
 |
 |
 |
 |
 |
Artigo
| Molecule design and properties of cyclic energetic materials based on nitroguanidine and 1,1-diamino-2,2-dinitroethene |
|
Xiao MenghuiI; Jin XinghuiI,*; Zhou JianhuaI; Hu BingchengII
I. School of Chemistry and Pharmaceutical Engineering and Qilu University of Technology (Shandong Academy of Sciences), Ji'nan - 250353, China Recebido em: 10/09/2018 *e-mail: jingetiema0000@126.com A series of cyclic energetic derivatives based on nitroguanidine and 1,1-diamino-2,2-dinitroethene were theoretically designed. The spatial structures, infrared spectrometry, heats of formation, electronic structures, detonation properties, and thermal stabilities of these designed compounds were fully investigated by density functional theory. It is found that all the designed compounds have moderate stabilities (bond dissociation energies range from 11.3 to 99.0 kJ mol-1), high crystal densities (from 1.9589 to 2.00188 g cm-3), high positive heats of formation (from 649.6 to 1060.8 kJ mol-1) and high positive heats of detonation (from 1074.77 to 1332.06 cal g-1) which lead to the excellent detonation properties (detonation velocities range from 8.71 to 9.05 km s-1 while detonation pressures range from 35.47 to 38.55 GPa). Electronic structures such as electrostatic potentials on the surface, electronic densities, highest occupied molecular orbitals, lowest unoccupied molecular orbitals and their energy gaps were also simulated to give a better understanding of chemical and physical properties of these compounds. All the data may shine lights on the explosive searching and synthesis. INTRODUCTION High energy density materials have been receiving a continuous interests due to their high positive heats of formation (HOFs), excellent detonation properties and acceptable thermal stabilities which lead to the wide applications both in military and civilian.1-5 For instance, hexahydro-1,3,5-trinitro-1,3,5-triazine (Figure 1, RDX) and 1,3,5,7-tetranitro-1,3,5,7-tetraazacyclooctane (Figure 1, HMX) are of this type of energetic materials with positive heats of formation (HOF, 79 kJ mol-1 and 102.4 kJ mol-1, respectively),6 acceptable impact sensitivities (h50, 26 cm and 29 cm, respectively),7 excellent detonation velocities (D, 8.75 km s-1 and 9.10 km s-1, respectively) and detonation pressures (P, 34.0 GPa and 39.0 GPa, respectively)6. The main reasons were summarized as follows: (1) There exists a large amount of C―N or N―N chemical bonds in the molecule. (2) The cyclic structure will also release lots of energy due to the ring-opening reaction. (3) The high nitrogen content. All these described reasons will make great contribution to the energetic properties during the thermal decomposition or detonation process of these explosives.
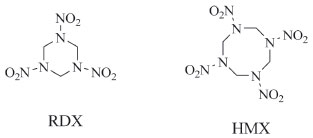 Figure 1. Molecular structure of RDX and HMX
Not surprisingly, 1,1-diaminoethene and guanidine (Figure 2) consist of this type of structure with high nitrogen content of 48.23% and 71.14% respectively, and exist a large amount of C―N or N―N chemical bonds. To our knowledge, 1,1-diaminoethene and guanidine as energetic materials were mainly expressed in the following form: 1,1-diamino-2,2-dinitroethene,8,9 1,2,3-triaminoguanidine,10 1,2-dinitroguanidine,11 energetic salts12-17 and 1,1-diamino-2,2-dinitroethene or guanidine based individual explosives (Figure 2).18,19 Although there are many species of 1,1-diaminoethene or guanidine based energetic materials, each has different advantages and disadvantages with respect to the stabilities and deto nation properties. For example, 1,2-dinitroguanidine, whose detonation properties were superior to RDX, was a strong acid which in turn limited its applications. This is because the hydrogen atom located in ― NHNO2 group was easily to be dissociated due to the strong electron withdrawing effect of nitro group.20
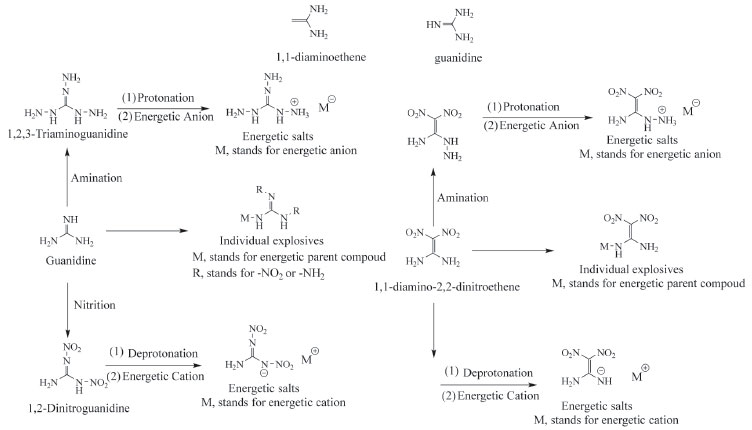 Figure 2. Some guanidine based explosives
Since cyclic structure may release lots of energy due to the ring-opening reaction during the thermal decomposition or detonation process of an explosive, it led to the idea of designing a series of guanidine based cyclic energetic parent compounds (Figure 3, compounds 1-5 and 1'-5'). Till now, little research was done on cyclic energetic materials based on nitroguanidine and 1,1-diamino-2,2-dinitroethene.21,22 On the other hand, it was also found that the increasing number of nitro groups into the cyclic compound can lead to an increase in mass densities, heats of formation and detonation properties. And finally, aiming at looking for new high energy density materials with better detonation properties, some cyclic nitroamines were designed (Figure 4, A1-A5 and B1-B5).
