
 |
 |
 |
 |
 |
Nota Técnica
|
|
| Avaliação de diferentes métodos de extração para a identificação de resíduos de macrolídeos em alimentos infantis industrializados à base de carne por cromatografia a líquido acoplada à espectrometria de massas sequencial (lc-ms/ms) Evaluation of different extraction methods for identification of macrolides residues in meat-based canned baby food by liquid chromatography-tandem mass spectrometry (lc-ms/ms) |
|
Jônatas V. Grutes*; Rosana G. Ferreira; Mychelle A. Monteiro; Mararlene U. Pereira; Bernardete F. Spisso
Instituto Nacional de Controle de Qualidade em Saúde, Fundação Oswaldo Cruz, Rio de Janeiro - RJ, Brasil Recebido em: 02/03/2018 *e-mail: jvgrutes@gmail.com The interest in a better quality of life raised the concern about food safety. The administration of veterinary drugs in livestock may lead the occurrence of residues in food that may be harmful to consumers. Processed products are being consumed more and more, which increases concern for the safety of infants and children because they are more susceptible physiologically. Among these products stand out canned baby foods, defined by Ordinance 34/1998 issued by Brazilian Ministry of Health. Eggs and meat-based salty products for infants and children, legally designated as soups, fit into this definition and can not contain residues of hormones, antibiotics, as well as other pharmacologically active substances. Several techniques are used as identification and confirmatory assays of veterinary drug residues in food, although the main used technique is liquid chromatography - tandem mass spectrometry (LC-MS/MS). The aim of this work was to make a comparative assessment between different extraction methods in order to establish the best condition within the tested ones for the identification of macrolide residues in meat-based canned baby food by LC-MS/MS. A simple and effective extraction method, with good recoveries (65-93%) and satisfactory relative standard deviations (0.4-11%), was achieved using QuEChERS procedure for sample preparation. INTRODUÇAO Nos dias de hoje é cada vez maior a preocupaçao com a segurança dos alimentos, sobretudo devido à busca da sociedade por uma melhor qualidade de vida.1 Sabe-se que os medicamentos veterinários ocupam papel significativo no incremento da produçao no setor pecuário.2 Entretanto, a contaminaçao química devido à essa utilizaçao pode se tornar um grande problema para a saúde dos consumidores devido à presença dos resíduos de medicamentos veterinários, definidos como todos os compostos ou seus metabólitos presentes em qualquer porçao comestível de um produto de origem animal.3,4 Diversos órgaos de regulamentaçao estabelecem Limites Máximos de Resíduos (LMR) permitidos para alimentos. Concentraçoes acima desses LMR para substâncias autorizadas e a presença de substâncias proibidas podem elevar as chances do desenvolvimento de reaçoes alérgicas, discrasias sanguíneas e carcinogenicidade nos consumidores. Além disso, no caso dos antimicrobianos, mesmo concentraçoes abaixo desses limites podem induzir o surgimento de bactérias resistentes.5 Ressalta-se que os efeitos prejudiciais à saúde anteriormente citados podem ser ainda mais pronunciados em lactentes (crianças de zero a doze meses incompletos) e crianças de primeira infância (crianças de um a três anos),6 tanto por serem mais suscetíveis fisiologicamente, quanto por estarem, proporcionalmente, mais expostos aos alimentos do que adultos, levando-se em consideraçao a taxa de consumo por unidade de peso.7 Inseridos no contexto dos antimicrobianos de uso veterinário, os macrolídeos se destacam devido à sua ampla utilizaçao na pecuária no tratamento de micoplasmose, doença digestiva hemorrágica, infecçoes respiratórias e abscessos hepáticos, e por serem classificados como criticamente importantes para a induçao do surgimento de bactérias resistentes pela Organizaçao Mundial da Saúde e Organizaçao Mundial de Saúde Animal.8,9 Os macrolídeos se caracterizam por apresentarem em suas estruturas químicas um anel lactona macrocíclico contendo 14 (eritromicina, roxitromicina, claritromicina, oleandomicina e troleandomicina), 15 (azitromicina) ou 16 (espiramicina, tilosina e tilmicosina) átomos de carbono, com moléculas de açúcares ligadas por ligaçoes glicosídicas.10 A Figura 1 apresenta as estruturas químicas da tilosina e da espiramicina, respectivamente, dois dos principais macrolídeos empregados na medicina veterinária.
