
 |
 |
 |
 |
 |
Revisão
|
|
| Nanomateriais plasmônicos: Parte II. química de coordenação de superfície e sua aplicação em sensores e catalisadores Plasmonic nanomaterials: Part II. surface coordination chemistry and its application to sensors and catalysts |
|
Diego P. dos SantosI; Daniel GrasseschiII,*
I. Departamento de Físico-Química, Instituto de Química, Universidade Estadual de Campinas, 13083-970 Campinas - SP, Brasil Recebido em 31/03/2020 *e-mail: dgrasseschi@iq.ufrj.br In this second part we will discuss how surface chemistry properties of plasmonic nanomaterials can be tailored by applying coordination chemistry, supramolecular and materials chemistry approaches, in which nanoparticles with well-controlled features conjugated with properly chosen molecules are the basis of functional organized structures that can be applied as high sensitivity sensors and high efficient photocatalysts. To achieve this level of control the interactions between the nanomaterials surface and the chemical environment surrounding it, the metal-ligand interactions on the materials surface and the electrons and energy transfers at the interface should be well understood. Therefore, we will show how the Surface Enhanced Raman Effect (SERS) can be explored to study the nanomaterials interface. Finally, we will discuss the role of plasmonic nanomaterials in sensors, more specifically SERS sensors, and photocatalysis. INTRODUÇÃO Conforme discutido na primeira parte deste artigo, as propriedades diferenciadas de nanomateriais, principalmente as provenientes da interação da luz com os mesmos, têm grande impacto em diferentes áreas da ciência, englobando a química, física, biologia, engenharia, medicina, dentre outras. A aplicação de nanomateriais em biomedicina,1-3 fotocatálise e eletrocatálise,4-7 sensoriamento,8-10 fotônica e computação óptica,11-13 geração de energia,14-16 dentre outras, vêm crescendo constantemente e é de grande interesse da comunidade científica, por apresentarem alta eficiência e levarem a questão da miniaturização de sistemas ao extremo. Uma vez que a aplicação eficiente de nanopartículas metálicas em diferentes sistemas envolve a comunicação das mesmas com moléculas em sua superfície, o estudo das propriedades espectroscópicas de nanopartículas funcionalizadas com ligantes, a interação destes com o ambiente químico ao seu redor, a elucidação dos processos de transferência de elétrons e/ou de energia entre as espécies envolvidas são de suma importância. Desta forma, essa segunda parte do artigo será focada na química de superfície de nanopartículas plasmônicas, como estas interagem com o ambiente ao seu redor e como suas propriedades ópticas, discutidas na primeira parte do artigo, podem ser exploradas em diferentes tipos de aplicações.
QUÍMICA DE SUPERFÍCIE DAS NANOPARTÍCULAS A aplicação das nanopartículas requer um controle fino de suas propriedades como tamanho, forma, composição e propriedades superficiais. A síntese de nanopartículas utiliza ligantes capazes de controlar a nucleação e o crescimento dos cristais, proporcionando estabilidade ao coloide. Tióis são os ligantes mais utilizados para essa finalidade pois adsorvem em superfícies metálicas formando monocamadas automontadas conferindo estabilidade à superfície e controlando suas propriedades químicas e físico-químicas. Além disso, o uso de ligantes pontes multifuncionais pode ser explorado na coordenação simultânea de metais de transição na superfície das partículas,17,18 estendendo os conceitos de química de coordenação para o design de novos materiais híbridos com propriedades supramoleculares, ópticas, químicas e físico-químicas diferenciadas. A formação de uma forte ligação covalente entre o átomo de enxofre e o metal diminui a energia livre superficial das nanopartículas e efeitos eletrostáticos e estéricos previnem a agregação, conferindo estabilidade às nanopartículas. Já as características dos grupos terminais na interface molécula/solvente determinam as propriedades físico-químicas e a reatividade superficial das partículas e, sendo mais acessível a técnicas experimentais, suas características e propriedades são mais bem conhecidas e controladas. Em contrapartida a natureza e a estrutura das interações metal-ligante na interface molécula-partícula ainda não foram completamente elucidadas. Contudo, esse conhecimento é crucial para a aplicação eficiente das nanopartículas nas diferentes áreas. Em busca de um entendimento mais completo acerca das propriedades interfaciais de nanopartículas funcionalizadas e suas aplicações, diversos grupos de pesquisas vêm desenvolvendo estudos teóricos e experimentais. Dentre os estudos teóricos a utilização de Teoria do Funcional de Densidade independente do tempo (DFT) e dependente do tempo (TD-DFT) vem se destacando no entendimento das propriedades dos estados fundamental e excitados de sistemas envolvendo pequenos clusters metálicos e moléculas orgânicas simples, como piridina e benzenotiol e seus derivados.19-21 Experimentalmente, o estudo da natureza dos complexos superficiais é um tópico desafiador, uma vez que poucas técnicas são capazes de fornecer informações diretas a respeito da interação metal-ligante em superfícies metálicas. Além disso, a complexidade da análise dos dados e a falta de reprodutibilidade são problemas constantemente enfrentados na caracterização das propriedades dos complexos formados. Dessa forma, muitos trabalhos se concentram principalmente na síntese e caracterização das NPs, faltando uma discussão mais detalhada a respeito das naturezas das interações e das propriedades do sistema. Nas próximas seções vamos mostrar como a Espectroscopia Raman Intensificada por Superfície é uma ferramenta poderosa para estudar a superfície de nanopartículas. Existem inúmeros protocolos para sua funcionalização e aplicação. Em um artigo de revisão Beloglazkina e colaboradores22 mostram diferentes métodos de funcionalização de nanopartículas de ouro com ligantes apresentando grupos multifuncionais (fluoróforos, cromóforos, receptores, ativos eletroquimicamente) ligados a um grupo tiol; e sua aplicação em sensores e catalisadores. Nele os autores agrupam os métodos de modificação de NPs em dois tipos: por troca de ligantes na superfície e por modificação de um já existente na superfície. A primeira abordagem consiste em substituir o ligante já existente na superfície das partículas por diferentes ligantes, que apresentam a atividade desejada. Portanto, a labilidade do ligante presente na superfície influenciará a cinética de troca de ligantes e, consequentemente, a estrutura do complexo final. Dessa forma é preferível a utilização de nanopartículas lábeis, normalmente sintetizadas pelo método de Turkevich, através da redução de um sal de metálico com citrato de sódio.17,23 O segundo método consiste em utilizar as nanopartículas como plataformas para realização de reações orgânicas convencionais, gerando grupos terminais com a funcionalidade desejada. Em ambos os métodos deve-se levar em consideração a estabilidade do coloide durante todo processo de funcionalização, evitando a agregação das partículas, a diminuição do rendimento das reações e da atividade do ligante. Apesar da grande variedade de métodos para funcionalização de nanopartículas, os efeitos da formação da ligação ligante-metal na estrutura eletrônica, atividade e reatividade das moléculas ainda é pouco conhecido.
EFEITO SERS E A QUÍMICA DE SUPERFÍCIE Até o momento, a intensificação observada da luz espalhada tem como base dois mecanismos principais: o mecanismo eletromagnético (EM) e o mecanismo químico (CM).19 O mecanismo EM, discutido detalhadamente na primeira parte do artigo, já é bem estabelecido, apresenta natureza clássica e surge devido aos plasmons de superfície, em ressonância com a radiação incidente. Nessa segunda parte vamos focar na discussão dos mecanismos químicos e como estes podem ser aplicados na elucidação das interações na superfície das nanopartículas. Tendo origem na sobreposição das funções de onda do metal e do ligante, o mecanismo CM apresenta três tipos de contribuições diferentes: i) Transferência de carga dinâmica (CT), na qual há uma condição de ressonância entre o laser incidente e transições do tipo de transferência de carga nanopartícula-ligante e ligante-nanopartícula envolvendo os estados próximo ao nível de Fermi da estrutura plasmônica; ii) Raman Ressonante (RR), em que há a excitação de transições eletrônicas internas da molécula adsorvida. Essas transições podem ser intensificadas pela presença da nanoestrutura quando apresentam energia próxima da energia da banda plasmônica, dando origem ao efeito SERS ressonante ou SERRS; iii) Mecanismo químico não ressonante (CHEM) que é causado devido à relaxação eletrônica na molécula, pela presença da estrutura metálica. As três contribuições diferentes do mecanismo CM podem contribuir com um fator de intensificação da ordem de 10 a 106.24 Os mecanismos EM e CM ocorrem mutuamente, de forma a produzir um efeito SERS global. A Figura 1 esquematiza os processos que ocorrem no efeito SERS.
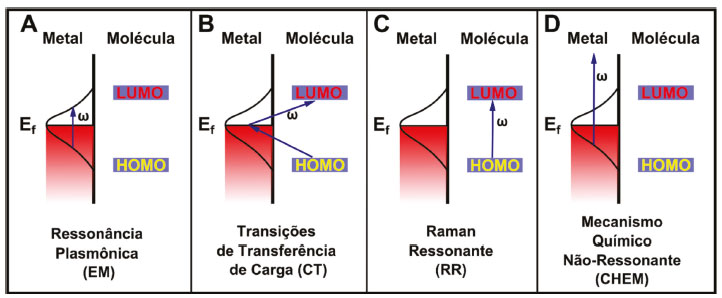 Figura 1. Esquema ilustrando as quatro contribuições para o efeito SERS. (A) mecanismo eletromagnético, (B) mecanismo químico ressonante, (C) Efeito Raman ressonante e (E) Mecanismo químico não ressonante
Nas seções seguintes serão discutidos com mais detalhes os fatores que influenciam a intensidade do espalhamento Raman em cada um dos mecanismos químicos de intensificação descritos acima.
