
 |
 |
 |
 |
 |
Artigo
| New applications for amoxicillin determination in pure form and pharmaceuticals based on iodate-iodide mixture: titrimetry and spectroscopy studies |
|
Nagib A. S. Qarah*,I; Sameer A. M. AbdulrahmanII; Faisal K. Algethami#,III; Kanakapura BasavaiahIV; Ezzouhra El-MaaidenV
I. Department of Chemistry, Faculty of Education-Zabid, Hodeidah University, Hodeidah, Yemen Recebido em 28/07/2019 *e-mail: nagib2015qarah@gmail.com Three simple methods are described for the determination of amoxicillin (AMX) in bulk drug and dosage forms using iodate-iodide mixture reagent. These methods employed the well-known analytical reaction between iodate-iodide mixture in the presence of acid solution. The iodide oxidized by iodate to iodine in an amount equivalent to the -COOH group present in amoxicillin (AMX) and the liberated iodine determined by titrimetry and spectrophotometry. In the titrimetric method (method A), the liberated iodine was reacted with a measured excess of thiosulphate and the residual thiosulphate was determined by titration with a standard iodine solution using starch indicator, the method was applicable over 2 - 16 mg of AMX. In the second and third methods, the absorbance of the liberated iodine was measured at 370 nm (method B) and also iodine was reacted with starch and starch-iodine complex was measured at 570 nm (method C). In spectrophotometric methods (B and C), Beer's law was obeyed over the concentration range of 5 - 50 and 5 - 60 µg mL-1 AMX with a molar absorptivity values of 5.55×103 L mol-1 cm-1 and 4.76×103 L mol-1 cm-1 for the method A and method B, respectively. The limits of detection (LOD) and quantification (LOQ) were calculated for method B and method C. The proposed methods were found to yield reliable results when applied to bulk drug and dosage forms analysis, and hence they can be applied in quality control laboratories. INTRODUCTION Amoxicillin (AMX) is chemically known as [(6R)-6-(α-p-hydroxyphenyl-D-glycyl-amino)-penicillanic acid] (Figure 1) is abroad spectrum antibiotic belonging to the synthetic penicillins which are widely used in therapy, and has the molecular formula C16H19N3O5S. Its molecular weight is 365.41. It was synthesized by Nayler and Smith.1
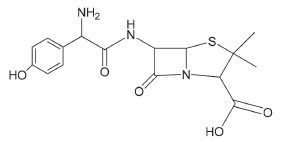 Figure 1. The chemical structure of amoxicillin (AMX)
The British Pharmacopoeia and the United States Pharmacopoeia have described spectrophotometric and titrimetric methods for the analysis of AMX in bulk drug and in the common dosage forms, respectively.2,3 The drug is also official in Indian Pharmacopoeia.4 AMX is used for the treatment of common bacterial infections both in humans and animals.5 AMX is one of the most common antibiotics prescribed for children. It is on the World Health Organization (WHO)'s List of Essential Medicines. They are normally the only penicillins added to feedstuffs at the maximum level of 500 mg kg−1.6 Many methods have been developed to determine amoxicillin in bulk and dosage forms. Various methods for the determination of AMX have been reviewed by Flory.7 Analytical methods for determination of penicillins in general and AMX, in particular, have also been reviewed by Llena et al.8 Recently, Gupta et al.9 have published a review article focusing on various methods available to the determination of AMX in bulk and dosage forms. A literature survey regarding the quantitative analysis of AMX revealed that there are six reports on the titrimetric determination of AMX.10-15 AMX in pharmaceuticals has been assayed by a host of other techniques and include, visible spectrophotometry,16-35 UV-spectrophotometry,36-38 kinetic spectrophotometry,39,40 fluorimetry,41,42 flow injection analysis (FIA) with chemiluminescence detection,43,44 near infrared (IR) reflectance spectroscopy,45 plasma resonance spectroscopy,46 resonance Rayleigh scattering spectrometry,47 voltammetry,48-52 HPLC53-63 and LC-MS/MS.64 From the survey presented above, we found that most titrimetric and spectrophotometric methods reported for AMX are not simple and straightforward requiring a scrupulously anhydrous medium, pre-hydrolysis step at elevated temperature, rigid optimum