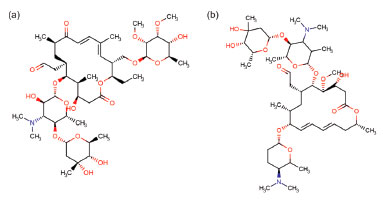 Figura 1. Estrutura química de dois dos principais macrolídeos empregados na medicina veterinária. (a) tilosina, (b) espiramicina11
Quanto à regulaçao de resíduos em alimentos infantis, especificamente em alimentos de transiçao, ou seja, aqueles industrializados utilizados como complemento do leite materno e que tem como objetivo promover uma adaptaçao progressiva aos alimentos comuns, a Portaria nº 34 de 1998 explicita que nao é admissível a presença de resíduos de hormônios, antibióticos, bem como resíduos de substâncias farmacologicamente ativas em tais produtos. Enquadram-se nessa definiçao os alimentos utilizados como refeiçoes salgadas à base de carnes e ovos para lactentes e crianças de primeira infância, legalmente designados como sopinhas.6 A aplicaçao do conceito de tolerância zero exige que sejam empregados nas análises métodos com alta sensibilidade e, devido à complexidade e variedade das matrizes desses alimentos, um processo adequado de extraçao das amostras se faz necessário.12 As principais dificuldades analíticas na análise de resíduos como, por exemplo, os baixos níveis de concentraçao e o grande número de substâncias pesquisadas podem ser superados com o emprego da técnica analítica adequada, em que se destaca a cromatografia a líquido acoplada à espectrometria de massas sequencial (LC-MS/MS).13 Entretanto, uma matriz tao complexa, como as sopinhas, requer procedimentos de preparo de amostras apropriados que garantam a desproteinizaçao, a remoçao de gordura e açúcares e a extraçao dos fármacos, evitando interferências da matriz na análise e permitindo a aplicaçao da legislaçao relativa a esses alimentos.10,14 Além disso, a combinaçao entre uma técnica de extraçao simples e o uso da LC-MS/MS possibilita que o método seja mais rápido e econômico que outros.7 Nesse contexto, o método QuEChERS (Quick, Easy, Cheap, Effective, Rugged Safe) para extraçao e limpeza de extratos atraiu grande interesse nos últimos anos. Originalmente baseado na extraçao/partiçao prévia da amostra utilizando acetonitrila, sulfato de magnésio e cloreto de sódio com posterior etapa de limpeza empregando extraçao por fase sólida dispersiva combinando amina primária e secundária e sulfato de magnésio.15 Este método apresenta vantagens sobre mé todos tradicionais de preparo de amostra por proporcionar altos percentuais de recuperaçao para um grande número de substâncias de diferentes polaridades e volatilidades. Além disso, permite a realizaçao de determinaçoes rápidas com pequenos volumes de solventes e pode ser realizado por um único analista, sem a necessidade de utilizaçao de muitos materiais e equipamentos.16 Pelas vantagens e número de estudos que o utilizam pode-se afirmar que o método QuEChERS repre senta o estado da arte na determinaçao multirre síduos em alimentos.17 Embora ainda em pequeno número, pesquisas mostraram ser possível a determinaçao com sucesso de diversos medicamentos veterinários em alimentos de transiçao por LC-MS.7,18 O objetivo desse trabalho foi realizar uma avaliaçao comparativa entre diferentes métodos de extraçao, com o intuito de se estabelecer a melhor condiçao entre os testados, para a obtençao de um método qualitativo de identificaçao de resíduos de macrolídeos em alimentos industrializados à base de carne por LC-MS/MS.
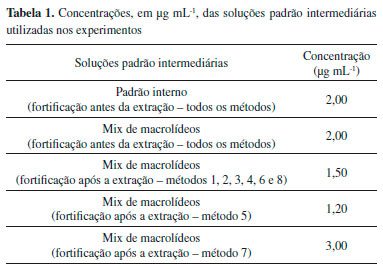
PARTE EXPERIMENTAL Amostras As amostras dos alimentos infantis (sopas) foram adquiridas em redes de supermercados e farmácias da cidade do Rio de Janeiro e armazenadas em temperatura ambiente. Padroes e reagentes Os padroes analíticos de estearato de eritromicina (ERI), tartarato de tilosina (TIL) e troleandomicina (TROL) foram obtidos da Farmacopeia Americana (Rockville, EUA). Com exceçao da TROL (83% de pureza) as substâncias possuíam purezas maiores que 99%. A espiramicina (ESPI) foi adquirida da Sigma-Aldrich (St. Louis, EUA) e possuía pureza de 87,7%. A claritromicina (CLA) (100% de pureza) foi obtida da Farmacopeia Brasileira (Santa Maria, Brasil). A oleandomicina (OLE), a tilmicosina (TILM) e a roxitromicina (ROX), todas como purezas superiores a 96,5%, foram adquiridas da Dr. Ehrenstorfer (Augsburg, Alemanha), sendo a ROX escolhida como padrao interno. Os reagentes acetato de sódio anidro (NaOAc), acetonitrila (ACN) para cromatografia a líquido, ACN para LC-MS, ácido acético (HOAc), ácido fórmico (FOA), carbonato de potássio (K2CO3), cloreto de sódio (NaCl), sulfato de magnésio (MgSO4) e sulfato de sódio (Na2SO4) foram adquiridos da Merck (Darmstadt, Alemanha) e possuíam purezas superiores a 99% com exceçao do FOA (98-100%). O metanol (MeOH) para cromatografia a líquido foi obtido na J. T. Baker (Phillipsburg, EUA) e possuía pureza superior a 99,8%. A amina primária e secundária (PSA) foi adquirida da Agilent Technologies (Santa Clara, EUA) e possuía pureza superior a 99%. A água purificada tipo I foi obtida por sistema Milli-Q (Millipore, Bedford, EUA). Soluçoes estoque e intermediárias Cada padrao foi pesado e solubilizado em MeOH a fim de se obter soluçoes padrao estoques com concentraçoes de aproximadamente 1000 µg mL-1. As soluçoes foram transferidas para microtubos e armazenadas em temperaturas inferiores a -70 °C. Para a fortificaçao das amostras foram preparadas soluçoes padrao intermediárias contendo um mix dos analitos a partir das soluçoes padrao estoques. As soluçoes foram preparadas visando atingir concentraçoes finais dos extratos iguais em todos os métodos. As concentraçoes das soluçoes padrao intermediárias encontram-se descritas na Tabela 1.