EFEITO QUÍMICO RESSONANTE O tratamento mais aceito atualmente para o efeito químico, foi proposto inicialmente por Lombardi e Birke25-27 e consiste em uma expansão do formalismo de Albrecht28 para a intensidade do espalhamento Raman normal, em função da perturbação causada pela variação das coordenadas nucleares. A intensidade do espalhamento Raman tem sua origem em um "roubo" de intensidade de transições eletrônicas permitidas, entre estados eletrônicos da molécula que são acoplados por um modo normal de vibração da molécula (Qk). Esse processo é conhecido como acoplamento Herzberg-Teller e a extensão desse acoplamento é proporcional a integral de acoplamento entre os estados (), ao modo normal Qk e inversamente proporcional a diferença de energia entre os estados acoplados.29 No formalismo de Albrecht28 o momento de transição de polarizabilidade pode ser dividido em quatro termos (A, B, C e D). Em condições fora de ressonância, o termo A descreve basicamente o espalhamento Rayleigh, pois depende das integrais <vf│vr ><vr│vi>, que serão diferente de zero somente se os estados vibracionais finais (vf) e iniciais (vi) forem iguais (vf = vi), ou seja não haverá variação da frequência da radiação espalhada em relação a incidente. Porém, em condição de ressonância, tais integrais não seriam zero, já que estaríamos considerando estados vibracionais de estados eletrônicos diferentes e estas integrais estariam relacionadas as intensidades das linhas associadas a transições vibrônicas, caracterizando o efeito Raman Ressonante. O termo B considera o acoplamento entre os dois estados excitados da molécula, o termo C entre os estados excitados e fundamentais, e o termo D considera o acoplamento entre múltiplos estados excitados. Para o caso convencional, considera-se que o acoplamento só ocorre entre os estados excitados da molécula pois a diferença de energia entre os estados excitados e fundamental é muito maior que a diferença de energia entre os estados excitados. Portanto, os termos C e D podem ser desconsiderados. Uma discussão mais aprofundada sobre a origem e consequências de cada termo pode ser encontrada no material suplementar. Porém quando se considera o sistema ligante-nanopartícula como um todo, essa aproximação não é mais válida. Devido à grande densidade de estados da partícula metálica haverá estados das bandas de condução e valência próximos ao nível de Fermi (eM) com energia adequada para que o acoplamento entre os estados excitados e fundamental do sistema ligante-partícula sejam observados, aumentando a contribuição dos termos B e C na equação de polarizabilidade. Tal acoplamento também pode levar a origem a transições de transferência de carga nanopartícula-ligante ou ligante-nanopartícula que contribuem para a intensificação do espalhamento Raman. Experimentalmente observa-se que o nível de Fermi (EF) de nanopartículas metálicas (por exemplo, AuNP EF ~ -4 eV)30 encontra-se acima do HOMO da maioria dos ligantes utilizados (e.g. piridina HOMO = -9.26 eV) e abaixo do LUMO, para cada transição HOMO-LUMO do ligante há uma transição de transferência de carga (CT) correspondente envolvendo o nível de Fermi (EF→LUMO ou HOMO→EF). A energia das transições CT deve ser menor em relação à energia das transições eletrônicas puramente moleculares e por essa razão o mecanismo CT pode levar a uma grande intensificação do efeito SERS na região do visível do espectro eletromagnético. O estudo do mecanismo químico em nanopartículas plasmônica é uma tarefa complicada que envolve uma análise minuciosa dos espectros SERS. Porém, de maneira simplificada, pode-se inferir a sua existência através da análise do acoplamento Herberg-Teller, no qual a intensificação de modos normais não simétricos é maior em relação aos modos simétricos. Em experimentos utilizando a técnica de espectro-eletroquímica, nos quais é possível ajustar o nível de Fermi da nanoestrutura metálica através de um potencial elétrico aplicado, as transições CT EF→LUMO e HOMO→EF podem ser comprovadas a partir da dependência do sinal Raman em função do potencial aplicado.25,26,31-34 Alternativamente, a existência de estados de transferência de carga tem sido comprovada por cálculos teóricos de DFT e TD-DFT. Esses estudos correlacionam as propriedades de sistemas pequenos, envolvendo clusters de normalmente até 20 átomos metálicos e uma molécula de ligante, com as propriedades dos complexos formados na superfície das partículas, através da comparação entre os perfis e intensidade do espectro SERS calculado e experimental. Os fatores de intensificação teóricos calculados para o mecanismo CT variam na faixa de 103 a 106, devido à existência de transições de transferência de carga. Jensen et al., utilizando clusters com 20 átomos de prata, mostraram que a coordenação da piridina apresenta forças de ligações diferentes quando ocorre nas faces ou nas pontas do cluster, sendo que na última a força de ligação é maior.35 Além disso, há um desdobramento dos estados eletrônicos da piridina, devido à quebra de simetria causada pela interação com os orbitais do cluster metálico. Porém, nesse caso o desdobramento é maior quando a coordenação ocorre nas faces. Dessa forma, há o surgimento de transições de transferência de carga entre o HOMO, centrado no cluster de Ag20 e os dois primeiros orbitais desocupados de menor energia, que apresentam contribuições predominantemente da piridina. Estendendo o estudo para um cluster de Au20, os mesmos autores mostraram que a intensificação do espalhamento Raman proveniente do efeito EM é menor no ouro, em relação a prata. Contudo, analisando as contribuições do mecanismo CM chegam à conclusão que no complexo formado entre a piridina e o cluster de ouro, a força do oscilador para transições de transferência de carga metal-ligante são maiores em relação ao complexo com prata.36 Hongxing et al. introduziram a utilização de um método baseado na diferença de densidade de cargas entre os estados excitado e fundamental, para a visualização dos estados de transferência de cargas. Com esse método, mostraram que considerando um complexo piridina-Ag20, quando a excitação é feita acima de 725 nm não há ressonância com nenhuma transição eletrônica e a intensificação do espectro Raman ocorre devido ao aumento de polarizabilidade causado pela interação metal-ligante no estado fundamental. Quando a excitação é feita entre 725 a 503 nm e as transições internas do cluster são excitadas, a intensificação ocorre devido à transferência de energia do cluster para a piridina através do mecanismo de Förster, sendo análogo ao mecanismo EM observado em nanopartículas. Já para excitações feitas entre 300-500 nm, as transições entre os estados próximos ao nível de Fermi e o LUMO da piridina são excitadas e a intensificação do espectro Ramam ocorre pelo mecanismo CT.37 Em nanopartículas metálicas as propriedades químicas são determinadas pelos átomos superficiais e pela interface metal-ligante formada na superfície.17,18,38 Esses átomos superficiais apresentam sítios de coordenação livres e tendem a adsorver ou ligar moléculas para reduzir a energia livre de superfície. Como consequência, as propriedades da interface das partículas são modificadas, influenciando sua estabilidade através de interações metal-ligante eletrostáticas, covalentes ou estéricas.39-43 Como já foi mencionado o mecanismo CT de intensificação do sinal SERS envolve transições eletrônicas entre metal-ligante na superfície das nanoestruturas metálicas. Portanto a natureza da ligação química na interface nanopartícula-ligante se torna extremamente relevante e a aplicação de conceitos clássicos de química de coordenação auxilia o seu entendimento. Transições CT são facilitadas quando a interação ocorre entre a nanopartícula com característica de ácido mole44,45 e ligantes cujo sítio de coordenação apresenta características de base mole (e.g tióis)44,45 e com ligantes aceptores π. Isso se dá pela simetria dos orbitais do ligante e pelo aumento da polarizabilidade local. Quando essas interações são maximizadas e a probabilidade de ocorrência de transições CT aumenta, e o espectro SERS pode ser observado mesmo na ausência de hot-spots.24,46,47 Apesar das interações ácido-base mole-mole entre metal e ligante serem o ponto de partida para a funcionalização superficial de nanopartículas, o comportamento das interações metal-ligante na nanoescala ainda não são completamente entendidos. A cinética de substituição e os modos de coordenação específicos do ligante ditiooxamida (dto) na superfície de nanopartículas é um bom exemplo de como a interação metal-ligante na superfície metálica é determinante para os processos de transferência de elétrons e de intensificação do espectro SERS.48 A coordenação da dto na AuNP apresenta um isomerismo de ligação, conforme mostrado na Figura 2a, no qual à baixas concentrações de dto a espécie bidentada, correspondente à ligação bis-tiona-Au na forma cis, {Au-S(NH2)CC(NH2)S-Au}, é predominante (Figura 2d). Mesmo em altas concentrações, a espécie cis é observada no início, porém, conforme a competição com as moléculas de dto não coordenadas procede, o modo de ligação muda gradualmente para o modo monodentado tiona-Au, {AuS(NH2)CCS(NH2)}, (Figura 2c). Nesse modo, o ligante adota a forma termodinamicamente favorável na conformação trans, produzindo mudanças drásticas no espectro SERS (Figura 2a e c). Tal comportamento dinâmico do ligante na superfície plasmônica é de importância crucial, podendo ser um dos fatores associados à falta de reprodutibilidade do espectro SERS frequentemente discutido na literatura.
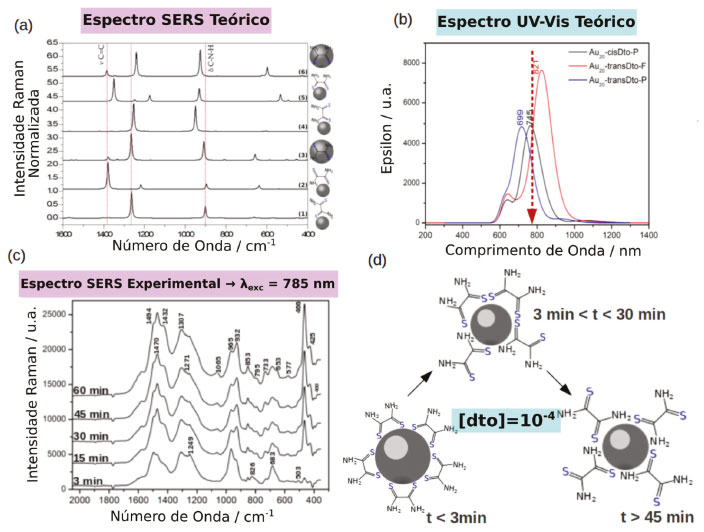 Figura 2. Dinâmica de coordenação da ditiooxamida na superfície de nanopartículas de ouro. Espectro SERS (a) e de absorção no UV-Vis (b) teórico para os diferentes isômeros do sistema dto-AuNP. (c) Espectro SERS experimental de uma suspensão de dto-AuNP. (d) Representação da reação de isomerização da dto na superfície de AuNP. Adaptado da referência 48
Estudos teóricos no sistema dto-AuNP mostram que cada modo de coordenação do ligante apresenta transições de transferência de carga metal-ligante de intensidades e energias diferentes (Figura 2b), sendo que a forma monodentada tiona-Au, {AuS(NH2)CCS(NH2)}, apresenta a maior probabilidade de transição e se encontra em ressonância com o comprimento de onda de excitação de 785 nm utilizado nos experimentos.49 Portanto, conforme a espécie cis bidentada é convertida na espécie trans monodentada a contribuição do mecanismo CM para o efeito SERS aumenta gradualmente, levando ao aumento do sinal SERS observado em função do tempo (Figura 2c).
