmética no trabalho do profissional Cabeleireiro, 1st ed., Senac: Sao Paulo, 1999. 3. Bouillon, C.; Clinics in Dermatology 1996, 14, 113. 4. https://www.abihpec.org.br/, acessado em novembro de 2016. 5. Abramovits, W.; Kennedy, A. J.; SKIN: Dermatology for the Clinician 2004, 3, 95. 6. Salvador, A.; Pascual-Martí, M.C.; Aragó, E.; Chisvert, A.; March, J.G.; Talanta 2001, 51, 1171. 7. Miller, H. E.; Arch. Dermatol. Syph. 1935, 31, 516. 8. Leyden, J. J.; McGinley, K. J.; Mills; O. H.; Kyriakopoulos, A. A.; Kligman, A.M.; Cutis 1987, 39, 557. 9. Nitschke, M.; Pastore, G. M.; Quím. Nova 2002, 25, 772. 10. Bamford, J. T. M.; J. Am. Acad. Dermatol. 1983, 8, 211. 11. Andrew, N.; Lin, M. D.; Richard J. Reimer, B. A.; Martin C. M. D.; J. Am. Acad. Dermatol. 1988, 18, 553. 12. http://www.fda.gov/, acessado em março de 2017. 13. Nunes, T. S.; Muller, C. C.; Balestrin, P.; Muller, A. L. H.; Mesko, M. F.; Mello, P. A.; Muller, E. I.; Anal. Methods. 2015, 7, 2129. 14. Lobinski, R.; Marczenko, Z.; Crit. Rev. Anal. Chem. 1992, 23, 55. 15. Galo, A. L.; Colombo, M. F. M.; Quim. Nova 2009, 32, 488 16. Tabatai, M. A.; Environ. Lett. 1974, 7, 237. 17. Krug, F. J.; Rocha, F. R. P.; Métodos de preparo de amostras para análise elementar, 3rd ed., EditSBQ: Sao Paulo, 2016. 18. Demanze, C.; Rugroff, L.; Karleskind, A.; Parfums, cosmet., aromes 1984, 58, 69. 19. Pathare, M. N.; Sawant A. D.; Anal. Lett. 1995, 28, 317 20. Burguera, M.; Burguera, J. L.; Quim. Anal. 1996, 15, 112. 21. Smith, F. E.; Arsenault E. A.; Talanta 1996, 43, 1207. 22. Hoenig, M.; Talanta 2001, 54, 1021. 23. Azcue, J.; Mudroch, A.; Environ. Anal. Chem. 1994, 12, 211. 24. Melo, L. C. A.; Silva, C. A.; Quím. Nova 2008, 31, 556. 25. Mesko, M. F.; Picoloto, R. S.; Ferreira, L. R.; Costa, V. C.; Pereira, C. M. P.; Colepicolo, P.; Muller, E. I.; Flores, E. M. M.; J. Anal. At. Spectrom. 2014, 30, 260. 26. Flores, E. M. M; Microwave-Assisted Sample Preparation for Trace Element Determination, 1st ed., Elsevier: Amsterdam, 2014. 27. Itoh, H.; Shinbori, Y.; Bull. Chem. Soc. Jpn. 1987, 60, 1327. 28. López-Ruiz, B.; Journal of Cromatography A 2000, 881, 607. 29. Corazza, G.; Henn, A. S.; Mesko, M. F.; Duarte, F. A.; Flores, E. M. M.; Mello, P. A.; J. Braz. Chem. Soc. 2016, 27, 1569. 30. Vogel, A. R.; Análise química quantitativa, 6th ed., LTC - Livros Técnicos e Científicos Ed. S.A.: Rio de Janeiro, 2002. 31. http://www.inmetro.gov.br/, acessado em dezembro de 2016. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access