conditions, liquid-liquid extraction step and/or too many reagents. Further, although diverse titrants and chromogenic agents were used to develop assay methods for AMX, iodate-iodide mixture do not seem to have been used before. Using the reagent (iodate-iodide mixture), the authors have succeeded in developing new titrimetric and two spectrophotometric methods for the determination of AMX in bulk and dosage forms. EXPERIMENTAL Apparatus A Systronic model 166 digital spectrophotometer (Systronic Ahmedabad, Gujarat, India) with matched 1 cm quartz cells was used for absorbance measurements. Materials Pure amoxicillin: Pharmaceutical grade AMX (anhydrous) (99.9% pure) was received from Astrazeneca, Bangalore, India, as gift, and was used as received. Perssmox tablets (Polaris Health Care, India) and bigmex capsules (Astra Zeneca, Bangalore, India) both containing 250 mg AMX, were purchased from local drugstores. Chemicals and Reagents Spectroscopic grade organic solvents (Merck, Mumbai, India) were used in the study. Doubly distilled water and analytical reagent grade chemicals were used. Potassium iodate solution: A saturated solution was prepared by stirring approximately 25 g potassium iodate KIO3 (Merck Ltd., Mumbai, India) in a beaker containing 100 mL water for 1 hour, then the solution was filtered using quantitative filter paper. Potassium iodide: A saturated solution of potassium iodide (Merck Ltd., Mumbai, India) was prepared just before use in order to prevent atmospheric oxidation to iodine. A 2% KI solution was prepared by dissolving 800 mg of the chemical in 40 mL of water for use in preparation of iodine solution. Sodium thiosulphate (0.01 M): This solution was prepared by dissolving about 1.0 g chemical (S.D. Fine Chem., Mumbai, India) in 1 L water. Iodine solution (5 mM): A 5 g of iodate-free KI was placed in 25-30 mL of water in stoppered calibrated flask of 500 mL. About 0.63 g of resublimed iodine was placed in the 500 mL flask containing concentrated potassium iodide solution by means of a small dry funnel and mixed well in the cold condition until iodine was dissolved. Allowed the solution to acquire room temperature, and made up to mark with water in a 500 mL calibrated flask and standardized as usual.65 Saturated borax: it was prepared by dissolving 30 g of borax (S.D. Fine Chem., Mumbai, India) in 100 mL water and stirred with the help of magnetic stirrer for 20 minutes, and then the solution was filtered (pH between 8 and 9). Starch indicator (1%): This was prepared by adding a paste of 1 g starch in water to 100 mL boiling water, boiled for 1 min, and cooled. Standard drug solution A stock standard solution equivalent to 2 mg mL-1 AMX was prepared by dissolving 500 mg of pure drug in water with the aid of heat and diluted to 250 mL in a calibrated flask with water and used in titrimetry (method A). The stock solution was diluted appropriately to get a working concentration of 200 µg mL-1 with water for spectrophotometric methods. Assay procedures Method A (Titrimetry) A 10 mL aliquot of pure AMX solution containing 2-16 mg of AMX was taken in an Erlenmeyer flask. Four milliliters each of saturated solution of KIO3 and KI, and 10 mL of 0.01 M sodium thiosulphate (accurately measured) were added and the flask was let for 15 min with occasional swirling. Then, the unreacted thiosulphate was titrated against a 5 mM standard iodine solution after adding 1 mL of starch indicator to a blue colour endpoint. The amount of AMX in the measured aliquot was calculated. Method B (Spectrophotometry where I2 is measured) Aliquots (0.25-2.5 mL) of 200 µg mL-1 AMX solution were transferred into a series of 10 mL calibrated flasks by means of a micro burette and diluted to 2.5 mL with water. To each flask, one milliliter of each KIO3 and KI were added, flasks are stoppered, content mixed and let stand for 15 min. Then, 1 mL of saturated borax was added and made up to the mark with water. The absorbance of each solution was measured at 370 nm against the reagent blank Method C (Spectrophotometry where I2-starch complex is measured) Volumes in the range 0.25-3.0 mL of 200 µg mL-1 AMX were taken in a series of 