EFEITO QUÍMICO NÃO RESSONANTE (CHEM) O efeito químico não ressonante vem sendo estudado teoricamente por métodos de primeiros princípios, através de cálculos de DFT e TD-DFT,19,20,50-52 porém as evidências experimentais ainda não levam a conclusão sobre seu efeito real. É um mecanismo estático que surge a partir da sobreposição das funções de onda dos estados fundamentais do ligante e da nanopartículas. Tais interações levam a uma relaxação dos estados eletrônicos do ligante devido à interação com a superfície metálica, aumentando a polarizabilidade do sistema. Dessa forma, esse efeito está correlacionado com as características do complexo formado na superfície, sendo sensível à geometria do ligante na superfície da partícula; a simetria do complexo ligante-partícula e o ambiente químico ao redor da molécula. De uma maneira simplificada, ligantes que apresentam maior força de interação com a superfície da partícula, maior deslocalização eletrônica e possibilidade de retrodoação, podem levar a uma contribuição elevada do mecanismo CHEM para o efeito SERS.47 Nesse mecanismo, o fator de intensificação pode ser estimado como sendo a razão entre a menor energia de excitação da molécula livre e a menor energia de transferência de carga no complexo formado na superfície da nanopartícula. Simplificadamente, para sistemas com alta sobreposição das funções de onda, o fator de intensificação será proporcional a razão entre a diferença de energia HOMO-LUMO do ligante livre e do complexo ligante-cluster. Na Equação 1 ΩX é a diferença de energia HOMO-LUMO na molécula isolada e ΩX-Au é a energia do primeiro estado excitado do complexo. Os valores calculados para diferentes sistemas variam na faixa de 5 a 1000, dependendo do funcional, do conjunto de bases e das aproximações para os potenciais da camada interna utilizados no cálculo DFT. Sendo que os valores mais aceitos estão na faixa de 5 a 100.47 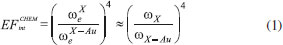 Schatz, Jensen e Zhao53 mostraram por TD-DFT que a coordenação de piridina em um adatom na superfície de um cluster metálico de Ag20 é mais forte que a coordenação em um face perfeita, nessa situação a polarizabilidade do complexo formado na superfície no eixo da ligação Ag-N é maior que a soma das polarizabilidades do cluster de Ag20 e da piridina isolados. Implicando uma modificação da seção de choque Raman devido a variações nas propriedades eletrônicas do complexo na superfície. Os mesmos autores expandiram seus estudos para dímeros de Ag20-piridina-Ag20, e observaram um aumento ainda maior da polarizabilidade no eixo de ligação.53 Lang e colaboradores realizaram uma análise detalhada das propriedades de ligação e do espalhamento Raman de ligantes fotosensíveis dimercaptoazobenzeno (dmab) adsorvidos em nanoclursters de ouro utilizando DFT. Os autores observaram que as conformações cis e trans do dmab apresentam efeito CHEM diferentes e conforme o tamanho do cluster de ouro era aumentado a ligação Au-S apresentava um caráter iônico maior.54,55 A maioria dos sistemas com nanopartículas metálicas explora a funcionalização seletiva com ligantes que apresentam sítios de coordenação com enxofre ou nitrogênio, o que permite uma ampla gama de aplicações. Apenas alguns ligantes alternativos que apresentam forças de interações comparáveis com superfícies metálicas têm sido reportados. Um exemplo são sistemas utilizando carbenos N-heterocíclicos em nanopartículas de ouro.56 Tais sistemas apresentam resistência a calor e reagentes químicos consideravelmente maior quando comparados com os sistemas análogos utilizando tióis. O aumento em estabilidade está relacionado a forte ligação Au-C, devido ao modo de coordenação distinto do ligante carbeno. Como consequência, uma vez ligado ao ouro os carbenos não são deslocados por tióis ou tioéteres, e são estáveis em altas temperaturas, em diferentes solventes orgânicos e valores extremos de pH.57 Em particular, carbenos derivados de benzoimadazóis proporcionam a formação de filmes altamente estáveis, exibindo certa organização a curtas distâncias. Estima-se que a ligação carbeno-Au seja 90 kJ mol-1 mais forte que a ligação Au-P e duas vezes mais forte que a ligação Au-S correspondentes. Por essa razão, a funcionalização com carbenos proporciona uma nova gama de ligantes que apresentem efeitos químicos ressonantes e não ressonantes para o efeito SERS maximizados. Um exemplo onde a formação da ligação Au-C leva a uma maximização da contribuição dos efeitos químicos para o sinal SERS foi observado em nanopartículas de ouro sintetizadas pelo método de Turkevich utilizando diferentes condições experimentais.23,58 Na presença de grande excesso de citrato (cit) a camada superficial de moléculas que garante estabilidade as nanopartículas é formada por grupos carboxílicos ligados eletrostaticamente aos átomos de ouro na superfície. Nessas condições, o sinal SERS associado as partículas esféricas de cit-AuNP geralmente é fraco devido à ausência de hot-spots e a influência negligenciável dos mecanismos químicos ressonante e não ressonante. A observação do sinal SERS da cit-AuNP é possível somente após a indução da agregação das partículas, através da quebra a estabilidade coloidal ocasionada através do aumento da força iônica (Figura 3a). A agregação facilita a formação dos hot-spots, levando ao aumento do sinal SERS devido à maximização do efeito eletromagnético (Figura 3b).
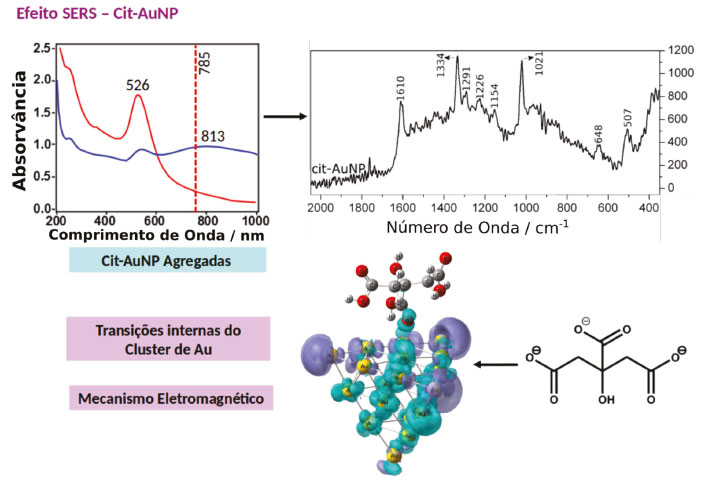 Figura 3. Efeito SERS em AuNP estabilizadas com citrato. (a) Espectro de extinção de uma suspensão de cit-AuNP antes (curva vermelha) e após a agregação (curva azul). (b) Espectro SERS de agregados de cit-AuNP. (c) Estado excitado teórico para um complexo Au20-citrato considerando excitação em 785 nm. Superfícies ciano representam a localização de elétrons e superfícies roxas a localização de buracos.Adaptado da referência 23
Foi demonstrado que através da diminuição da concentração de citrato durante a síntese das nanopartículas o produto de oxidação do citrato, a acetonadicarboxilato (ket), permanece fortemente ligado a superfície das nanopartículas. Surpreendentemente, o espectro SERS de uma suspensão de ket-AuNP, mesmo em condições onde não se observar nenhuma agregação, exibe um alto fator de intensificação (Figura 4a), contrastando com o comportamento observado para a suspensão de cit-AuNP.23 Adicionalmente, a dependência do perfil do espectro SERS com o comprimento de onda de excitação para a suspensão de ket-AuNP não pode ser explicada somente pelo mecanismo EM uma vez que o fator de intensificação é máximo quando a excitação é feita em 633 nm, estando fora da região de excitação dos plasmons da nanopartícula de ouro (Figura 4b). A contribuição do mecanismo RR também pode ser descartada, uma vez que a molécula de acetonadicarboxilato não apresenta transições eletrônicas na região do visível. Por essas razões o alto fator de intensificação do sinal SERS foi descrito a partir da contribuição do mecanismo CT e CHEM que ocorre na interface da ket-AuNP. A existência de um equilíbrio ceto-enólico (Figura 4c), para a acetonadicarboxilato favorece a interação do enol com a superfície da nanopartícula formando uma ligação Au-C estável e contribuindo para intensificação dos mecanismos químicos do efeito SERS.23 De acordo com cálculos teóricos para a estrutura Au20-enol, a ligação Au-C pode ser formada através do orbital molecular localizado na ligação C=C (HOMO). A alta polarizabilidade do complexo Au20-enol, em relação às espécies livres, facilita a ocorrência do mecanismo químico não-ressonante. A ocorrência do mecanismo CT foi discutida com base em cálculos de TD-DFT para o sistema Au20-enol, que mostram a existência de transições de transferência de carga em 652 nm, envolvendo o orbital HOMO (localizado na ligação C=C do enol) e o LUMO localizado no cluster de Au (Figura 4c). Portanto, o alto fator de intensificação observado para a suspensão de ket-AuNP, quando a excitação é feita em 633 nm, pode ser atribuído às contribuições simultâneas dos mecanismos EM, CT e CHEM.