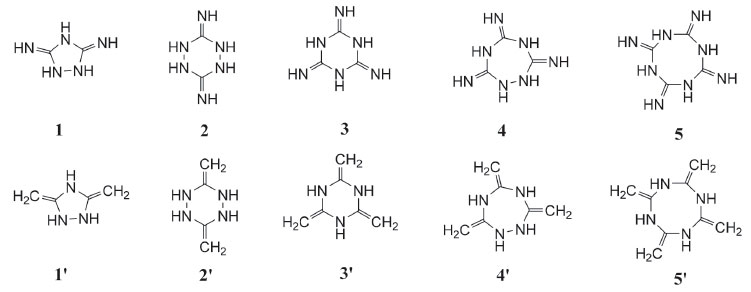 Figure 3. Guanidine based cyclic energetic parent compound
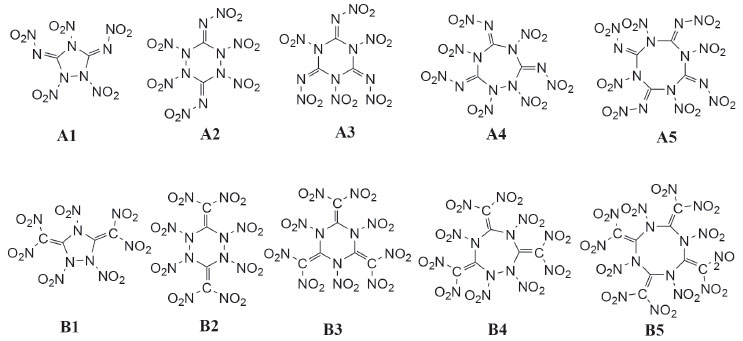 Figure 4. Structures of the designed compounds
In this work, a series of nitramines were designed based on the structures of nitroguanidine and 1,1-diamino-2,2-dinitroethene. The related properties such as heats of formation (HOFs), energetic properties, bond dissociation energies (BDE) and electronic structures were fully investigated. All the presented research may provide useful information for the laboratory synthesis of these designed molecules.
COMPUTATIONAL APPROACH The optimized structures and vibrational analyses of the designed molecules were carried out by Gaussian 03 software23 combined with B3LYP method at 6-31G (d, p) level of density functional theory (DFT) which have demonstrated as a reliable way to calculate the accurate energy.24-27 The optimizations were performed without any symmetry restrictions using the default convergence criteria in the Gaussian program. All of the optimized structures were characterized to be true local energy minima on the potential energy surfaces without imaginary frequencies. Heat of formation (HOF), which means the energy release during the decomposition of an energetic material, is of an essential data to predict the heat of detonation, detonation velocity and detonation pressure. Consequently, isodesmic reactions were employed to calculate the accurate heats of formation of the designed compounds in this work. It has demonstrated that this is a convenient and reliable method since the electronic circumstances of the related reactants and products in the isodesmic reactions are very similar which in turn will reduce the errors of the calculated HOF greatly.28 The designed isodesmic reactions (Figure 5) and related equations (Equation 1-2) were expressed in the following form:
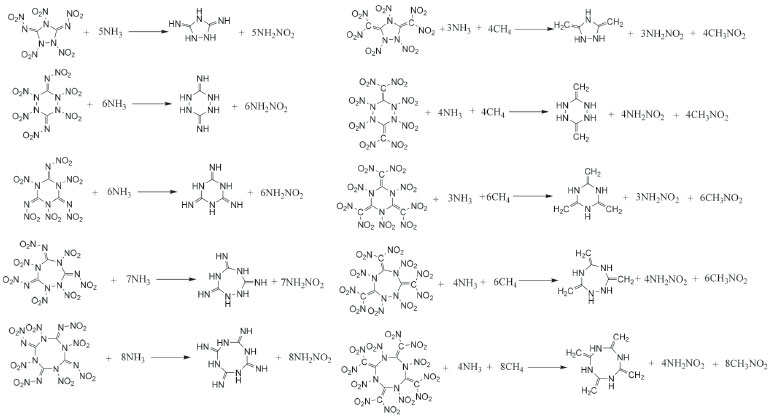 Figure 5. Isodesmic reactions for the designed compounds
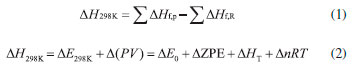 where n means the number of the energetic groups, ΔHf,p means the HOFs of products; ΔHf,R means the HOFs of reactants, ΔE0 means the energy changes between products and reactants, DZPE means the difference between the zero-point energy (ZPE) of products and reactants, ΔHT means the thermal correction from 0 to298 K, Δ(PV) equals to ΔnRT in the reaction. It is found that HOFs of NH3 and NH2NO2 can be obtained either from CRC Handbook of Chemistry and Physics or references. However, accurate HOFs of compounds 1-5 were unavailable and thus, atomization reaction CaHbNc→aC(g)+bH(g)+cN(g) combined with CBS-Q method were employed to predict the accurate HOFs of these unknown compounds.29 It should be also noted that heats of formation obtained via isodesmic reactions were in gas-phase rather than solid-phase while most of the energetic materials exist as solid state instead of gas state. Therefore, solid-phase heats of formation were determined using the gas-phase heats of formation and heat of phase transition according to