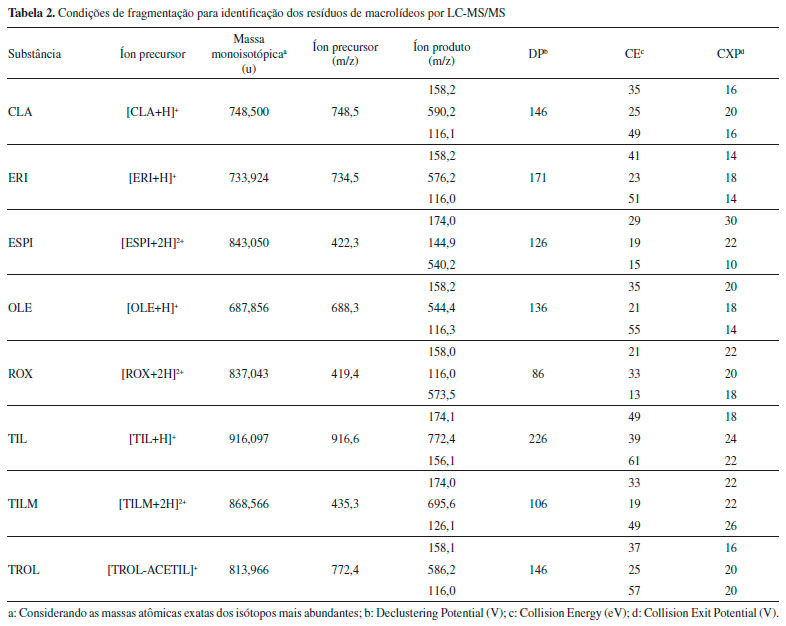
Instrumentaçao LC-MS/MS A separaçao cromatográfica foi realizada utilizando um Cromatógrafo a Líquido de Alta Eficiência, modelo Prominence (Shimadzu, Kyoto, Japao) com módulo de aquecimento para coluna cromatográfica e dispositivo de resfriamento de amostra. A separaçao das substâncias foi realizada com coluna cromatográfica Pursuit® C18 (Agilent Technologies, Amsterda, Holanda) (2,0 x 100 mm, 3 µm de tamanho de partícula) equipada com coluna de guarda Pursuit® C18 (Agilent Technologies, Amsterda, Holanda) e temperatura do forno de 35 °C. A fase móvel utilizada foi 0,1% (v/v) de FOA em água (fase A), 0,1% (v/v) de FOA em acetonitrila (fase B) e 0,1% (v/v) de FOA em metanol (fase C). A eluiçao do gradiente foi: 41% de A e 59% de C até 4 min (fluxo de 0,25 mL min-1); 50% de B e 50% de C em 4,10 min, com manutençao até 10 min (fluxo de 0,30 mL min-1); 41% de A de 59% de C em 10,10 min, com manutençao até 14 min (fluxo de 0,30 mL min-1) e restabelecimento do gradiente inicial com 41% de A de 59% de C em 14,10 min, com manutençao até 16 min (fluxo de 0,25 mL min-1), resultando em uma corrida total de 16 min. O volume de injeçao foi de 25 µL e as injeçoes foram efetuadas em duplicata. A detecçao foi realizada em espectrômetro de massas sequencial triplo quadrupolo API 5000 (Applied Biosystems/MDS Sciex, CA, EUA), com interface eletrospray trabalhando no modo de ionizaçao positivo. Os parâmetros espectrométricos foram: temperatura da fonte TurboIonSpray = 550 °C; voltagem do potencial de entrada = 10 V; voltagem do ionspray = 5000 V; voltagem do detector = 2300 V; voltagem do defletor = -100 V. Nitrogênio foi aplicado como gás de nebulizaçao (50 psi), secagem (55 psi), cortina (10 psi) e colisao (4 psi). O dwell time foi definido como 30 ms. A avaliaçao dos dados foi realizada com auxílio do software Analyst® V1.4.2. As condiçoes de monitoramento de reaçoes múltiplas (MRM) encontram-se descritas na Tabela 2.