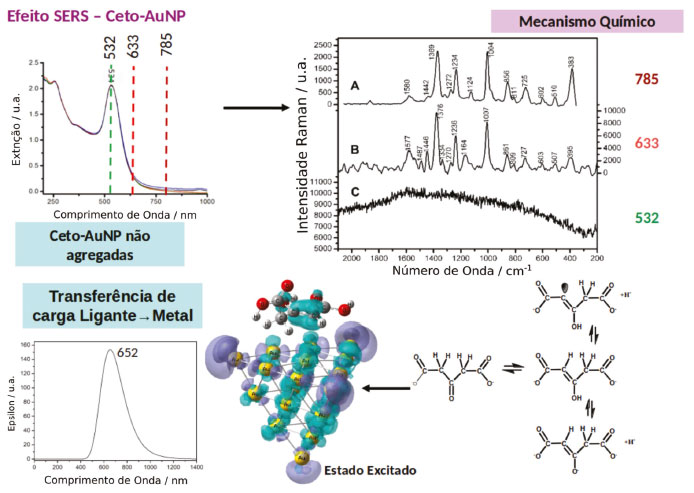 Figura 4. Efeito SERS em AuNP estabilizadas com acetonadicarboxilato. (a) Espectro de extinção de uma suspensão de ket-AuNP. (b) Espectro SERS de nanopartículas isoladas de ket-AuNP adquiridos com diferentes comprimentos de onda de excitação. (c) Equilíbrio ceto-enólico presente na acetona dicarboxilato e representação do estado excitado teórico para um complexo Au20-ket considerando excitação em 633 nm. Superfícies ciano representam a localização de elétrons e superfícies roxas a localização de buracos. Adaptado da referência 23
UNIFICANDO O EFEITO SERS Considerando o efeito Raman Ressonante, a Ressonância Plasmônica de Superfície e as transições de transferência de carga partícula-ligante, discutidas anteriormente, uma visão unificada introduzida por Lombardi e Birke,59 em extensão ao formalismo de Albrecht,28 para o efeito SERS pode ser equacionada. Nesse caso, o momento de transição de polarizabilidade é descrito pela Equação 2, onde os termos do denominador consideram a contribuição de cada mecanismo. O mecanismo EM é considerado no primeiro termo, que inclui as funções dielétricas da nanopartícula metálica [ε1(Ω) e ε2], do ambiente ao redor da partícula (εm), e atinge seu valor mínimo na ressonância plasmônica, maximizando o efeito. Esse termo surge da equação da seção de choque de extinção calculada por Mie, que racionaliza os processos de espalhamento e absorção de radiação para uma partícula esférica isolada, conforme discutido na primeira parte deste artigo. Além disso o campo elétrico oscilante de uma nanopartícula isolada é proporcional a r-3, onde r é a distância de um ponto em relação à superfície da partícula, estando essencialmente confinado na superfície. Portanto, pelo efeito EM, apenas os modos vibracionais das moléculas próximas à superfície serão intensificados.60,61 O segundo termo do denominador envolve as interações de transferência de carga entre os estados eletrônicos do metal, representado pelo nível Fermi da partícula (eF) e os estados eletrônicos excitados (er) da molécula ligada à superfície, e condição de ressonância ocorre quando a frequência da radiação incidente (Ω1) é igual a diferença de energia entre os estados eletrônicos acoplados do ligante e da partícula metálica 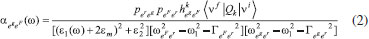 O numerador é o produto dos momentos de transição de dipolo da molécula adsorvida ( Os quatro termos devem ser diferentes de zero para ocorra a intensificação de um determinado modo normal através do efeito SERS. Essa equação apresenta uma utilidade enorme por explicar com certa precisão os resultados observados experimentalmente, porém, ainda existem pontos abertos para discussão. Por exemplo, para sistemas mais complexos envolvendo estruturas anisotrópicas ou condições de acoplamento interpartículas, uma expansão do termo referente ao efeito EM deveria ser considerada. Além disso, efeitos recentemente descobertos, como o efeito CHEM, ainda não apresentam uma forma unificada para que possam ser considerados na equação.
SENSORES Sensores plasmônicos Nanopartículas plasmônicas podem ser empregadas como plataformas colorimétricas de sensoriamento do ambiente químico ao redor utilizando basicamente duas estratégias. Na primeira a mudança do índice de refração do meio ou da camada ao redor da nanopartícula é utilizada como forma de quantificação das mudanças observadas.62 Como foi visto nas seções anteriores, a posição e intensidade da banda plasmônica depende da função dielétrica do meio no qual a nanopartícula se encontra. Liu e colaboradores mostram por microscopia de campo escuro que o deslocamento do espectro de espalhamento de AuNP pode ser usado para quantificar a quantidade de 1-propanol, 1-octanol ou óleo em água. Através da correlação do espectro de espalhamento e a composição RGB das imagens de campo escuro de AuNP de depositadas em sílica foi possível quantificar variações de 10 a 45% dos álcoois em água.63 Raschke e colaboradores mostraram que o espectro de espalhamento de nanopartículas de Au funcionalizadas com biotina é deslocado pela presença de streptavidina no meio, criando um sensor baseado no reconhecimento molecular de biomoléculas.64 Martín-Sánchez e colaboradores demostraram a utilização de nanobastões de ouro para detectar a mudança de índice de refração da água em altas pressões, até 9 GPa. Os autores acompanharam a variação de posição e largura da banda plasmônica longitudinal dos nanobastões em função da pressão e mostraram que existem descontinuidades no índice de refração devido a mudanças de fase de líquido → geloVI → geloVII.65 Este é um dos primeiros relatos de dependência das propriedades ópticas de nanopartículas plasmônicas com a pressão e uma das primeiras medidas do índice de refração da água em pressões elevadas. Mais exemplos de sensores que utilizam o deslocamento da banda de LSPR em função de mudanças no meio podem ser encontrados nos artigos de revisão.10,39,62,66 A segunda estratégia para utilização de nanopartículas plasmônicas como sensores colorimétricos é explorar as mudanças no espectro de extinção e consequentemente de cor devido ao acoplamento plasmônico durante o processo de agregação das nanopartículas.62 A coordenação de moléculas neutras67 ou a funcionalização das partículas com moléculas que reconheçam determinada espécie em solução que induzam a agregação das partículas pode ser explorada como métodos de sensoriamento.17 Liu e colaboradores mostraram que quando duas suspensões de nanopartículas funcionalizadas com oligonucleotídeos diferentes são misturadas na presença de metais de transição há a agregação das nanopartículas devido à coordenação dos metais em solução com ambas partículas. Apenas na presença de Hg2+ a agregação é irreversível com o aumento de temperatura, tornando o método seletivo para detecção de mercúrio.62,68 Lu et al. utilizaram nanopartículas de ouro funcionalizadas com sequências de DNA com diferentes aptâmeros para controlar a agregação das nanopartículas. Contudo quando o aptâmero interagia com a molécula alvo, cocaína nesse caso, a agregação das nanopartículas era quebrada e era possível observar a intensa coloração vermelha das nanopartículas isoladas.69 Métodos colorimétricos são simples e de fácil manipulação por pessoas com treinamento mínimo, facilitando sua implementação. A utilização das nanopartículas plasmônicas para aumentar a sensibilidade e seletividade do método os tornam extremamente atrativos principalmente para aplicação em testes clínicos. Ligantes multidentados como sondas SERS Uma das espécies mais simples capaz de ser utilizado como sonda SERS para detecção de metais é o ligante cianeto. Íons CN- são ligantes extremamente fortes para metais de transição, incluindo nanopartículas metálicas. Ele se liga através do átomo de C, deixando os átomos de N terminais livres para interagir com o solvente, H+ ou outros íons metálicos, gerando espécies em ponte NP-CN-Metal. Devido sua característica de ligante π aceptor, essas interações alteram a densidade eletrônica nos orbitais π antiligante do CN-, alterando a frequência de vibração do modo de estiramento da ligação C-N de 2074 cm-1, no KCN, para 2126 cm-1 quando se encontra ligado à superfície de nanopartículas metálicas.70 O espectro SERS de nanopartículas de ouro funcionalizadas com CN- é sensível à interação com compostos orgânicos voláteis e diferentes íons de metais de transição e foi explorado por Kim e colaboradores no desenvolvimento de sensores SERS para detecção de 2,6-dimetilfenilisocianeto, Mn2+, Fe2+, Co2+, Ni2+, Fe3+e Cr3+.70 A coordenação de ligantes multifuncionais na superfície de nanopartículas metálicas, como o ligante 2,4,6-trimercapto-1,3,5-triazina (tmt) foi explorado por Zamarion et al.71 como uma interface interativa para o monitoramento de íons metálicos através de suas reações de complexação (Figura5a). Na presença do tmt, devido aos seus múltiplos sítios de ligação, ligantes como citrato ou CTAB podem ser removidos da superfície de nanopartículas metálicas sem que ocorra agregação das partículas. Para maximizar a contribuição do efeito EM para o sinal SERS, a floculação das partículas foi induzida pela adição de eletrólitos, como NaCl, levando ao aparecimento da banda de acoplamento plasmônico na região de 730 nm. Na condição de floculação, o sistema se mantém ativo quimicamente e pode ser explorado para fins analíticos. Dessa forma, um sinal SERS intenso e reprodutível pode ser obtido de suspensões de tmt-AuNP, onde os picos característicos do tmt deslocados devido à interação com a superfície metálica podem ser observados (Figuras 5c, e).
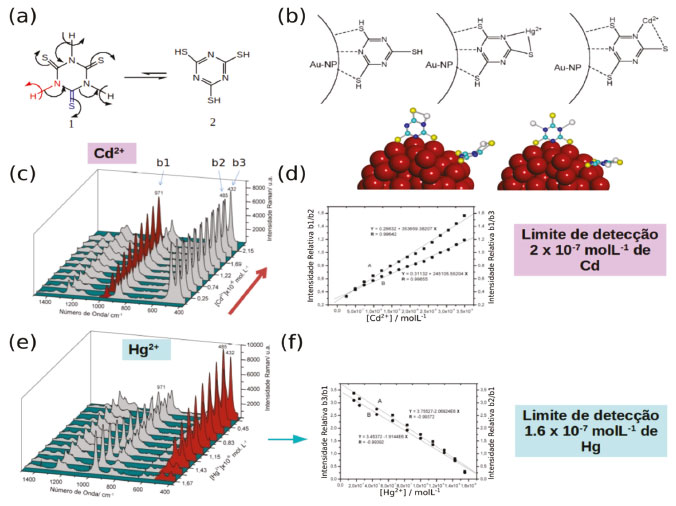 Figura 5. AuNP funcionalizadas com tmt para detecção de metais pesados. (a) Equilibrio tiol-tiona presenta no tmt. (b) Modos de coordenação do tmt na superfície de AuNP e sua interação com metais pesados. (c) Espectro SERS de tmt-AuNP na presença de Hg2+ em diferentes concentrações. (d) Curva de calibração para tmt-AuNP na presença de Hg2+mostrando o limite de detecção. (e) Espectro SERS de tmt-AuNP na presença de Cd2+ em diferentes concentrações. (f) Curva de calibração para tmt-AuNP na presença de Cd2+mostrando o limite de detecção. Adaptado da referência 71
No espectro SERS de tmt-AuNP os picos mais intensos são associados com os modos vibracionais exibindo maiores contribuições dos grupos C-S e S-H, em 1259, 867, 872, 485 e 432 cm-1, com exceção para os picos em 1218 e 971 cm-1 que são modos associados às vibrações do anel. O espectro pode ser explicado pela coordenação do tmt através de dois átomos de S e um N heterocíclico, conforme mostrado na Figura 5b. Nessa geometria o terceiro átomo de enxofre e os outros dois N heterocíclicos se encontram livres para interagir com outros íons metálicos, permitindo que partículas de tmt-AuNP sejam utilizadas como sondas SERS para Hg2+ e Cd2+ (Figura 5b). Observa-se um decréscimo sistemático dos picos νC-S em 485 e 432 cm-1 e do modo de respiro de anel em 973 cm-1 em função da concentração de Hg2+, apresentando um limite de detecção na faixa de ppb (Figura 5d). Essas alterações no espectro são consistentes com a coordenação do íon Hg de modo bidentado através de um átomo de S e um H heterocíclico do tmt (Figura 5b). No caso da interação entre tmt-AuNP e íons Cd2+ as mudanças no modo de estiramento de anel são mais acentuadas indicando uma coordenação mais efetiva com o N heterocíclico (Figura 5b). Outro exemplo é a interação de ditizona com AuNP. A ditizona (dtz) é um reagente complexante utilizado em testes analíticos colorimétricos para metais de transição. Sua forma tautomérica preferencial apresenta três sítios de coordenação em átomos de N e um sítio contendo uma tiona. Nas condições usuais dos testes colorimétricos, a ditizona é usada em uma concentração de 100 mmol L-1 e apresenta bandas de absorção em 620 e 440 nm que são sensíveis à coordenação com metais de transição. Em concentrações menores que 10-5 mol L-1 a solução se torna incolor e apresenta sinal Raman negligenciável. Porém, quando adsorvidas na superfície de nanopartículas de ouro é possível observar o espectro SERS da ditizona em concentrações de até 10-8 mol L-1 (Figura 6a, b e c).9 Os picos mais intensificados no espectro SERS são compostos principalmente pelos modos de estiramento C=S, N=N e C-N em 1588, 1339, 1305, 1221, 1164 e 998 cm-1,72,73 indicando uma coordenação na superfície do ouro pelos átomos de N e S (Figura 6c).