Hess' law.30 The related equations were expressed in the following form: 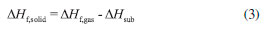 where ΔHsub means the heat of sublimation proposed by Politzer et. al:31 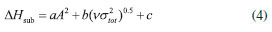 where A means the surface area of the 0.001 e bohr-31 isosurface of electronic density of the molecule, n means for the degree of balance between positive and negative potential on the isosurface, means the measure of variability of the electrostatic potential on the molecular surface which can be performed on Multiwfn program,32 a, b and c are coefficients from reference.33 Empirical Kamlet-Jacob equations (5-6) 34 which were widely employed to calculated the energetic properties were also used in this work to predict the detonation velocity and detonation pressure of the designed compounds. 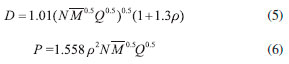 where N means the moles of detonation gases per-gram explosive (mol g-1), In order to get the accurate crystal density (r) of these designed molecules, an improved equation proposed by Politzer et al was introduced:35 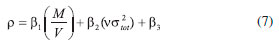 where V means the volume of a molecule, n means the degree of balance between positive and negative potential on the isosurface, σ tot 2 means the measure of variability of the electrostatic potential on the molecular surface while β1, β2, and β3 are coefficients. Bond dissociation energy (BDE), which can be defined as the difference between total energies of products and reactants, is a fundamental parameter to investigate the thermal stability and decomposition mechanism of an energetic material. The related equation was written as follows: 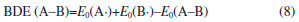 where BDE(A-B) stands for the bond dissociation energy of the neutral molecule, E0(A·) and E0(B·) stand for the energy of the product radicals after bond dissociation, E0(A-B) stands for the total energy of the neutral molecule
RESULTS AND DISCUSSION Heat of formation Table 1 illustrates the total energies (E0), thermal corrections (HT), zero point energies (ZPEs), and heats of formation (HOFs) of the related compounds that referred in the isodesmic reaction. It is seen that all the designed parent compounds have high positive heats of formation (from 196.3 to 554.6 kJ mol-1) which in turn will make great contribution to the heats of formation of the designed compounds.
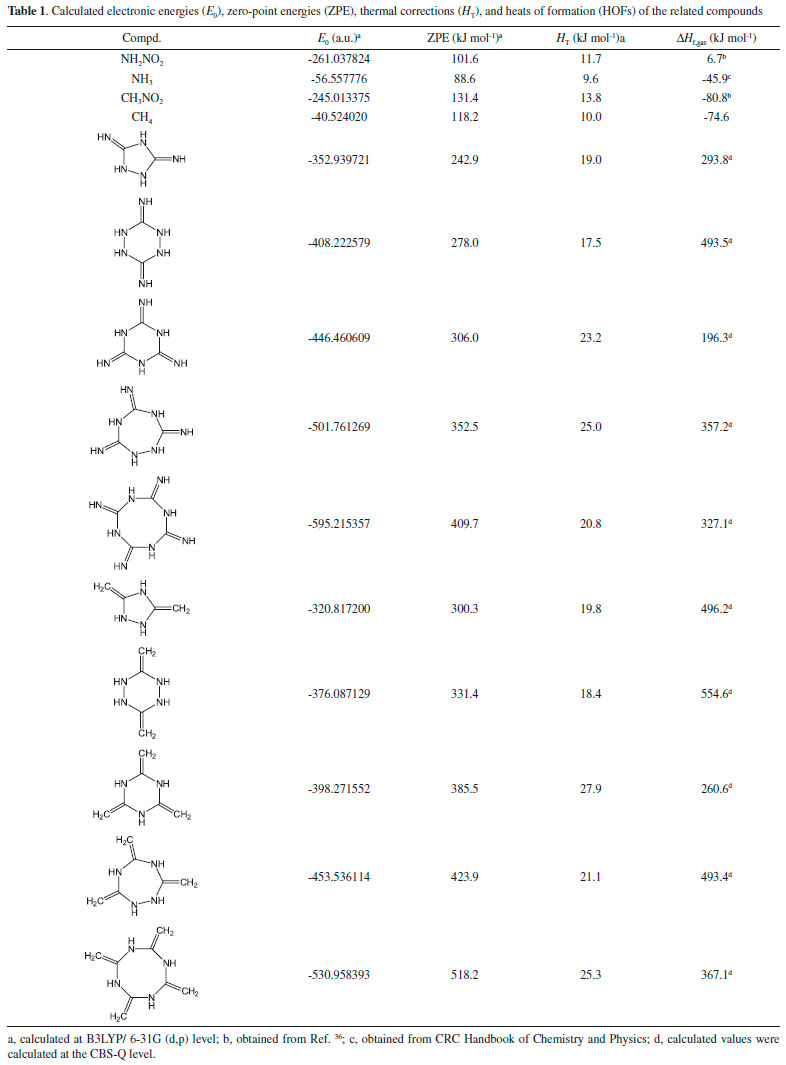
Table 2 summarized E0, HT, ZPE, gas-phase heat of formation (Hf,gas) and solid-phase heat of formation (Hf,solid) of the designed compounds at the B3LYP/6-31G (d, p) level. Obviously, solid-phase heat of formation of compounds that substituted by nitro group were higher than that of the parent compounds. HOFs of compounds A1-A5 were from 710.8 (compound A1) to 1060.8 kJ mol-1 (compound A5) while compounds B1-B5 were from 581.7 (compound B3) to 756.7 kJ mol-1 (compound B4). These high positive heats of formation may be profited from the large amounts of N-N and C-N bonds and the strain energy of the ring in the molecule.