10 mL calibrated flasks and the volume brought to 3.0 mL with water. To each flask, 2 mL each of KIO3 and KI solutions were added, flasks were stoppered and content mixed. The flasks were let stand for 15 min. Then, 1 mL of each saturated borax and starch indicator were added to each flask and made up to the mark with water. The absorbance of each flask was measured at 570 nm against the reagent blank. A standard graph was established by plotting the measured absorbance values versus the concentration values of AMX, and the concentration of the unknown was computed using the regression equation derived from the Beer's law data. Procedure for tablets/capsules Twenty tablets or content of 20 capsules were weighed accurately and ground into a fine powder separately. An accurately weighed amount of the powdered tablet or capsule equivalent to 200 mg of AMX was transferred into a 100 mL beaker and dissolved in 50 mL of water with the aid of heat. After cooling, the content was transferred quantitatively to a 100 mL calibrated flask and diluted to the mark with water and mixed well and filtered using a Whatman No. 42 filter paper. An aliquot (5 mL) of the filtrate (2 mg mL-1 AMX) was used for method A, and the filtrate was diluted to 200 µg mL-1 AMX and used for the assay by method B and method C as described above. Procedure for placebo blank and synthetic mixture A placebo blank containing starch (40 mg), acacia (30 mg), sodium citrate (35 mg), hydroxyl cellulose (40 mg), magnesium stearate (45 mg), talc (35 mg) and sodium alginate (35 mg) was prepared by uniform mixing. A 100 mg of the placebo blank was accurately weighed and its solution prepared as described under procedure for tablets/capsules, and then subjected to analysis following the assay procedures. To 50 mg of the placebo blank prepared, 100 mg of AMX was added and mixed well and transferred to a 50 mL calibrated flask and its extract was prepared as described under procedure for tablets/capsules, and then subjected to analysis by following the procedures described above.
RESULTS AND DISCUSSION The acidic property of AMX (-COOH) served to release iodine from the iodate-iodide mixture solution and allowed the determination of AMX drug using titrimetric and spectrophotometric techniques. Iodide present is oxidized by iodate in an amount equivalent to the -COOH and the liberated iodine is determined. In titrimetric method, the released iodine was treated with a measured excess of thiosulphate and the unreacted thiosulphate was titrated with a standard iodine solution and the end point was detected visually using a starch indicator. In spectrophotometric methods, AMX was determined by two different colour reactions and involve the measurement of the absorbance of either the liberated iodine at 370 nm or the iodine-starch complex at 570 nm. Method development In the titrimetric method (method A), the reaction between AMX and the iodate-iodide mixture reagent was tested by reacting 2-16 mg pure AMX with a measured excess of iodate-iodide mixture reagent (Eq. 1) followed by the addition of a known excess of sodium thiosulphate to determine the released iodine (Eq. 2). For the range (2-16 mg) of the drug studied in 10 mL solution, 5.0 mL each of saturated solutions of iodate and iodide were found adequate. The reaction was found to be complete and quantitative when the reaction mixture was allowed to stand for 15 min and any delay in the back titration with a standard iodine solution up to 30 min had no effect on the reaction stoichiometry for the range studied. Then unreacted Na2S2O3 was titrated against a standard iodine solution using starch as indicator to a blue colour end point (Eq. 3).65 The releasing of iodine was not rapid enough to allow direct titration with thiosulphate. Hence, the indirect titrimetric method was followed. 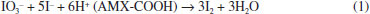 Before the end point: 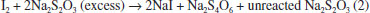 After the end point: 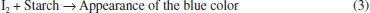 In method B and method C, the absorbance of either liberated iodine (method B) or starch-iodine complex (method C) was measured at 370 nm or 570 nm (Figure 2 and Figure 3).