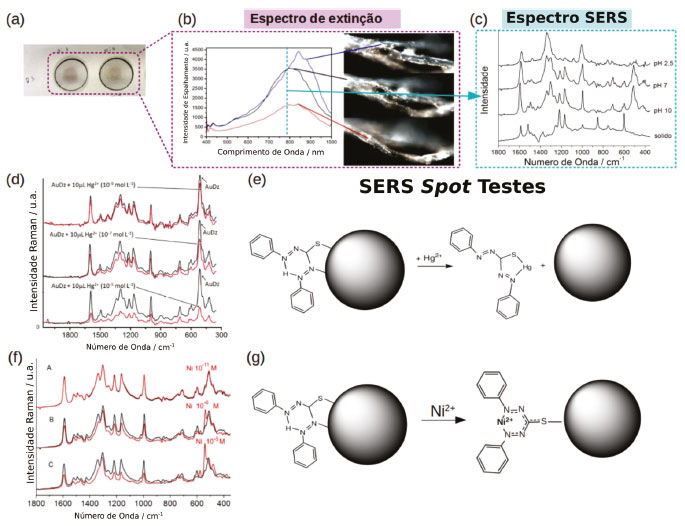 Figura 6. SERS spot testes com AuNP funcionalizadas com ditizona: (a) Imagem do papel filtro impregnado com dtz-AuNP. (b) Imagem de microscopia hiperespectral de campo escuro e espectro de espalhamento das fibras de celulose do papel filtro mostrando a presença de agregados de dtz-AuNP. (c) Espectro SERS do spot mostrado em (a) para diferentes valores de pH e comparação com o espectro Raman da dtz sólida. (d) SERS spot teste para detecção de Hg2+.(e) Esquema da reação de coordenação de ions Hg com a dtz. (f) SERS spot teste para detecção de Ni2+.(e) Esquema da reação de coordenação de ions Ni com dtz-AuNP. Adaptado da referência 9
As nanopartículas funcionalizadas com ditizona podem ser depositadas em um papel de filtro e utilizadas em condições reais de spot testes. Nessas condições a intensidade do sinal SERS é suficientemente alta para suprimir o sinal Raman da celulose (Figura 6c). O perfil do espectro SERS das AuNP impregnadas em papel é similar ao observado em suspensão, porém exibindo uma mudança significativa no modo relacionado a deformação de ângulo Ph-N, sugerindo que a ditizona adquire uma conformação planar na superfície das nanopartículas. Ao aplicar uma microgota de solução diluída de sais metálicos (< 10-4 mol L-1) mudanças significativas no espectro SERS da ditizona são observadas, conforme mostrado nas Figuras 6d e f. Um aspecto importante da utilização de celulose impregnada com nanopartículas plasmônicas como substratos SERS é a maior reprodutibilidade e a observação de mudanças sistemáticas no espectro em função da concentração de analito, diferentemente do comportamento observado para suspensão de nanopartículas. Outra vantagem desse tipo de substrato é o confinamento da solução de analito em uma região específica do substrato, fazendo com que a concentração local de analito aumente com a evaporação do solvente, em contraste com os efeitos de diluição observados quando os testes são realizados em solução. A diminuição gradativa do sinal SERS da ditizona em função da adição de íons Hg2+ observado na Figura 6d é consistente com o deslocamento da ditizona da superfície das nanopartículas de ouro e a formação do complexo ditizona-Hg em solução (Figura 6e). Essa observação explica a maior seletividade para íons mercúrio(II), uma vez que envolve uma competição direta pelos sítios de coordenação da ditizona, seguindo a tendência esperada pelas constantes de estabilidades. O limite de detecção dos spot tests SERS com ditizona para Hg2+ é de 2 x 10-9 mol L-1, correspondendo a 0.5 pg de mercúrio.9 Contudo, utilizando um metal menos mole, como Ni2+, a ditizona ancorada na superfície das nanopartículas de ouro é capaz de coordenar ao metal, através da liberação do átomo de H inter, e se manter coordenada à superfície metálica pelo átomo de S (Figura 6g). As mudanças espectrais observadas no modo de estiramento simétrico N=C-N, em 1341 cm-1, são indicativas da coordenação do Ni (Figura 6f). Adicionalmente, o surgimento de uma nova banda de estiramento C-S em 520 cm-1, pode ser interpretada como sendo decorrente da conversão gradual do modo de ligação bidentado da ditizona na superfície do ouro para uma coordenação monodentada mais simétrica através do átomo de enxofre. A utilização do efeito SERS em spot tests aumentou a sensibilidade para detecção de níquel em, pelo menos, quatro ordens de magnitude em relação ao teste colorimétrico convencional.74
EFEITO SERS DE COMPLEXOS DE METAIS DE TRANSIÇÃO A funcionalização de nanopartículas plasmônicas com complexos de metais de transição pode ser explorada de forma a construir estruturas supramoleculares automontadas que possuem propriedades distintas das espécies separadas. Nesse sentido, o centro metálico na superfície da nanopartícula pode atuar coordenando a ligação de diferentes ligantes, formando estruturas organizadas. Um exemplo dessa estratégia é mostrado na Figura 7a, nele AuNPs foram funcionalizadas com complexos de Ru com ligantes dicarboxybipiridina (dcbpy) e tmt ([Ru(dcbpy)2(tmt)]).75-77 Complexos de Ru são corantes inorgânicos comumente aplicados em células solares sensibilizadas por corantes (DSSC). Nessas células solares o corante ancorado na superfície de um eletrodo de TiO2 absorve energia da radiação solar e seus elétrons são transferidos para o eletrodo e em seguida para um circuito externo gerando uma corrente elétrica.78 Nesse caso, nanopartículas plasmônicas podem ser utilizadas para intensificar a absorção de radiação na região do visível aumentando a eficiência da célula solar.79-82 Portanto, complexos como o [Ru(dcbpy)2(tmt)] podem ser utilizados para se coordenar na superfície de eletrodos de TiO2 e a AuNP simultaneamente, conforme mostrado na Figura 7a.77 Esse sistema ilustra a ocorrência do efeito SERRS, mesmo em condições onde não se observa agregação, evidenciando o papel da ressonância de plasmon em promover a intensificação do mecanismo Raman Ressonante no complexo.76 Nesse caso o efeito de transferência de energia da partícula para o complexo de Ru através do acoplamento do dipolo elétrico da AuNP com o momento de dipolo de transição Ru→dcbpy em 528 nm (Figura 7f) é responsável pela intensificação do efeito Raman ressonante levando a um maior fator de intensificação do espectro SERS quando a excitação é feita em 532 nm (Figura 7e).
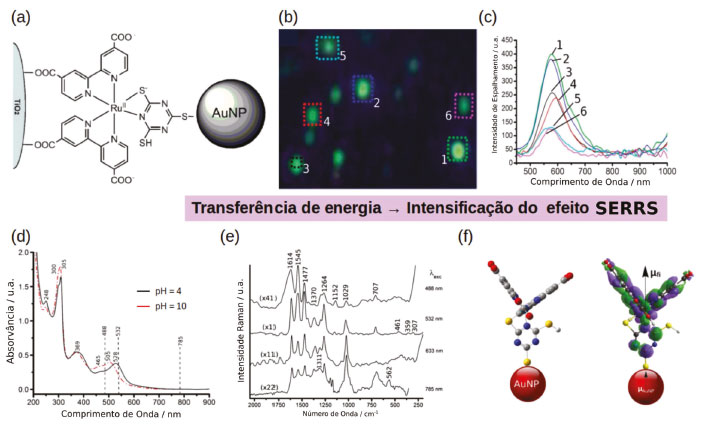 Figura 7. Nanopartículas funcionalizadas com complexos de Ru para aplicação em DSSC. (a) Representação da coordenação do complexo [Ru(dcbpy)2(tmt)] em nanopartículas de Au e TiO2. (b) Microscopia hiperespectral de campo escuro mostrando a presença de nanopartículas de ouro isoladas funcionalizadas com [Ru(dcbpy)2(tmt)]. (e) Espectro de espalhamento das partículas mostradas em (b). (d) Espectro de absorção no UV-Vis do complexo [Ru(dcbpy)2(tmt)] em diferentes valores de pH. (e) Espectro SERS de AuNP funcionalizadas com [Ru(dcbpy)2(tmt)] utilizando diferentes comprimentos de onda de excitação. (f) Representação do acoplamento entre o dipolo elétrico da AuNP e do diplo de transição para transição eletrônica no complexo [Ru(dcbpy)2(tmt)]. Adaptado das referências 76 e 77
Resultados similares foram observados por Theil et al. para uma série de complexos tiocianato-Ru-carboxibipiridina comercialmente disponíveis (N719, N749 e Z907) na superfície de nanobastões de ouro e nanoestrelas de Au/Pt/Au.83,84 Vohlídal et al. tentam explicar o efeito da interação entre nanopartículas de prata e complexos de ferro(II) terpiridina nos níveis eletrônicos do complexo e, como essa perturbação altera a espalhamento Raman do sistema, em relação ao complexo de ferro isolado.85 Através do perfil de excitação do espectro Raman do complexo isolado e na superfície das nanopartículas de prata, os autores mostram que não ocorre perturbação dos níveis eletrônicos do complexo, contudo, nesse caso, não há a formação de uma ligação covalente entre a terpiridina e a prata. Apesar de se tratar de apenas uma adsorção física, esse artigo mostra uma abordagem interessante para o estudo dos efeitos da interação na interface partícula-molécula. Paine e colaboradores mostraram por XPS que a interação do ligante TPASH@C8, Figura 8, ocorre através da formação de ligações covalente entre as espécies Au0 e S-1. Os autores mostram um aumento da atividade catalítica dos complexos de ferro(II) formados na superfície das partículas funcionalizadas com TPASH de cerca de 8 a 10 vezes para reações de decarboxilação do benzilformiato,86 além disso, observaram um aumento no número de ciclos catalíticos. Os autores justificam essas mudanças apenas em função do aumento da área superficial e da auto-organização dos complexos na superfície do ouro, o que facilitaria o acesso do substrato ao sítio ativo do complexo, deixando de lado os efeitos da interação eletrônica entre as nanopartículas e o complexo.