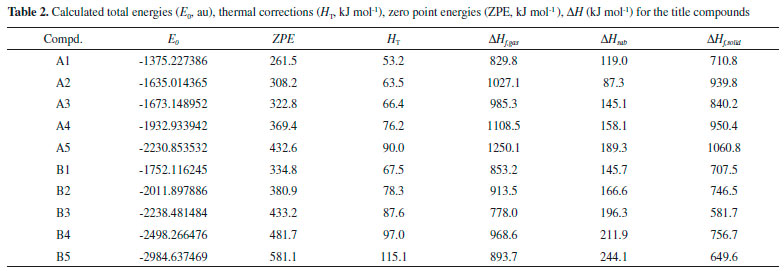
In order to give an evident variation in solid-phase heat of formation, Figure 6 shows the changing trend of heats of formation of the designed compounds. It is seen that heats of formation of compounds A1-A5 were in the following order: A5>A4>A2>A3>A1 while no changing trends were presented for compounds B1-B5. The results were consistent well with previous reports that the heat of formation of an energetic material will increase with the increasing number of nitro groups.37 It also should be noted that heat of formation of compound A2 was higher than that of compound A3 which was caused by the increasing number of N-N chemical bonds in compound A2. It may be concluded that N-N chemical bonds may make a higher contribution to heat of formation to C-N chemical bonds. Compared series A with B, it is found that compounds with the similar parent structure in series A have higher heat of formation than series B since C=N chemical bond is more effective in improving heat of formation of an energetic material than C=C chemical bond. Additionally, compound A5 possesses higher heat of formation than that of compound B5 though the number of nitro group in compound B5 is more than that of compound A5. It may lead to the conclusion that C=N chemical bonds will make more contribution to heat of formation than nitro groups.
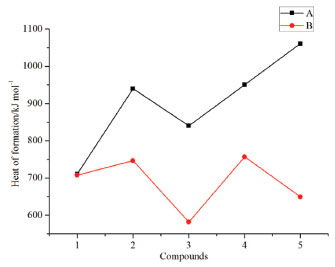 Figure 6. HOFs of the designed compounds
Detonation properties Detonation properties such as heat of detonation (Q), density (ρ), detonation velocity (D) and detonation pressure (P) were important indicators to reflect the energetic performance of an energetic material. These parameters were summarized and listed in Table3.
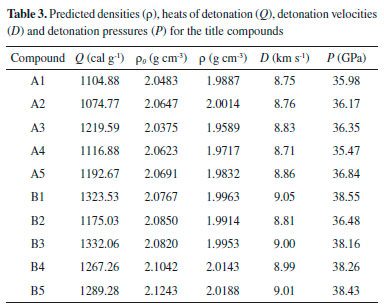
Figure 7 illustrates a comparison of the Q, ρ, D and P for the designed compounds. It is found that all the designed molecules have high densities which range from 1.9589 (compound A3) to 2.0188 g cm-3 (compound B5) and obviously, all the values were higher than those of RDX (1.82 g cm-3) and HMX (1.91 g cm-3). For each series, values of r displays an irregular changing trend. However, density of compounds with the similar parent structure in series B is higher than those in series B. For example, compounds A5 and B5 have the similar parent structure, but the density of compound B5 (2.0188 g cm-3) is higher than that of compound A5 (1.9832 g cm-3). This result was in accordance with the previous research that nitro group is an effective functional group to improve density of an energetic material. Values of D were from 8.71 to 9.05 km s-1 while values of P were from 35.47 to 38.55 GPa and all the values were superior to that of RDX (D, 8.75 km s-1; P, 34.0 GPa). It also should be pointed out that detonation properties of compound A5 have similar values to HMX (D, 9.10 km s-1; P, 39.0 GPa) which may be benefitted by the large amount of N-N or C-N chemical bonds and nitro groups. In view of Q, values of series B were higher than those of series A except compounds B2 since the number of nitro groups in series B were larger than those in series A.