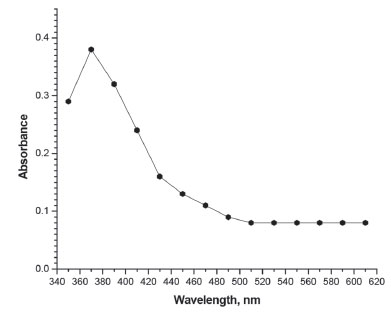 Figure 2. Absorption spectrum of method B: iodine released (20 µg mL-1 AMX)
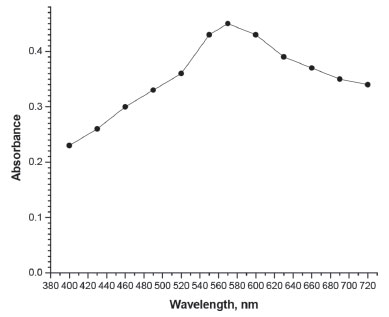 Figure 3. Absorption spectrum of method C: iodine-starch complex (20 µg mL-1 AMX)
In both the methods, the reaction was relatively fast in the beginning but, iodine continued to be liberated even after 15 min. The reaction was stopped after most iodine was liberated during the first 15 min by adding the saturated borax solution to the reaction mixture. The absorbance was remained constant for 30 and 40 min in method B and method C, respectively. The possible reaction pathways for the formation of coloured species in method B (Eq. 4) and in method C (Eq. 5) are presented below: 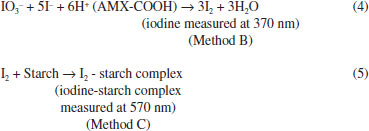 Method validation After optimizing the experimental variables for maximum product formation the proposed methods were validated according to the current ICH66 guidelines. Linearity, LOD and LOQ Under the experimental optimum conditions, the titrimetric method was applicable for 2-16 mg AMX range and slightly irregular stoichiometries were observed beyond these limits. In spectrophotometric methods, under the optimum conditions, a linear relation was found between absorbance and concentration of AMX in the ranges given in Table 1. The limits of detection (LOD) Eq. 6 and limits of quantification (LOQ) Eq. 7 values were calculated according to the equations: 
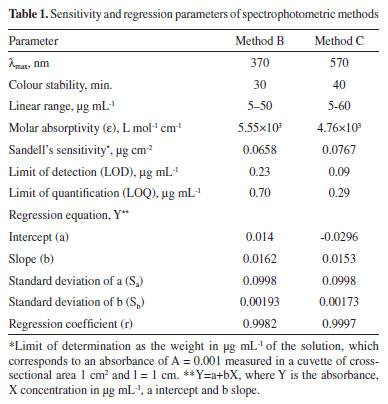
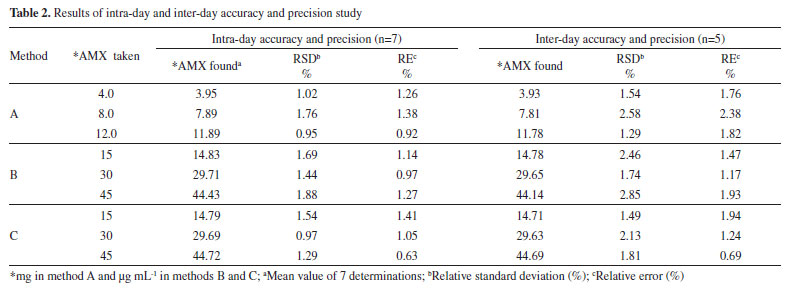
where Sblank is the standard deviation of replicate (n=7) blank absorbance, and b is the slope of the calibration curve. The sensitivity and regression parameters of methods B and C are compiled in Table 1. Accuracy and precision The intra-day and inter-day accuracy and precision were assessed by subjecting the pure drug solution to replicate analysis at three different concentrations in seven replicates during the same day (intra-day precision) and five consecutive days (inter-day precision). The precision expressed as percentage relative standard deviation (%RSD) and accuracy expressed as percentage relative error (%RE) and these values are summarized in Table 2. The results indicate adequate accuracy and precision of the proposed methods.