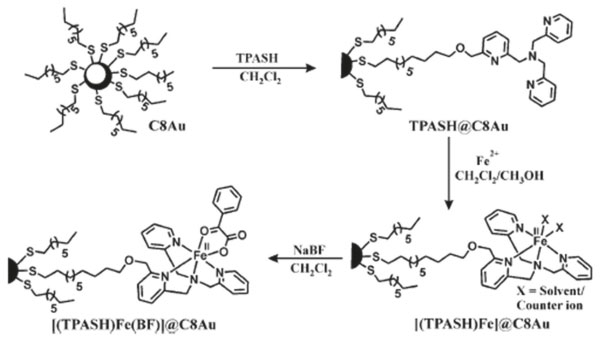 Figura 8. Funcionalização de AUNP com ligante TPASH e complexos de ferro
Outro efeito interessante observado a partir da interação entre nanopartículas de ouro e complexos superficiais é a alteração das propriedades magnéticas de complexos de disprósio utilizando tiocianato como ligantes pontes. Habib et al. comprovaram por medidas de magnetização que não há mudanças significativas das propriedades magnéticas do complexo livre e na superfície das AuNPs, quando aplicado um campo contínuo. Porém, as propriedades magnéticas dinâmicas, quando aplicado um campo alternado, mudam drasticamente.87 Contudo, a explicação é somente devido a mudanças na anisotropia local do complexo, e não foram explorados os efeitos da influência das nanopartículas de ouro nas propriedades do complexo. Li et al. mostraram uma alteração no perfil de fluorescência de porfirinas, base livre e metaladas, coordenadas à superfície de nanobastões de ouro, porém, não chegaram em nenhuma conclusão sobre a causa da alteração.88 Scaiano et al. discutem os efeitos da interação entre nanoestruturas metálicas e ligantes com base em três mecanismos: o aumento de geração de estados excitados, transferências de pares elétrons buracos e o aquecimento local, causado pela interação da nanoestrutura com a luz. Os dois primeiros mecanismos estão diretamente correlacionados com o grau de sobreposição dos estados eletrônicos da nanoestrutura e do ligante, ou seja, com o grau de covalência da interação entre os mesmos.6,89 Os exemplos dados evidenciam a exploração de diferentes propriedades de moléculas e complexos na superfície de nanopartículas, tais como propriedades espectroscópicas, fotoquímicas, magnéticas e catalíticas, no entendimento da natureza e do efeito das interações na interface partícula-molécula. Contudo, as conclusões ainda não são concretas e algumas de natureza especulativa. Isso mostra a necessidade de um entendimento mais fundamental a respeito da natureza das interações na superfície das nanopartículas, revisitando conceitos clássicos de química de coordenação, espectroscopia, química analítica, fotoquímica e fotofísica, e aplicando-os no estudo da influência que um ligante causa na superfície das partículas e vice-versa.
NANOCOMPÓSITOS DE NANOPARTÍCULAS PLASMÔNICAS COM MATERIAIS BIDIMENSIONAIS Além das nanopartículas, uma nova classe de nanomateriais vem surgindo e despertando grande interesse. São materiais cristalinos constituídos por uma única camada atômica de um elemento ou de compostos, sendo denominados bidimensionais (materiais 2D). A partir da obtenção do grafeno em 2004 por exfoliação mecânica de um cristal de grafite, reportado por Geim e colaboradores,90 diferentes classes de materiais 2D vêm sendo descobertas, dentre as quais destacam-se os mono e dicalcogenetos de metais de transição (DCMT)91,92 e os semicondutores monoelementares como fosforeno,93,94 siliceno,95 germaneno e estaneno.96 Assim como as nanopartículas, os materiais 2D apresentam propriedades diferenciadas dos seus respectivos materiais bulk, devido ao alto grau de confinamento quântico dos elétrons e a maior área superficial.96,97 Podem apresentar propriedades características de materiais isolantes, metálicos, semicondutores e até supercondutores dependendo da composição, do número de camadas, da aplicação de campos elétricos e tensões. Em geral, possuem altas mobilidades de carregadores de cargas. Por exemplo, no fosforeno a mobilidade de elétrons e buracos pode chegar a 5000 cm2 V-1 s-1,96 sendo 4 ordens de grandeza maior que a de nanopartículas de TiO2, comumente utilizado em células solares e fotocatalisadores. Juntamente às propriedades diferenciadas a possibilidade de controlar finamente a estrutura eletrônica e as propriedades ópticas a partir do empilhamento das camadas do elemento ou de elementos diferentes, criando heteroestruturas,96 faz com que essa classe de materiais seja ideal para aplicações em óptica,98 fotônica, eletrônica99,100 armazenamento e produção de energia,101,102 levando a miniaturização de dispositivos à escala atômica. Nos materiais 2D a química de superfície também tem grande impacto e abre um amplo campo de estudos com muito potencial a ser explorado. A manipulação das propriedades eletrônicas, como a magnitude do bandgap, a posição do nível de Fermi 95,103,104 e das propriedades magnéticas,105-107 podem ser alcançadas por meio do controle das interfaces entre os materiais e o meio. Dessa forma, a deposição de nanopartículas plasmônicas na superfície de materiais 2D pode levar a novos materiais compósitos para aplicações como substratos SERS, fotocatalisadores e sensores. Do ponto de vista espectroscópico, materiais 2D também apresentam efeitos de intensificação do espalhamento Raman em sua superfície, porém diferentemente de nanopartículas plasmônicas os mecanismos de transferência de carga são a principal origem da intensificação do sinal Raman observado, e tem sido observado em substratos a base de grafeno108-110 e DCMT111-114 para detecção de diferentes moléculas como cristal violeta, ftalocianinas, rodamina 6G, rodamina B e protoporfirina IX. Materiais compósitos de nanopartículas plasmônicas e materiais 2D são de especial interesse como substratos SERS, uma vez que proporcionam maiores fatores de intensificação devido à somatória das contribuições do mecanismo EM, CM e CHEM das nanopartículas metálicas com o mecanismo CM nos materiais 2D.108,109,111 Além disso, do ponto de vista de química de superfície, tais materiais possibilitam a exploração de novos meios de funcionalização ampliando a gama da analitos que podem ser estudados. Por exemplo, Hu e colaboradores, reportaram a utilização de um compósito de óxido de grafeno (GO)/nanobastões de ouro (AuNR) para detecção de corantes aromáticos, demonstrando a intensificação seletiva de certas moléculas sondas devido a interações preferenciais com a superfície do GO.115 Adicionalmente, tem sido reportado que o GO auxilia na remoção de ligantes e surfactantes, incluindo o CTAB (brometo de cetiltrimetilamônio) da superfície de nanopartículas de ouro116 e prata.117,118 Vianna e colaboradores introduziram a utilização de compósitos de GO/AuNR como substrato SERS capaz de suprimir o efeito de blinking do sinal observado.119 A estabilidade temporal do substrato SERS foi estuda através do coeficiente de variação do espectro integrado, demonstrando que a deposição de AuNR estabilizados com CTAB na superfície de GO diminui o coeficiente de variação do espectro SERS em 5 vezes, quando comparado às nanopartículas puras. Esse efeito foi atribuído à remoção do CTAB dos hot-spots plasmônicos devido a interação do grupamento polar do surfactante com os grupamentos oxigenados com cargas negativas na superfície do GO. O nanocompósito apresentou sensibilidade e estabilidade 4 vezes maiores para detecção de rodamina 640 em relação à nanopartícula pura. Essa alta estabilidade temporal possibilitou a aplicação desse tipo de nanocompósito em substratos opto-fluídicos para detecções em tempo real com uma taxa de aquisição de 20 espectros por segundo e uma sensibilidade de até 75 femtogramas de analito (Figura 9).120 Tais substratos são baseados em fibras ópticas de microcapilar de vidro, em que a parede interna da fibra é modificada com o nanocompósito e o analito é passado em fluxo pelo interior da fibra junto ao fluxo de luz (Figura 9).120
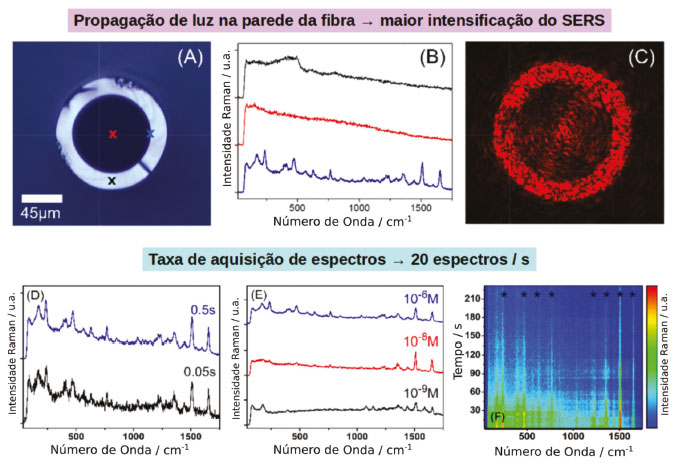 Figura 9. Nanocompósitos de GO@AuNR como substratos SERS. (a) Imagem do microscópio óptico da seção transversal da fibra capilar. Os pontos marcados referem-se a uma posição de aquisição de espectro relacionada à mesma curva de cor em (b). (b) O gráfico vermelho foi adquirido com uma potência de laser de 105,2 µW na interface sílica/capilar e as curvas em preto e azul com potência de laser de 4,82 mW foram adquiridas incidência de luz diretamente na parede de sílica e no buraco capilar, respectivamente. (c) Característica de guia de fibra para uma luz de 633 nm lançada exatamente na interface sílica / furo capilar com 23,7 µW. (d) Espectros SERS de RH640 em concentrações de 10-6 M e tempos de integração de 0,5 s (azul) e 0,05 s (preto). Potência do laser de 105 µW. (e) Espectros SERS de RH640 em concentrações de 10-6 M (azul), 10-8 M (vermelho) e 10-9 M (preto). Potências de laser de 105 µW (azul) e 1,85 mW (vermelho / preto). Tempo de integração de 0,5 s. (f) Séries temporais de espectros SERS para RH640. Potência do laser de 105 µW e tempo de integração de 0,5 s. Adaptado da referência 120
CATÁLISE PLASMÔNICA A energia dos plásmons excitados pode ser aproveitada de diferentes maneiras, devido aos processos de decaimentos radiativos e não-radiativos. O processo de amortecimento radiativo está relacionando ao decaimento dos elétrons em fótons, sendo dominante em partículas maiores que 50 nm.121,122 Já os não-radiativos estão relacionados aos processos de espalhamento elétron-fônon, elétron-elétron, elétron-defeitos, elétron-superfície e a criação de pares elétron-buraco, devido à excitação de transições inter e intrabandas. E são mais eficientes em partículas menores que 30 nm.123 Assim, há basicamente três formas de utilizar a energia absorvida através da excitação dos plasmons de superfície: através da geração de estados excitados, transferência de pares elétron-buraco ou aquecimento localizado na superfície, exemplificadas na Figura 10.