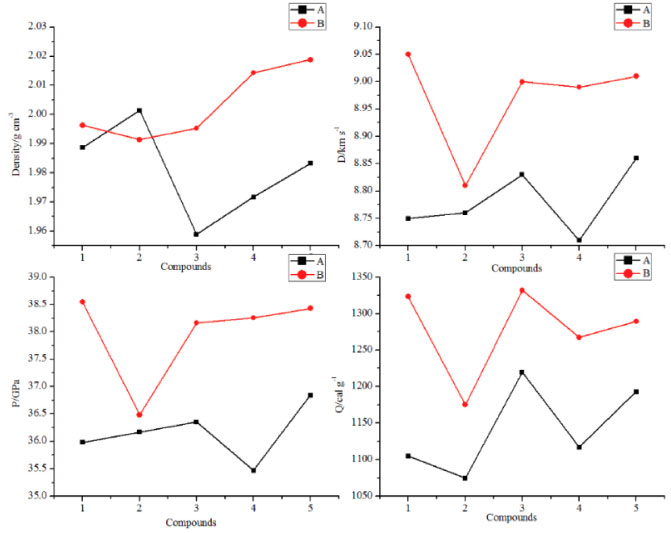 Figure 7. ρ, D, P and Q of the designed compounds
Thermal stability Bond dissociation energy (BDE) can provide useful information in thermal decomposition process and stabilities of an energetic material. In general, the smaller is the energy for breaking a bond, the weaker the bond is, and the easier the bond becomes more unstable. On the other hand, a consensus has been reached that nitro groups attached to the ring often represent as the trigger bonds in thermal decomposition process of organic polynitro compounds and thus, the weakest C-NO2 and N-NO2 bonds, which were screened based on bond order (BO), were selected as the breaking bonds for the calculation of the BDEs. Table 4 lists the values of BO and BDE of the designed compounds.
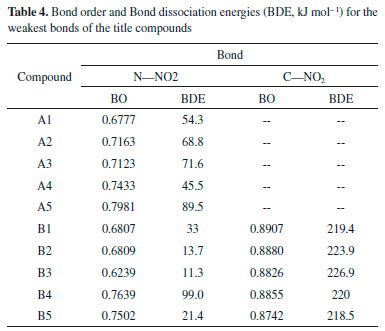
Figure 8 displays the variation in BO and BDE of C-NO2 and N-NO2 bonds, respectively. It is found that BO of C-NO2 (range from 0.8742 to 0.8907) is higher than those of N-NO2 (series A, range from 0.6777 to 0.7981, series B, range from 0.6239 to 0.7639) and consequently, BDE of C-NO2 (range from 218.5 to 226.9 kJ mol-1) is higher than those of N-NO2 (series A, range from 45.5 to 0.89.5 kJ mol-1, series B, range from 13.7 to 99.0 kJ mol-1). It is predicted that N-NO2 bond will be firstly dissociated out from the ring rather than C-NO2 bond during the decomposition process of these designed compounds. Based on the BDE values, the thermal stable order of series A can be defined in the order of A5>A3>A2>A1>A4 while series B were in the order of B4>B1>B5>B2>B3.
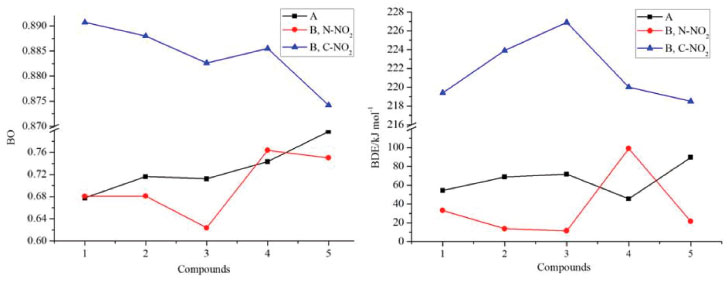 Figure 8. BO and BDE of the designed compounds
Frontier molecular orbitals The highest occupied molecular orbitals (HOMOs) and the lowest unoccupied molecular orbitals (LUMOs) are two most important aspects for frontier molecular orbitals (FMO). The energy of HOMO, LUMO and their gap can also supply useful information in the kinetic stability, optical polarizability and chemical reactivity of a molecule. Therefore, HOMO, LUMO and their energy gap were depicted in Figure 9.