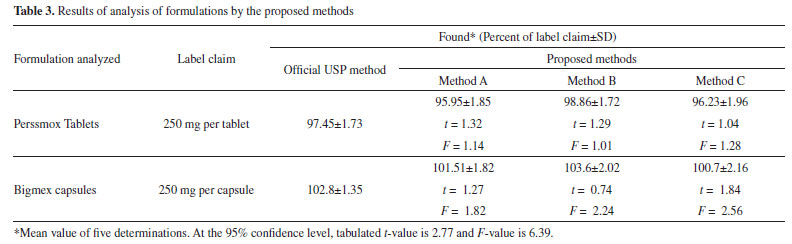
Selectivity This was investigated by observing the effect of co-formulated substances present in tablets by performing placebo blank and synthetic mixture analyses. When the placebo blank was analyzed by the proposed methods, the measured responses were equal to that generated by the blanks. Application of the methods to synthetic mixture analysis yielded 98.55 ± 0.86, 101.54 ± 1.33 and 102.5 ± 1.74 recoveries of AMX by method A, method B and method C, respectively. This suggested the non-interference of tablet/capsule additives and excipients. Application to tablets and capsule analysis The proposed methods were applied to the determination of two brands of tablets and capsule containing AMX. The results of the assay are shown in Table 3 and in good agreement with the label claim and with those obtained using the official USP method.3 Statistical analysis of the results obtained applying Student's t-test and variance-ratio F-test showed that all the calculated t- and F-values are less than the tabulated t- and F-values. These results revealed that no significant difference in the performance of the proposed methods and the official method, with respect to accuracy and precision.67
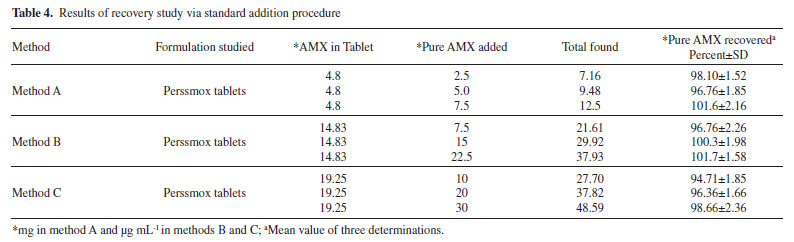
Accuracy by recovery experiment To ascertain the accuracy of the proposed methods, recovery experiment via standard-addition procedure was performed. Pre-analyzed tablet powder was spiked with pure AMX at three levels and the total was found by applying the proposed methods. Each determination was done in triplicate. The results of this study presented in Table 4 and demonstrate that the proposed methods are not affected by the co-formulated substances in the tablets.
CONCLUSIONS This article devoted to develop new analytical methods for titrimetric and spectrophotometric assay of AMX in pharmaceuticals using of iodate-iodide mixture. The titrimetric method is simple to perform compared to reported methods, which requires non-aqueous medium, per-hydrolysis step at elevated temperature, commercially available oxidant and drastic experimental conditions. The method is applicable over a wide range permitting determination of AMX at different levels. Similar advantages can be claimed where to spectrophotometric methods, which employ a single and simple chromogenic agent in contrast to multi-step reactions/multiple reagents employed in many previously reported methods. The proposed methods does not depend on the use of liquid-liquid extraction step or organic solvent. Sensitivity parameters of the proposed methods are comparable to those offered by the published methods. However, some of the published methods have some disadvantages such as longer contact time and elevated temperature.
ACKNOWLEDGEMENTS Authors are thankful to Astrazeneca, Bangalore, India, for gifting the pure sample of amoxicillin. Prof. K. Basavaiah wishes to thank the University Grants Commission, New Delhi, India, for the award of BSR faculty fellowship. The first author (NASQ) wishes to express his thanks to Hodeidah University, Republic of Yemen for awarding research fellowship.