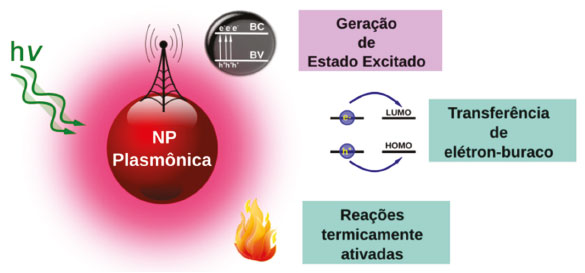 Figura 10. Possíveis tipos de catálise utilizando nanopartículas plasmônicas
A geração de estados excitados ocorre devido às interações do campo eletromagnético intensificado na superfície da partícula com os reagentes. Nesse mecanismo o decaimento radiativo dos elétrons levando a emissão de fótons é o principal responsável pela transferência de energia entre a nanopartícula e os substratos. Esse decaimento gera um efeito antena que amplifica a intensidade da radiação incidente na vizinhança da superfície, a partir da qual é gerado um número maior de estados excitados. Na Figura 10 exemplifica-se uma situação na qual a interação entre NPs e um semicondutor, que apresenta um bandgap na mesma região de frequência de absorção da nanopartícula plasmônica, o que leva a uma intensificação da geração de estados excitados na região onde o campo da partícula metálica é intensificado. A Figura 11 apresenta resultados de simulações computacionais baseadas no método DDA ("discrete dipole approximation") para a intensificação do campo elétrico local (E) em relação ao campo elétrico da radiação incidente (E0). As simulações foram feitas para sistemas híbridos de nanopartículas de Ag e de TiO2 considerando radiação incidente com comprimento de onda em 434 nm. A excitação plasmônica ocorre próxima à borda de absorção do TiO2 e leva à amplificação do campo elétrico no interior do semicondutor (Figura 11A). A utilização de uma camada de SiO2 (4 nm) em torno das nanopartículas de Ag (Figura 11B) acarreta uma menor concentração de campo no semicondutor. As diferenças de intensificação são refletidas na performance para a degradação de fenol sobre estes materiais, sendo esta mais eficiente no sistema de maior amplificação de campo.124
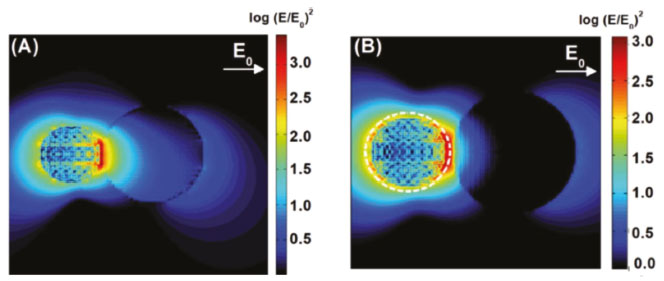 Figura 11. Mapas de intensificação de campo elétrico em sistemas híbridos de nanopartículas de Ag (30 nm de diâmetro) e de TiO2 (50 nm de diâmetro): (A) Ag + TiO2, com separação de 1 nm entre as superfícies; (B) sistema caroço@casca de Ag@SiO2, com espessura de casca de 4 nm. As simulações foram realizadas com comprimento de onda em 434 nm. Reproduzido da Ref. 124 com permissão da Royal Society of Chemistry
Esse processo pode ser empregado em células solares sensibilizadas por corantes, por exemplo. Uma vez que os corantes utilizados e as partículas metálicas absorvem na mesma região do espectro, o efeito antena das estruturas plasmônicas pode ser utilizado para geração de um maior número de moléculas de corante no estado excitado.15,81,125 A geração de estados excitados está relacionada à distância da molécula à superfície da nanopartícula. A intensidade da fluorescência de uma molécula pode ser utilizada como parâmetro para avaliar o efeito de intensificação na geração de estados excitados de um sistema, quanto maior esse efeito, mais intensa será a fluorescência. Scaiano e colaboradores verificaram a intensidade de fluorescência de uma molécula em um substrato fixo em função da distância com relação a uma ponta de AFM modificada com AuNP de 80 nm.6 Existem três regiões distintas que mostram a influência das nanopartículas na geração de estados excitados, conforme pode ser visto na Figura 12. Na primeira região, onde o fluoróforo se encontra a distâncias menores que 2 nm da superfície da nanopartícula, há uma diminuição da fluorescência com a diminuição da distância entre a AuNP e a molécula, devido à trocas de elétrons. Tais transferências também dependem da diferença de energia entre os estados da nanopartícula e da molécula e do grau de interação dos orbitais das moléculas com a superfície da partícula, conforme discutido anteriormente.
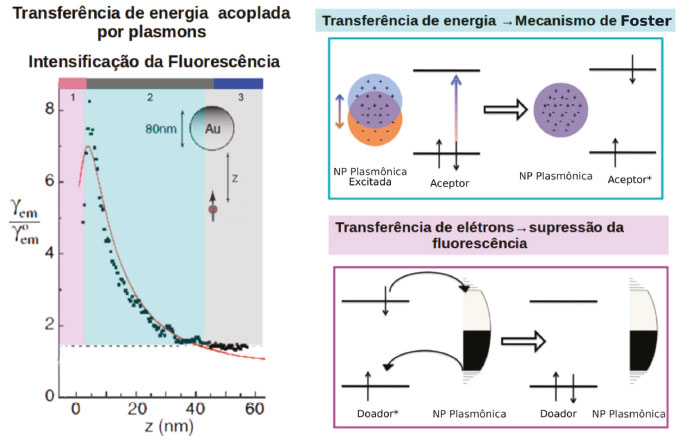 Figura 12. Tranferência de energia acoplada por plasmons. (a) Fator de intensificação da intensidade de fluorescência em função da distância do fluoróforo para a superfície de uma AuNP de 80nm. (b) Mecanismo de transferência de energia de Förster. (c) Mecanismo de supressão da fluorescência através da transferência de elétrons. Adaptado da referência 125
A segunda região, onde a distância entre molécula e a superfície da nanopartícula varia de 3 a 30 nm, a molécula é excitada pelo campo elétrico dos plasmons e a transferência de energia pode ser explicada pelo mecanismo de Förster (Figura 12).125 Segundo esse mecanismo, a molécula pode ser excitada por qualquer campo elétrico oscilante de frequência apropriada e em ressonância com as transições eletrônicas moleculares. Portanto, para que ocorra transferência de energia através do mecanismo de Förster deve haver uma sobreposição da banda de absorção da molécula ou do semicondutor e a banda plasmônica da nanoestrutura. Já na terceira região (distâncias > 30 nm) a AuNP não exerce influência na fluorescência da molécula, uma vez que nessas distâncias o campo elétrico da nanopartícula tende a zero (Figura 12). Um exemplo de reação catalisada por transferência de energia foi dado por Stamplecoskie et al., onde nanopartículas de prata foram utilizadas para intensificar a geração de fotodecomposição do 2,20-azo-bis-isobutironitrila (AIBN) gerando radicais iniciadores da polimerização do monômero triacrilato (trimetilolpropano triacrilato, TMPTA). Nesse caso, a fraca interação do AIBN com a superfície da prata e a sobreposição da banda de absorção do composto com a banda plasmônica das AgNP favorecem a transferência de energia pelo mecanismo de Förster, intensificando a geração de radicais e consequentemente aumentando o rendimento da polimerização.126 No segundo mecanismo, a excitação de transições eletrônicas inter e intrabandas levam à geração de pares elétron-buraco na nanoestrutura. Estes podem ser utilizados diretamente para reações de oxirredução com o substrato, através da transferência do elétron ou do buraco para os orbitais das moléculas.6,127 Outra possibilidade é a transferência do elétron excitado para banda de condução de uma molécula ou de um semicondutor. Nesse caso a reação de redução pode ocorrer no semicondutor e a reação de oxidação na superfície metálica. A partícula plasmônica atua como um sensibilizador, diminuindo o bandgap do semicondutor, que por sua vez atua como um separador de cargas (Figura 13). Nesse mecanismo é essencial que a sobreposição das funções de onda da nanoestrutura e da molécula ou do semicondutor, seja suficientemente grande para que a velocidade de transferência de elétrons seja maior que a velocidade de recombinação do par elétron-buraco, diminuindo assim a resistência na interface.7 Essa estratégia pode ser aplicada em sistemas fotocatalíticos para geração de H2128,129 e fotodegradação de poluentes, por exemplo.