Métodos para extraçao das amostras Fortificaçao das amostras antes e após o processo de extraçao para avaliaçao da recuperaçao A fortificaçao inicial da amostra seguiu o mesmo processo em todos os métodos. Pesou-se 2 g da amostra em 4 tubos de centrífuga de 50 mL distintos, os quais foram identificados como amostra controle conforme fortificada com padrao interno (ACCPI), amostra controle nao conforme com fortificaçao no final (ACNCFF), amostra controle nao conforme 1 (ACNC1) e amostra controle nao conforme 2 (ACNC2). Um tubo de centrífuga de 50 mL para avaliaçao dos reagentes foi identificado como amostra controle branco de reagentes (ACBR). Aos tubos da ACBR e da ACNCFF foram adicionados 100 µL de MeOH. A ACCPI foi fortificada com 75 µL de MeOH e 25 µL da soluçao intermediária do padrao interno (ROX) a 2 µg mL-1. A ACNC1 e ACNC2 foram fortificadas com 50 µL de MeOH, 25 µL da soluçao intermediária do padrao interno e 25 µL da soluçao intermediária do mix de macrolídeos para fortificaçao da amostra no início do processo de extraçao a 2 µg mL-1. Após agitaçao em vortex por 10 s os tubos foram mantidos em repouso por 10 min. Métodos distintos de extraçao foram testados (métodos 1 a 8). Após os processos de extraçao, retirou-se 250 µL de sobrenadante de cada um dos tubos, transferiu-se para tubos de centrífuga de 15 mL e evaporou-se até a secura sob fluxo de N2, à temperatura máxima de 47 °C. Ressuspendeu-se o extrato seco da ACBR, ACCPI, ACNC1 e ACNC2 com 1 mL de MeOH:H2O (65:35, v/v), agitou-se em vortex por 15 s e filtrou-se a soluçao com filtro de PVDF de 0,22 µm para um vial. Nos métodos identificados como 1, 2, 3, 4, 6 e 8, paralelamente, ressuspendeu-se o extrato seco da ACNCFF com 1 mL da soluçao padrao intermediária com mix de macrolídeos para fortificaçao da amostra após processo de extraçao, agitou-se em vortex por 15 s e filtrou-se a soluçao com filtro de PVDF de 0,22 µm para um vial. O mesmo procedimento foi adotado para os extratos secos da ACNCFF dos métodos 5 e 7, entretanto a fortificaçao da mesma foi realizada com 1 mL das soluçoes padroes intermediárias com mix de macrolídeos para fortificaçao da amostra após o processo de extraçao com as concentraçoes especificas para esses métodos. Procedimentos de extraçao Os métodos descritos como 1, 2, 3, 4 e 8 foram baseados em ensaios desenvolvidos previamente no laboratório,19-23 enquanto os métodos 5, 6 e 7 basearam-se em métodos descritos por diversos autores, selecionados após levantamento bibliográfico.18,7,24 Método 1: ACN Adicionou-se a todos os tubos duas porçoes de 4 mL de ACN, com agitaçao por 1 min no agitador múltiplo após a adiçao de cada porçao. Agitou-se os tubos na mesa agitadora por 20 min, em velocidade de 240 rpm, e centrifugou-se a 4 °C, 5000 rpm, por 5 min. Método 2: ACN + 0,2 g de NaOAc + 0,8 g de MgSO4 (QuEChERS) Adicionou-se a todos os tubos duas porçoes de 4 mL de ACN, com agitaçao por 1 min no agitador múltiplo após a adiçao de cada porçao. Adicionou-se a cada tubo 0,2 g de NaOAc e 0,8 g de MgSO4. Agitou-se por 1 min no agitador múltiplo e centrifugou-se a 4 °C, 5000 rpm, por 5 min. Método 3: 0,1% (v/v) de FOA em ACN + 0,2 g de NaOAC + 0,8 g de MgSO4 (QuEChERS) Adicionou-se a todos os tubos duas porçoes de 4 mL de 0,1% (v/v) de FOA em ACN, com agitaçao por 1 min no agitador múltiplo após a adiçao de cada porçao. Adicionou-se a cada tubo 0,2 g de NaOAc e 0,8 g de MgSO4. Agitou-se por 1 min no agitador múltiplo e centrifugou-se a 4 °C, 5000 rpm, por 5 min. Método 4: ACN + 0,2 g de NaOAC + 0,8 g de MgSO4 + 200 mg de PSA (QuEChERS) Adicionou-se a todos os tubos duas porçoes