REFERENCES 1. Nayler, J. H.; Smith, H.; British pat. 1964, 978, 178. 2. The British Pharmacopoeia, Volume I and II, Her Majesty's Stationary Office, London, UK, 1998. 3. The United States Pharmacopoeia, 21st Revision, National Formulary 16th ed., 1985. 4. The Pharmacopoeia of India, 3rd ed., Ministry of Health and Family Welfare, Government of India, New Delhi, 1985 5. Becker, M.; Zittlau, E.; Petz, M.; Anal. Chim. Acta 2004, 520, 19. 6. Injac, R.; Kocevar, N.; Strukeli, B.; Croatica Chem. Acta 2009, 82, 685. 7. Florey, K. G.; Trends Anal. Chem. 1985, 4, 129. 8. Llena, M.; Girona, V.; De Bolos, M.; Castillo, J.; Cienc. Ind. Farm. 1985, 4, 13. 9. Gupta, A.; Rajkumar, Y. V.; Rawat, S.; Drug Inven. Today 2011, 3, 33. 10. Kilic, E.; Koseoglu, F.; Akay, M. A.; J. Pharm. Biomed. Anal. 1994, 12, 347. 11. Hasan, H. M. I.; Habib, I. H.; Omar, S. K.; Der Pharmacia Sinica 2012, 3, 327. 12. Qureshi, S. Z.; Qayoom, T.; Helalet, M. I.; J. Pharm. Biomed. Anal. 1999, 21, 473. 13. Chaudhary, S.; Ali, S. K.; Singh, Y. P.; Orien. J. Chem. 2010, 26, 75. 14. Belal, F.; El-Brashy, A.; Ibrahim, F.; J. Assoc. Offic. Anal. Chem. 1990, 73, 896. 15. Shukla, I. C.; Vinod, K.; Brijesh, S.; J. Indian Chem. Soc. 2011, 88, 1383. 16. El-Didamony, A. M.; Saad, M. Z.; El-Shaprawy, D. S.; Main Group Chem. 2013, 12, 139. 17. Salem, H.; Saleh, G. A.; J. Pharm. Biomed. Anal. 2002, 28, 1205. 18. Mohamed, G. G.; Egyptian J. Chem. 2001, 44, 181. 19. Revanasiddappa, H. D.; Veena, M. A.; E-J. Chem. 2008, 5, 100. 20. Singh, D. K.; Maheshwari, G.; Drug Test Anal. 2010, 2, 503. 21. Shafique, A. A.; Rahman, N.; Islam, F.; J. Anal. Chem. 2004, 59, 119. 22. Farhadi, K.; Ghadamgahi, S.; Maleki, R.; Asgari, F. S.; J. Chinese Chem. Soc. 2002, 49, 993. 23. Nagaralli, B. S.; Seetharamappa, J.; Melwanki, M. B.; J. Pharm. Biomed. Anal. 2002, 29, 859. 24. Feng, S.; Jiang, J.; Fan, J.; Chen, X.; Chem. Anal. 2007, 52, 83. 25. Al-Abachi, M. Q.; Haddi, H.; Al-Abachi, A. M.; Anal. Chim. Acta 2005, 554, 184. 26. Nagaraja, P.; Shrestha, A. K.; E-J. Chem. 2010, 7, 395. 27. Basavaiah, K.; Tharpa, K.; Prasad, N. R.; Hiriyanna, S. G.; Vinay, K. B.; Indian J. Chem Techno. 2009, 16, 265. 28. Prasad, R. G. A.; Rao, S. V.; J. Pharm. Res. 2010, 3, 869. 29. Srinivas, C. K.; Babu, N. P.; Divakar, T. E.; Rasayan J. Chem. 2014, 7, 170. 30. Cao, W.; Yan-jing, F.; Yan, M.; Xue-min, M.; Jing-he, Y.; Phys. Test Chem. Anal. Part B: Chem. Anal. 2008, 44, 334. 31. Keskar, M. R.; Jugade, R. M.; World J. Pharmacy Pharm. Sci. 2014, 3, 1340. 32. Mohamed, G. G.; J. Pharm. Biomed. Anal. 2001, 24, 561. 33. Li, Q.; Yang, Z.; Anal. Lett. 2006, 39, 763. 34. El-Ashry, S. M.; Belal, F.; El-Kerdawy, M. M.; El-Wasseef, D.R.; Microchimica Acta 2000, 135, 191. 35. Freitas, S. K. B.; Lins, D. S. V.; Araujo, A. N.; Montenegro, M. C.; Reis, B. F.; Paim, A. P. S.; J. Brazilian Chem. Soc. 2011, 22, 279. 36. Giang, D. T.; Hoang, V. D.; J. Young Pharm. 2010, 2, 190. 37. Huong, V. T.; Hoang, V. D.; Int. J. PharmTech Res. 2009, 1, 1173. 