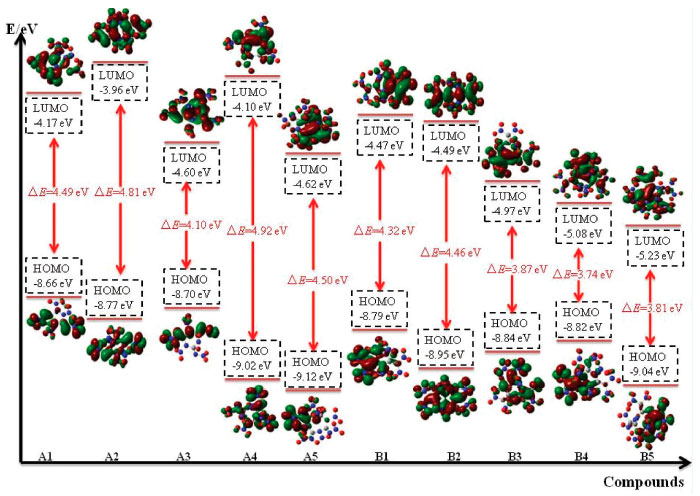 Figure 9. HOMO and LUMO of the designed compounds
From the figure, it is clear thatthe distribution of the HOMO and LUMO varies from different compounds: LUMOs and HOMOs of compounds A1, A3, A4, A5, B1, B3, B4 and B5 were mostly localized on the nitro groups while compounds A2 and B2 were localized both on the ring and nitro groups. In addition, the values of LUMO of these designed compounds range from -5.23 to -3.96 eV while values of HOMO range from -9.12 to -8.82eV. In view of the energy gap between HOMO and LUMO, the values range from 3.74 (compound B4) to 4.92 eV (compound A4) which means that compound A4 has the worst chemical reactivity while compound B4 will be easily reacted with other reagents under given conditions. Again, the reactivity of all the designed compounds can be drawn in the following order: B4>B5>B3>B1>B2>A3>A1>A5>A2>A4. Obviously, the reactivity of series B were higher than that of series A which may be due to the increasing number of nitro groups. Take compounds A1 (ΔE=4.49 eV) and B1 (ΔE=4.32 eV) for example, though they have similar parent chemical structure, compound B1 was more active than compound A1 since more nitro groups were located in compound B1. This phenomenon was consistent with the effect that the increasing number of nitro group will improve energetic properties effectively while it will decrease the stability of a compound evidently.38 Electronic density Electronic density is a fundamental indicator to express a variety of chemical and physical properties of a molecule. Thus, contour line map of the electronic density of the designed compounds were investigated and illustrated in Figure 10. It is seen from the picture that delocalization occurred in the ring (for example, Figure 10, A1, region A) and will improve the stability of the ring skeleton which in turn will improve the stability of the compound. Oppositely, electron densities were found to be concentrated in the bonding area (Figure 10, A1, region B) because of electron pair sharing between atoms bonded covalently. Also, the electron densities around the oxygen and nitrogen atoms were seen higher than those around other atoms (Figure 10, A1, region C) due to strong electro negativity. This is because high peaks correspond to the nuclear charge of the heavy nucleus, which will make great contribution to improve electron aggregation and display integral exponential attenuation towards all surrounding atoms. Finally, electronic density reduced in region D because of the repulsive interactions of lone pair electrons among the adjacent atoms. The reduction of repulsion between adjacent lone pairs were also found by involving the lone pairs of alternate nitrogen atoms in coordinate covalent bonding to carbon atoms which in turn will stabilize the designed compounds. For example, compound A5 possesses this type of structure and consequently, it has the highest BDE value among series A which means that compound A5 was more stable than compounds A1-A4.
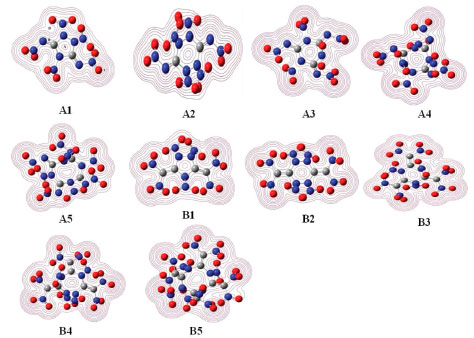 Figure 10. Electronic density of the designed compounds
Electrostatic potential Electrostatic potential (ESP) on molecular surface is a critical parameter since it can provide meaningful information in understanding the charge distributions and molecular reactivity.39,40 Figure 11 presents the ESP and surface area of these designed compounds at the B3LYP/6-31G(d, p) level. It also should be pointed out that the red color in the picture denotes the most negative potential while the blue color denotes the most positive potential.