de 4 mL de ACN, com agitaçao por 1 min no agitador múltiplo após a adiçao de cada porçao. Adicionou-se a cada tubo 0,2 g de NaOAc e 0,8 g de MgSO4. Agitou-se os tubos na mesa agitadora por 20 min, em velocidade de 240 rpm, e centrifugou-se a 4 °C, 5000 rpm, por 5 min. Para a limpeza dos extratos, de cada um dos tubos transferiu-se 2 mL do sobrenadante para tubos de centrífuga de 15 mL distintos contendo 200 mg de PSA. Agitou-se por 1 min no agitador múltiplo e centrifugou-se a 4 °C, 5000 rpm por 5 min. Método 5: 1% (v/v) de FOA em ACN Adicionou-se 2 mL de H2O aos tubos e agitou-se por 15 s. Posteriormente adicionou-se a todos os tubos duas porçoes de 4 mL de 1% (v/v) de FOA em ACN, com agitaçao por 1 min no agitador múltiplo após a adiçao de cada porçao. Agitou-se os tubos na mesa agitadora por 20 min, em velocidade de 240 rpm, e centrifugou-se a 4 °C, 5000 rpm, por 5 min. Método 6: 1% (v/v) de HOAc em ACN + 0,2 g de NaOAc + 0,8 g de MgSO4 (QuEChERS) Adicionou-se a todos os tubos duas porçoes de 4 mL de 1% (v/v) de HOAc em ACN, com agitaçao por 1 min no agitador múltiplo após a adiçao de cada porçao. Adicionou-se a cada tubo 0,2 g de NaOAc e 0,8 g de MgSO4. Agitou-se por 1 min no agitador múltiplo e centrifugou-se a 4 °C, 5000 rpm, por 5 min. Método 7: 1% (v/v) de HOAc em H2 O:ACN (16:84, v/v) + 2,4 g de Na2 SO4 + 0,58 g de NaOAc (QuEChERS) Adicionou-se a todos os tubos duas porçoes de 2 mL de 1% (v/v) de HOAc em H2O:ACN (16:84, v/v), com agitaçao por 1 min no agitador múltiplo após a adiçao de cada porçao. Adicionou-se a cada tubo 2,4 g de Na2SO4 e 0,58 g de NaOAc anidro. Agitou-se por 1 min no agitador múltiplo e centrifugou-se a 4 °C, 5000 rpm, por 5 min. Método 8: ACN + 0,8 g de Na2 SO4 + 0,2 g de NaCl + 0,4 g de K2 CO3 (QuEChERS) Adicionou-se a todos os tubos duas porçoes de 4 mL de ACN, com agitaçao por 1 min no agitador múltiplo após a adiçao de cada porçao. Adicionou-se a cada tubo 0,8 g de Na2SO4, 0,2 g de NaCl e 0,4 g de K2CO3. Agitou-se por 1 min no agitador múltiplo e centrifugou-se a 4 °C, 5000 rpm, por 5 min. Avaliaçao dos métodos Os métodos com melhores recuperaçoes globais (Rglobal) dos analitos e menores desvios padroes relativos (RSD) foram selecionados. Para os cálculos da Rglobal e do RSD, primeiramente calculou-se os desvios padroes relativos entre as injeçoes (equaçao 1) e a recuperaçao (equaçao 2) de cada substância. 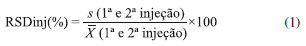 onde: s (1ª e 2ª injeçao) é o desvio padrao entre a área obtida na integraçao do pico da substância na primeira injeçao da ACNC (ou ACNCFF) e a área obtida na integraçao do pico da substância na segunda injeçao da ACNC (ou ACNCFF); 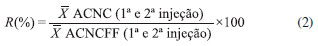 onde: As Rglobal (equaçao 3) e os RSD (equaçao 4) foram calculados segundo as equaçoes 3 e 4: 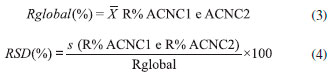 onde:
RESULTADOS E DISCUSSAO Avaliaçao comparativa dos métodos de extraçao Os alimentos infantis industrializados (sopas) à base de carnes sao matrizes complexas, com alto teor de proteínas, lipídeos e também de carboidratos quando legumes estao presentes na formulaçao. Portanto, sao necessárias etapas de limpeza das amostras antes da identificaçao dos analitos por LC-MS/MS. Os métodos se diferenciaram basicamente pelos solventes e sais utilizados para a extraçao, os quais estao descritos na Tabela 3.
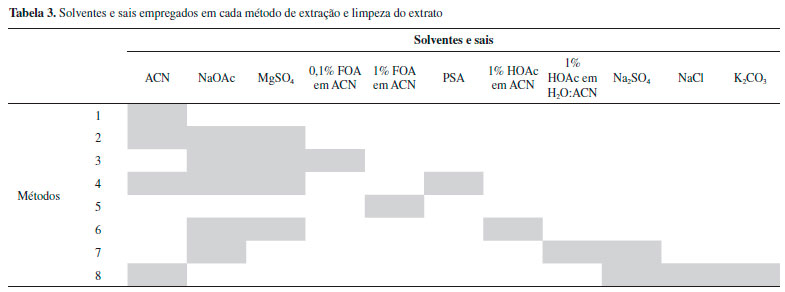
A ACN foi empregada, pura ou em mistura, em todos os métodos por ser reconhecidamente um solvente utilizado para a precipitaçao de proteínas.19 As Rglobal e os RSD dos métodos de 1 a 8 encontram-se nas Figuras 2 e 3 respectivamente.