38. Kemal, U.; Murat, P. I.; Elif, K.; Feyyaz, O.; Turkish J. Pharm. Sci. 2008, 5, 1. 39. Karpova, S. P.; J. Chem. Pharm. Res. 2014, 6, 1120. 40. Blazheevskyi, M. E.; Karpova, S. P.; Pharm. J. 2010, 5, 55. 41. Saini, A.; Singh, J.; Kaur, R.; Singh, N.; Kaur, N.; Sens. Actu. B Chem. 2015, 209, 524. 42. Hui-nan, T.; Wei-dong, G.; Mou-sheng, L.; Ya-ling, Y.; Chem. Res. Chinese Uni. 2011, 27, 190. 43. Wang, J.; Li, Y.; Zhang, X.; Chinese J. Pharm. 2014, 45, 269. 44. Chivulescu, A. I.; Badea, D. M.; Cheregi, M.; Danet, A. F.; Revue Roumaine de Chimie 2011, 56, 247. 45. Ying, L.; Wei-min, M.; Yue, C.; Li-ya, H.; Chinese J. Modern Appl. Pharm. 2012, 29, 941. 46. Fei, L.; Ying, L.; Shujun, Z.; Yuanfang, L.; Chinese J. Appl. Chem. 2012, 29, 1329. 47. Hong, J.; Ya-lin, S.; Xiu-ju, Z.; Phys. Test Chem. Anal. Part B: Chem. Anal. 2011, 47, 1063. 48. Fatma, A.; Anal. Lett. 2016, 49, 1366. 49. Kumar, B. P.; Ahmed, D. R.; Sadashiv, P. K.; Sens. Actu. B Chem. 2013, 176, 307. 50. Masoud, F.; Reza, H. M.; Ali, K. M.; Pourya, B.; Nader, T.; Hadis, B.; Int. J. Electrochem. Sci. 2011, 6, 1355. 51. Hong, J.; Mo, F.; Hua, Z.; Phys. Test Chem. Anal. Part B Chem. Anal. 2010, 46, 1447. 52. Behzad, R.; Sajjad, D.; Electroanalysis 2009, 21, 1577. 53. Hai-hong, Y.; Sheng-lian, T.; Hong-yan, F.; Qing, W.; Guo-wei, C.; Chinese J. Antibiotics 2014, 39, 923. 54. Sabry, S. M.; Abdel-Hay, M. H.; Belal, T. S.; Mahgoub, A. A.; Annales Pharmaceutiques Francaises 2015, 73, 351. 55. Shaheen, P.; Shahnaz, G.; Ismail, Y. R.; Harris, S. M.; Fatima, S. A.; World J. Pharm. Res. 2014, 3, 1662. 56. Min-wen, H.; Yao-zuo, Y.; Wen, Q.; Mei, Z.; Chinese J. Pharm. Anal. 2013, 33, 2171. 57. Zhi-hong, X.; Jie-hai, L.; Chinese J. Antibiotics 2013, 38, 749. 58. Ya-zhou, Z.; Lan-lan, F.; Xiao-sheng, Q.; Jie, F.; Yong-biao, W.; Med. Pharm. J. 2013, 17, 207. 59. Rele, R. V.; Mali, R. N.; Pharma Chemica 2013, 5, 273. 60. Chun-yang, Z.; Feng-qiu, W.; Xiang-jun, M.; Liaoning Chem. Ind. 2012, 41, 426. 61. Giang, D. T.; Hoang, V. D.; J. Young Pharm. 2010, 2, 190. 62. Madhura, D.; Vandana, G.; Pranav, J.; Int. J. Pharm. Pharm. Sci. 2010, 2, 129. 63. Zhongxiao, P.; Jie, T.; Chinese Pharm. Affairs 2008, 22, 682. 64. Hui, L.; Xi, X.; Yanan, X.; Shusheng, T.; Xilong, X.; Jiancheng, L.; Jianzhong, S.; J. Chromatogr. B Anal. Tech. Biomed. Life Sci. 2012, 900, 59. 65. Vogel, A. I.; A Text Book of Quantitative Inorganic Analysis Including Elementary Instrumental Analysis, London, 3rd ed., The English Language Book Society and Longman, 1961, pp. 355. 66. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH Harmonised Tripartite Guideline, validation of analytical procedures: text and methodology Q2(R 1), Complementary Guideline on Methodology dated 06 November 1996, incorporated in November 2005, London. 67. Inczedy, J.; Lengyel, T.; Ure, A.M.; IUPAC Compendium of Analytical Nomenclature: Definitive Rules, Blackwell Science Inc., Boston, MA, USA, 1998. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access