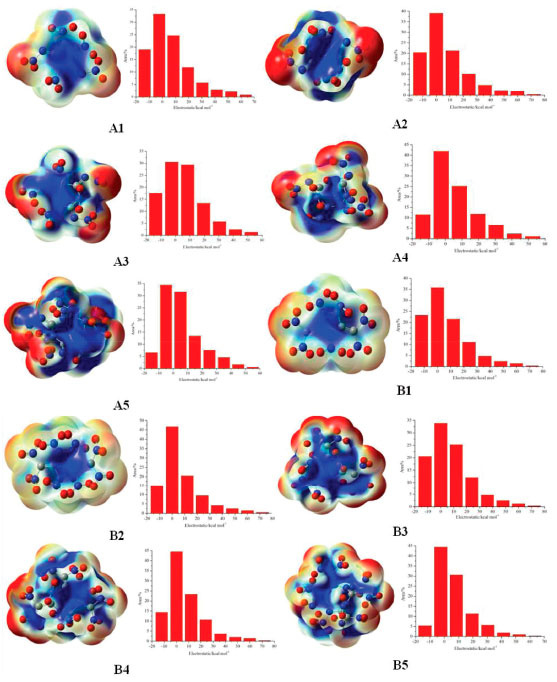 Figure 11. Electrostatic potential of the designed compounds
It can be seen from Figure 11 that the positive area was mainly focused on the ring while negative potentials were mostly distributed on the -NO2, =N-NO2 and =C-NO2 groups due to their higher electronegativity. It is to say that nucleophile attack might be easier happened in red areas while the blue areas were the primary electrophilic sites. On the other hand, positive areas on compounds A4,A5, B4 and B5 were more decentralized than compounds A1, A2, A3, B1, B2 and B3. It implies that compounds A1, A2, A3, B1, B2 and B3 will be more stable than compounds A4,A5, B4 and B5 based on the result proposed by Klapotke et al.41 electrostatic potential of positive areas on the molecular surface which were centralized and large enough will make great contribution to improving the stability of a molecule. Besides, the ratio of positive and negative area were also displayed in the figure. It is found that the area ratio of positive ESP appears on compounds A1, A2, A3, B1, B2 and B3 were larger and more centralized than those appear on compounds A4, A5, B4 and B5. Thermal dynamic properties Thermal dynamic parameters such as standard molar heat capacity (Cθp,m) , standard molar entropy (Sθm) and standard molar enthalpy (Hθm) from 200 to 600 K of these designed compounds were investigated based on vibrational analysis. It is found that all the parameters fit well with the equation X =a + bT + cT2 (X=Cθp,m, Sθm and Hθm). The related constants a, b and c were calculated and summarized in Table 5. From the table, it is seen that values of Cθp,m, Sθm and Hθm increased evidently as the temperature increasing. However, the growth rates of Cθp,m and Sθm decreased while growth rate of Hθm improved evidently as the temperature increased. This is because the translations and rotations of chemical bonds were the main influencing factors when the temperature is low while vibrational movement occurred and intensified at a high temperature. It is also found that values of Cθp,m, Sθm and Hθm increase as the number of nitro groups increasing due to the space steric effects and the strong interaction of nitro groups. Oppositely, the energy gaps between HOMO and LUMO decrease as the number of nitro groups increasing which also may be caused by the space steric effects of nitro groups. Take compounds A5 and B5 for example, Cθp,m, Sθm and Hθm of compound B5 were higher than those of compound A5 whereas the energy gap of compound A5 (4.5 eV)was higher than that of B5 (3.81 eV). In other words, it is the strong space steric effects of nitro groups in compound B5 that led to the higher values of Cθp,m, Sθm, Hθm and lower HOMO-LUMO energy gap. All the parameters may provide useful information in state equation, macroscopic properties and chemical reactions of the designed compounds.
CONCLUSIONS A series of nitroguanidine and 1,1-diamino-2,2-dinitroethene based cyclic energetic materials were designed and the spatial structures, infrared spectrometry, heats of formation, electronic structures, detonation properties and thermal stabilities of these designed compounds were fully investigated by density functional theory. It is found that all the designed molecules have high positive heats of formation (range from 649.6 to 1060.8 kJ mol-1), high heats of detonation (range 1074.77 to 1332.06 cal g-1). Detonation velocities and detonation pressures of the designed compounds range from 8.71 to 9.05 km s-1 and range from 35.47 to 38.55 GPa, respectively. It involves that all the designed compounds have superior detonation properties to those of RDX. Bond dissociation energy show that series B were less stable than those of series A since the former possess more nitro groups. In view of frontier molecular orbitals analysis, chemical reactivity of the designed compounds were summarized in the following order: B4>B5>B3>B1>B2>A3>A1>A5>A2>A4. Finally, compounds A1, A2, A3, B1, B2 and B3 will be more stable than compounds A4, A5, B4 and B5 based on the electrostatic potential analysis.
ACKNOWLEDGEMENTS This study was supported by the National Natural Science Foundation of China (Grant No. 11602121)