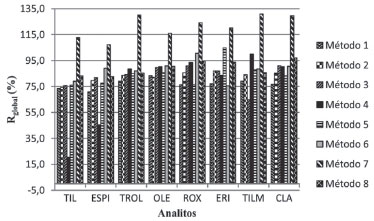 Figura 2. Rglobal(%) dos analitos nos métodos 1, 2, 3, 4, 5, 6, 7 e 8
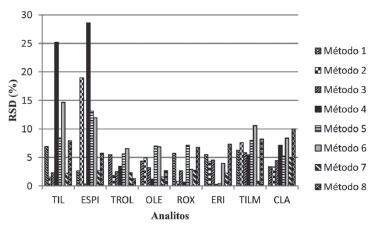 Figura 3. Desvio padrao relativo, RSD(%), entre ACNC1 e ACNC2 nos métodos 1, 2, 3, 4, 5, 6, 7 e 8
O conhecimento das propriedades físico-químicas das moléculas é fundamental para prever e explicar seus comportamentos frente às diferentes condiçoes de extraçao. Quimicamente, os macrolídeos apresentam em geral pKa entre 7,1 e 9,9, tendo propriedades de bases fracas lipofílicas e alguns sao sensíveis ao pH baixo sofrendo degradaçao em condiçoes ácidas.10 Embora os valores de pKa > 7 e do logaritmo do coeficiente de partiçao logP > 2 dos macrolídeos indiquem que a extraçao dessas substâncias em meio ácido nao seja favorecida, por estarem na forma de cátions hidrofílicos, devido à protonaçao dos grupamentos amino (Figura 2), alguns autores empregaram com sucesso acetonitrila ou misturas de acetonitrila e água adicionadas de ácido fórmico ou ácido acético, com ou sem adiçao de sais, para extrair esses antimicrobianos de alimentos infantis.7,18,24 Os ácidos sao geralmente utilizados para competir com o complexo analito-proteína, resultando em um aumento da recuperaçao. Considerando que alguns dos analitos pesquisados ligam-se fortemente a proteínas, tais como a eritromicina (95%) e roxitromicina (96%), a utilizaçao de solventes acidificados pode favorecer a recuperaçao. Entretanto, a presença de co-extrativos na fase orgânica obtida no processo de extraçao em meio ácido pode ocasionar um aumento do efeito de matriz, identificado muitas vezes pela pouca repetibilidade dos resultados de alíquotas verdadeiras ou de replicatas de injeçao. O método 1, que baseia sua extraçao unicamente na ACN, apresentou valores intermediários de Rglobal (71-83%) e valores de RSD de no máximo 7%. No método 5, a acidificaçao da ACN com 1% (v/v) de FOA proporcionou um aumento da Rglobal para todos os analitos, com exceçao da ROX, cuja recuperaçao manteve-se inalterada. Entretanto, ocorreu aumento nos RSD para TIL, OLE, ROX, TILM, CLA e ESPI, substância que apresentou considerável elevaçao do RSD, de 3% no método 1 para 13% no método 5. A partiçao de um analito entre uma fase aquosa e outra orgânica, com a adiçao de sais para promoçao do efeito de salting-out, fundamento básico do método de extraçao QuEChERS, depende da estrutura da molécula, da distribuiçao das microespécies em funçao do pH, a partir dos valores de pKa dos grupamentos ionizáveis, e das mudanças na solubilidade de acordo com a força iônica. Os perfis dos logaritmos dos coeficientes de distribuiçao (logD) versus valores de pH dos analitos estudados indicam que valores de logD> 0, correspondentes à lipofilia, com favorecimento da partiçao na fase orgânica, sao alcançados a partir de pH 4,9 (TIL) a 7,1 (ESPI), sendo que para TROL logD > 0 para todo o intervalo de pH.11 Isso indica que, exceto para a TROL, que independe do pH, em valores de pH mais elevados a recuperaçao seria favorecida. A utilizaçao dos sais secantes sulfato de magnésio e sulfato de sódio diminuem a quantidade de água na fase orgânica e os sais acetato de sódio, cloreto de sódio e carbonato de potássio promovem o efeito salting-out em diferentes valores de pH, reduzindo a solubilidade dos analitos na fase aquosa, com a obtençao de maiores percentuais de recuperaçao na fase orgânica. Ao se avaliar o método 2, cuja diferença para o método 1 é o emprego dos sais NaOAc e MgSO4, pode-se observar um aumento considerável na Rglobal dos analitos, exceto para OLE, cuja recuperaçao se manteve semelhante, e uma diminuiçao dos valores dos RSD para praticamente todos os analitos, com exceçao da ESPI, cujo RSD atingiu o valor de 19%. Por ter apresentado excelentes resultados foi proposta uma etapa de limpeza ao método 2 com emprego de PSA, dando origem ao método 4. Tal método, apesar da etapa de purificaçao, se mostrou inadequado, com a reduçao acentuada da Rglobal para TIL e ESPI, com grande aumento dos RSD para essas substâncias. TIL e ESPI, por serem os macrolídeos menos lipofílicos e mais polares dos estudados (logP 2,32 e 2,5, respectivamente), sofreram maior retençao pelo PSA, conhecido como quelante de açúcares, ácidos graxos e outras substâncias polares de amostras complexas. Ainda em comparaçao com o método 2, desenvolveu-se o método 3, cuja diferença foi a acidificaçao da ACN com 0,1% (v/v) de FOA. Excelentes resultados foram obtidos tanto para os RSD quanto para as Rglobal, sendo a TILM o único analito a apresentar diminuiçao da Rglobal (65%). Com relaçao ao método 7, baseado na extraçao com 1% (v/v) de HOAc em H2O:ACN, NaOAc e Na2SO4, o mesmo nao se apresentou adequado, visto que as Rglobal ficaram acima de 107% para todas as substâncias analisadas, embora os RSD tenham apresentado valores < 5%. Considerando que logP > 0 para todos os analitos pesquisados, a adiçao de água à acetonitrila nao se fez necessária para uma extraçao eficiente desses antimicrobianos, e ainda promoveu a extraçao de co-extrativos polares responsáveis pelo efeito matriz identificado pelas recuperaçoes acima de 100%. O método 6, que também empregou HOAc, apresentou RSD acima de 10% para três substâncias (TIL, ESPI e TILM), demonstrando uma falta de reprodutibilidade entre as injeçoes das ACNC1 e ACNC2, também sendo considerado inadequado. O método 8, diferentemente dos demais, utilizou NaCl e K2CO3, além de Na2SO4 em seu procedimento de extraçao, alcançando altos valores de Rglobal (83-97%) e baixos valores de RSD (≤10%), se mostrando adequado para a análise de alimentos infantis. A adiçao de K2CO3 elevou o pH a valores próximos ou superiores aos pKa dos analitos, favorecendo a partiçao das substâncias para a fase de acetonitrila. Com isso, os métodos 2, 3 e 8 foram selecionados para a repetiçao dos testes de extraçao (Figuras 4 e 5).
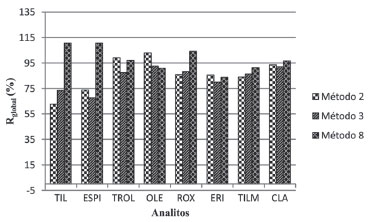 Figura 4. Rglobal(%) dos analitos nos métodos 2, 3 e 8
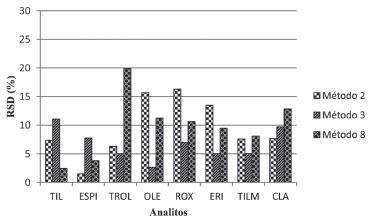 Figura 5. Desvio padrao relativo, RSD(%), entre ACNC1 e ACNC2 nos métodos 2, 3 e 8
Após a realizaçao de novos ensaios, observou-se que os métodos 2 e 8 apresentaram grandes variaçoes entre o primeiro e o segundo dia de ensaio, com valores de RSD de até 16% (ROX) e de até 20% (TROL), respectivamente para os métodos 2 e 8 no segundo dia de ensaio e Rglobal (> 100%) para as substâncias TIL, ESPI e ROX no segundo dia de ensaio no método 8. O método 3 apresentou os melhores valores de Rglobal e RSD, além de menor variaçao entre o primeiro e segundo dia de ensaios realizados, exceto para ESPI e TILM, demonstrando melhor precisao. A Figura 6 exibe os cromatogramas de íons extraídos referentes às transiçoes de quantificaçao da ROX e TROL em soluçao a 1,5 ng mL-1 (a, c) e na matriz fortificada (b, d) em concentraçao equivalente (25 µg kg-1) no método de extraçao 3.
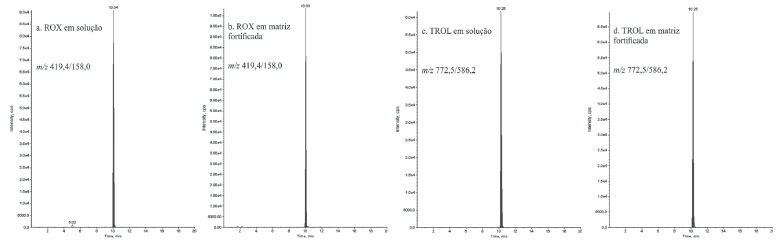 Figura 6. Cromatogramas de íons extraídos referentes às transiçoes de quantificaçao da ROX e TROL em soluçao a 1,5 ng mL-1 (a, c) e na matriz fortificada (b, d) em concentraçao equivalente (25 μg kg-1) no método de extraçao 3
CONCLUSAO Um método qualitativo rápido para identificaçao de resíduos de macrolídeos em alimentos infantis por LC-MS/MS foi obtido. O preparo das amostras foi baseado no método QuEChERS com acetonitrila. O método de extraçao 3, empregando 0,1% (v/v) de FOA em ACN, NaOAc e MgSO4, se mostrou simples e efetivo, pois nao foram necessárias etapas adicionais de limpeza. As recuperaçoes e desvios padroes relativos apresentaram valores entre 65 e 93% e 0,4 e 11%, respectivamente, para os analitos em questao em ensaios realizados em dois dias diferentes. Tais resultados sugerem que, após validado, o método selecionado, se aplicado à amostras de alimentos infantis, do tipo sopas, poderá subsidiar a avaliaçao da exposiçao de crianças e bebês aos resíduos dos medicamentos veterinários da classe dos macrolídeos em futuras açoes de vigilância sanitária.