REFERENCES 1. Politzer, P.; Murray, J. S.; Propellants, Explos. Pyrotech., 2016, 41, 414. 2. Politzer, P.; Lane, P.; Murray, J. S.; Cent. Eur. J. Energ. Mater., 2013, 10, 305. 3. Jin, X. H.; Zhou, J. H.; Hu, B. C.; Ma, C. M.; J. Phys. Org. Chem., 2017, 30, e3704. 4. Parbat, P.; Devi, A.; Ghule, V. D.; RSC. Adv., 2017, 7, 21585. 5. Qu, Y.; Babailov, S. P.; J. Mater. Chem. A, 2018, 6, 1915. 6. He, P.; Zhang, J. G.; Wang, K.; Yin, X.; Jin, X.; Zhang, T. L.; Phys. Chem. Chem. Phys., 2015, 17, 5840. 7. Guo Y. Y.; Chi, W. J.; Li, Z. S.; Li, Q. S.; Rsc. Adv., 2015, 5, 38048. 8. Latypov, N. V.; Johansson, M.; Holmgren, E.; Org. Process Res. Dev., 2007, 11, 56. 9. Vo, T. T.; Zhang, J. H.; Parrish, D. A.; Twamley, B.; Shreeve, J. M.; J. Am. Chem. Soc., 2013, 135, 11787. 10. Damse, R. S.; J. Hazard. Mater., 2009, 172, 1383. 11. Vasiliev, A. D.; Astachov, A. M.; Molokeev, M. S.; Kruglyakova, L. A.; Stepanov, R. S.; Acta Cryst., 2003, 59, 550. 12. Gao, H. X.; Shreeve, J. M.; Angew. Chem. Int. Ed., 2015, 54, 6335. 13. Vo, T. T.; Shreeve, J. M.; J. Mater. Chem. A, 2015, 3, 8756. 14. Guo, Y.; Gao, H. X.; Twamley, B.; Shreeve, J. M.; Adv. Mater., 2007, 19, 2884. 15. Zhang, Y.; Parrish, D. A.; Shreeve, J. M.; Chem. Eur.J., 2012, 18, 987. 16. Jin, X. H.; Zhou, J. H.; Hu, B. C.; Wang, L.; Sci. Tech. Energ. Mater., 2017, 78, 105. 17. Hu, B. C.; Jin, X. H.; Jia, H. Q.; Liu, Z. L.; Lv, C. X.; Aust. J. Chem., 2014, 67, 1037. 18. Hervé, G.; Jacob, G.; Latypov, N.; Tetrahedron, 2005, 61, 6743. 19. Fischer, N.; Klapötke, T. M.; Stierstorfer, J. ; Z. Naturforsch., B: J. Chem. Sci., 2012, 67, 573. 20. Altenburg, T.; Klapötke, T. M.; Penger, A.; Stierstorfer, J.; Z. Anorg. Allg. Chem., 2010, 636, 463. 21. Jin, X. H.; Zhou, J. H.; Wang, S. J.; Hu, B. C.; Quim. Nova, 2016, 39, 467. 22. Jin, X. H.; Hu, B. C.; Z. Anorg. Allg. Chem., 2016, 642, 635. 23. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgomery, J. A.; Stratmann, R. E.; Burant, J. C.; Dapprich, S.; Millam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q. K.; Morokuma, D. K.; Malick, A. D.; Rabuck, K.; Raghavachari, J. B.; Foresman, J. C.; Ortiz, J. V.; Baboul, A. G.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Andres, J. L.; Gonzalez, C.; Head-Gordon, M.; Replogle, E. S.; Pople, J. A.; Gaussian, Inc.: Pittsburgh, PA. Gaussian 03, 2003. 24. Politzer, P.; Lane, P.; Murray. J. S.; Cent. Eur. J. Energ. Mater., 2011, 8, 39. 25. Mounir, J.; Roy, S.; Abou-Rachid, H.; Lussier, L. S.; J. Hazard. Mater., 2010, 176, 165. 26. Moxnes, J. F.; Hansen, F. K.; Jensen, T. L.; Sele, M. L.; Unneberg, E.; Propellants, Explos., Pyrotech., 2017, 42, 204. 27. Türker, L.; Varıs. S.; Z. Anorg. Allg. Chem., 2013, 639, 982. 28. Jin, X. H.; Wang, L.; Tan, Y.; Ren, J.; Wang, Z. M.; Liu, Y. L.; Wang, L. Y.; Zhou, J. H.; Hu, B. C.; ChemistrySelect, 2018, 3, 1142. 29. Wang, F.; Wang, G. X.; Du, H. C.; Zhang, J. Y.; Gong, X. D.; J. Phys. Chem. A, 2011, 115, 13858. 30. Atkins, P. W.; Physical chemistry, 2nd edn. Oxford University Press: Oxford, 1982. 31. Politzer, P. Ma, Y.; Lane, P.; Concha, M. C.; Int. J. Quantum Chem., 2005, 105, 341. 32. Lu, T.; Chen, F.; J. Comput. Chem., 2012, 33, 580. 33. Byrd, E. F. C.; Rice, B. M.; J. Phys. Chem. A, 2006, 110, 1005. 34. Kamlet, M. J.; Jacobs, S. J.; J. Chem. Phys., 1968, 48, 23. 35. Politzer, P.; Martinez, J.; Murray, J. S.; Concha, M. C.; Toro-Labbe, A.; Mol. Phys., 2009, 107, 2095. 36. Wilcox, C. F.; Zhang, Y. X.; Bauer, S. H.; J. Mol. Struct.: THEOCHEM, 2000, 528, 95. 37. Chi, W. J.; Li, L. L.; Li, B. T.; Wu, H. S.; J. Mol. Model., 2012, 18, 3695. 38. Jin, X. H.; Hu, B. C.; Jia, H. Q.; Liu, Z. L.; Lv, C. X.; Quim. Nova, 2014, 37, 74. 39. Politzer, P.; Murray, J. S.; Theor. Chem. Acc., 2002, 108, AR134. 40. Bulat, F. A.; Toro-Labbé, A.; Brinck, T.; Murray, J. S.; Politzer, P.; J. Mol. Model., 2010, 16, 1679. 41. Hammerl, A.; Klapötke, T. M.; Nöth, H.; Warchhold, M.; Holl, G.; Propellants Explos. Pyrotech, 2003, 28, 165. |
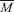 means the average molecular weight of these gases (g mol-1), Q means the heat of detonation (cal g-1), D means the detonation velocity (km s-1), P is the detonation pressure (GPa), ρ means the calculated density (g cm-3).
means the average molecular weight of these gases (g mol-1), Q means the heat of detonation (cal g-1), D means the detonation velocity (km s-1), P is the detonation pressure (GPa), ρ means the calculated density (g cm-3).
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access