AGRADECIMENTOS Ao Instituto Nacional de Controle de Qualidade em Saúde da Fundaçao Oswaldo Cruz (INCQS-FIOCRUZ) por todo o suporte.
REFERENCIAS 1. Spisso, B. F.; Nóbrega, A. W.; Marques, M. A. S.; Ciência & Saúde Coletiva 2009, 14, 2091. 2. Spinosa, H. S.; Palermo-Neto, J.; Górniak, S. L.; Medicamentos em Animais de Produçao, 1a ed., Guanabara Koogan: Rio de Janeiro, 2014. 3. Caballeto Torres, A. E. Em Temas de Higiene de los Alimentos; Sánchez, N. C., ed.; Ciencias Médicas: Ciudad de La Habana, 2008, cap. 22. 4. http://www.fao.org/fao-who-codexalimentarius/codex-texts/dbs/vetdrugs/glossary/en/, acessada em outubro de 2018. 5. Baynes, R. E.; Dedonder, K.; Kissell, L.; Mzyk, D.; Marmulak, T.; Smith, G.; Tell, L.; Gehring, R.; Davis, J.; Riviere, J. E.; Food Chem. Toxicol. 2016, 88, 112. 6. Portaria n. 34, de 13 de janeiro de 1998. Aprova o Regulamento Técnico referente a Alimentos de Transiçao para Lactentes e Crianças de Primeira Infância, constante do anexo desta Portaria. Diário Oficial da Uniao, Brasília, 16 de Janeiro de 1998. 7. Aguilera-Luiz, M. M.; Vidal, J. L. M.; Romero-González, R.; Frenich, A. G.; Food Chem. 2012, 132, 2171. 8. http://www.oie.int/fileadmin/Home/eng/Our_scientific_expertise/docs/pdf/Eng_OIE_List_antimicrobials_May2015.pdf, acessada em outubro de 2018. 9. http://apps.who.int/iris/bitstream/10665/255027/1/9789241512220-eng.pdf, acessada em acessada em outubro de 2018. 10. Sismotto, M.; Paschoal, J. A. R.; Reyes, F. G. R.; Quim. Nova 2013, 36, 449. 11. https://chemaxon.com/products/calculators-and-predictors#logp_logd, acessada em outubro de 2018. 12. Rodriguez, E.; Moreno-Bondi, M. C.; Marazuela, M. D.; Food Chem. 2011, 127, 1354. 13. Blasco, C.; Torres, C. M.; Picó, Y.; TrAC, Trends Anal. Chem. 2007, 26, 895. 14. Rodriguez, E.; Moreno-Bondi, M. C.; Marazuela, M. D.; J. Chromatogr. A 2008, 1209, 136. 15. Anastassiades, M.; Lehotay, S. J.; J. AOAC Int. 2003, 86, 412. 16. Prestes, O. D.; Friggi, C. A.; Adaime, M. B.; Zanella, R.; Quim. Nova 2009, 32, 1620. 17. Prestes, O. D; Adaime, M. B.; Zanella, R.; Scientia Chromatographica 2011, 3, 51. 18. Gómez-Pérez, M. L.; Romero-González, R.; Vidal, J. L. M.; Frenich, A. G.; Talanta 2015, 131, 1. 19. Spisso, B. F.; Ferreira, R. G.; Pereira, M. U.; Monteiro, M. A.; Cruz, T. A.; Costa, R. P.; Lima, A. M. B.; Nóbrega, A. W.; Anal. Chim. Acta 2010, 682, 82. 20. Pereira, M. U.; Spisso, B. F.; Jacob, S. C.; Monteiro, M. A.; Ferreira, R. G.; Carlos, B. S.; Nóbrega, A. W.; Food Chem. 2016, 196, 130. 21. Santos, J. R. M. P.; Trabalho de conclusao de curso de especializaçao, Instituto Nacional de Controle de Qualidade em Saúde, Fundaçao Oswaldo Cruz, Brasil, 2015. 22. Melo, J. M. M. C.; Trabalho de conclusao de curso de especializaçao, Instituto Nacional de Controle de Qualidade em Saúde, Fundaçao Oswaldo Cruz, Brasil, 2015. 23. Costa, R. P.; Spisso, B. F.; Pereira, M. U.; Monteiro, M. A.; Ferreira, R. G.; Nóbrega, A. W.; J. Sep. Sci. 2015, 38, 3743. 24. Jia, W.; Chu, X.; Ling, Y; Huang, J.; Chang, J.; J. Chromatogr. A 2014, 1347, 122. |
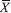 (1ª e 2ª injeçao) é a média entre a área obtida na integraçao do pico da substância na primeira injeçao da ACNC (ou ACNCFF) e a área obtida na integraçao do pico da substância na segunda injeçao da ACNC (ou ACNCFF).
(1ª e 2ª injeçao) é a média entre a área obtida na integraçao do pico da substância na primeira injeçao da ACNC (ou ACNCFF) e a área obtida na integraçao do pico da substância na segunda injeçao da ACNC (ou ACNCFF).On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access