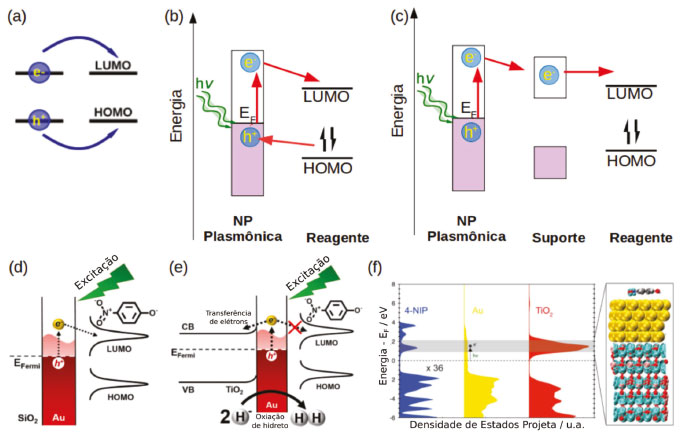 Figura 13. Catálise plasmônica por transferências de elétrons (a). (b) Mecanismo de transferência de elétrons e buracos diretamente entre a nanopartícula e o reagente. (c) Mecanismo de transferência de elétrons entre a nanopartícula plasmônica e um semicondutor de suporte e o reagente. (d) Exemplo de transferência de elétrons entre AuNP e p-nitrofenol. (e) Exemplo de transferências se elétrons em AuNP suportadas em nanopartículas de TiO2. (f) Densidades de estados calculadas por DFT para o sistema contendo AuNP, TiO2 e p-nitrofenol. Adaptado da referência 130
Ferrer et al. mostraram que nanopartículas de ouro funcionalizadas com complexos de ferro(II) com derivados de terpiridina (tpy), apresentam uma separação de cargas de tempo de vida longo sob excitação, e a natureza das espécies transientes depende do comprimento de onda de excitação. O autor propõe que com a excitação em 308 nm há a geração de espécies [Fe(tpyR)2]+3 e (AuNP)-1, devido à transferência de um elétron do complexo de ferro para a nanopartículas de ouro, provavelmente mediada pela formação da ligação entre a terpiridina modificada e a superfície do ouro.130 Adicionalmente, os autores mostram que essas cargas podem ser utilizadas para geração fotocatalítica de H2. Utilizando a reação de hidrogenação do p-nitrofenol com borohidreto de sódio catalisada por nanopartículas de ouro como prova de conceito Barbosa et al. mostraram que a catálise plasmônica apresenta diferentes caminhos reacionais quando as partículas estão isoladas ou suportadas em um semicondutor.131 Para partículas isoladas, após a excitação dos plasmons os elétrons excitados podem ser transferidos diretamente para o orbital desocupado do p-nitrofenol catalisando a reação de hidrogenação com o H adsorvido na superfície da AuNP, proveniente do borohidreto. Já para partículas suportadas em TiO2, devido à alta densidade de estados na banda de condução do TiO2 (Figura 13) a transferência de elétrons para o semicondutor é facilitada e observa-se a geração de H2 e a diminuição do rendimento da reação de hidrogenação do p-nitrofenol. Esse exemplo mostra de forma clara a importância de se conhecer a energia relativa dos níveis de energia da nanopartícula plasmônica, do semicondutor usado como suporte e dos reagentes no entendimento do mecanismo de reação da catálise plasmônica. As nanopartículas também podem ser suportadas em materiais bidimensionais, como o grafeno e seus derivados, nesse caso as propriedades catalíticas podem ser aprimoradas, devido à alta área superficial, estabilidades química e térmica e a influência dos bandgaps ajustáveis de materiais como o grafeno.132,133 O bandgap dos derivados químicos de grafeno pode ser modulado pelo seu grau de oxidação ou dependendo a qual metal ou óxido metálico está ligado,134 fornecendo melhores semicondutores para fotocatálise135 e dispositivos de armazenamento de energia, por exemplo.136 Nos materiais híbridos de óxido de grafeno e AuNP (GO@AuNP), a folha do óxido de grafeno pode atuar como receptor de elétron ou buraco, aumentando o número e o tempo de vida das cargas excitadas.133,137 Esse tipo de material híbrido apresenta maior atividade catalítica do que as AuNPs livres na redução da o-nitroanilina a 1,2-benzenodiamina, além de atuar como melhor substrato SERS para estas moléculas.138 Wang et al. relataram a atividade catalítica de nanopartícula de ouro suportadas em oxido de grafeno reduzido (RGO@AuNP) na oxidação do p-aminotiofenol em p,p'-dimercaptoazobenzeno, envolvendo a geração de espécies reativas de oxigênio.139 Pereira e colaboradores mostraram que o híbrido de RGO@AuNP pode ser utilizado para a decomposição de compostos policíclicos aromáticos como o 9-antraldeído levando à formação de ftalatos através da geração intensificada de espécies reativas de oxigênio.140 Utilizando o mesmo reagente (9-antraldeído) Scaiano et al. mostraram que a reação catalisada por nanopartículas de ouro suportadas em micropartículas de diamante leva à formação de uma dicetona como produto principal,127 evidenciando novamente que o mecanismo de reação é dependente do tipo de material utilizado como substrato. No caso das nanopartículas suportadas em óxido de grafeno reduzido o elétron excitado na AuNP é transferido para banda de condução do RGO, onde reage com o oxigênio do ar ou as funções oxigenadas do RGO gerando espécies reativas de oxigênio que atacam o 9-antraldeído.140 Já para as partículas suportadas em diamante, a transferência de elétrons ocorre diretamente entre o antraldeído e as AuNP gerando um radical que reagirá com oxigênio molecular dando origem a um peróxido. Catalisado pela geração de calor na superfície da partícula o peróxido se decompõe na dicetona, nesse caso o suporte de diamante atua como condutor de calor facilitando a decomposição do peróxido.127 O terceiro mecanismo de transferência de energia é baseado no decaimento não-radiativo elétron-fônon e o posterior decaimento térmico dos fônons excitados, levando à geração de um intenso fluxo de calor localizado em intervalos de tempo curtos, Figura 10. Dessa forma, pode-se ter o controle espacial e temporal da taxa de aquecimento em função da irradiação da nanoestrutura. Esse fluxo de calor pode ser utilizado para a realização de reações orgânicas convencionais com o controle mais preciso da taxa de aquecimento. Assim, reações secundárias podem ser minimizadas, aumentando o rendimento dos processos. Essa abordagem vem sendo utilizada para reações de decomposição de poluentes orgânicos,141 reações de retro Diels-Alder142 e reforma a vapor de etanol, gerando H2.143 A excitação dos plasmons de nanoestruturas apresenta uma série de vantagens, como a conversão mais eficiente de energia solar em energia química. Devido às altas estabilidades fotoquímica e térmica e as altas seções de choque de extinção das nanopartículas, que são em média cinco ordens de grandeza maiores que as seções de choque dos corantes utilizados usualmente.7 Além da fácil manipulação das propriedades químicas e físicas dos nanomateriais através da alteração da composição, tamanho, forma e funcionalização.144 Contudo, há certos contrapontos. Os mecanismos de transferências de elétrons e energia entre materiais plasmônicos e substratos não são completamente entendidos, dificultando a criação de sistemas específicos e que apresentem maior rendimento. Há uma carência de técnicas para determinar a temperatura da superfície, por exemplo, e de estudos cinéticos dos processos em superfície. Portanto, é um campo com uma ampla gama de estudos e possibilidades.
CONCLUSÕES O estudo sistemático das propriedades de nanopartículas plasmônicas como um todo, desde a síntese até a funcionalização das partículas, mostra que todos parâmetros experimentais de síntese estão correlacionados e suas variações levam às propriedades ópticas, químicas e físico-químicas diferentes. Dessa forma, um entendimento aprofundado das interações na superfície dos nanomateriais é essencial para aplicá-las de maneira mais eficiente. De uma maneira geral o maior grau de covalência da ligação metal-ligante na superfície da nanoestrutura aumenta a deslocalização eletrônica e, consequentemente, a polarizabilidade, contribuindo para um maior fator de intensificação do mecanismo químico fora de ressonância para o efeito SERS. Adicionalmente, transições de transferência de carga também são favorecidas, aumentando o fator de intensificação do mecanismo CT para o efeito SERS e a atividade fotocatalítica. As diferenças de labilidade de interações metal-ligante na superfície das NPs fazem com que a cinética de troca de ligante na superfície das partículas seja diferente, levando a um controle da geometria de coordenação e consequentemente à diferentes propriedades químicas e atividades. Esperamos que esse artigo tenha chamado a atenção para como as propriedades químicas de materiais plasmônicos podem ser controladas de forma que sejam gerados materiais com propriedades específicas para cada aplicação. Mostramos também como técnicas espectroscópicas e a interdisciplinaridade entre química e física se torna importante para o estudo mais aprofundado dos materiais plasmônicos.
AGRADECIMENTOS Agradecemos o apoio financeiro da FAPESP (2016/21070-5), CNPq (408985/2016-0), FAPERJ (26/211.281/2019) e Petrobras (2017/00367-2). Este trabalho foi financiado em parte pela CAPES-código 001.
REFERÊNCIAS 1. Aioub, M.; Austin, L. A.; El-Sayed, M. A.; J. Phys. Chem. Lett. 2014, 5, 3514. 2. Panikkanvalappil, S. R.; Hira, S. M.; Mahmoud, M. A.; El-Sayed, M. A.; J. Am. Chem. Soc. 2014, 136, 15961. 3. Au, L.; Zheng, D. S.; Zhou, F.; Li, Z. Y.; Li, X. D.; Xia, Y. N.; ACS Nano 2008, 2, 1645. 4. Xia, L.; Hu, X.; Sun, M.; Li, J.; Yang, D.; Wang, X.; Xu, H.; J. Raman Spectrosc. 2012, 43, 1024. 5. Gonzalez-Bejar, M.; Peters, K.; Hallett-Tapley, G. L.; Grenier, M.; Scaiano, J. C.; Chem. Commun. 2013, 49, 1732. 6. Scaiano, J. C.; Stamplecoskie, K.; J. Phys. Chem. Lett. 2013, 4, 1177. 7. Xiao, M.; Jiang, R.; Wang, F.; Fang, C.; Wang, J.; Yu, J. C.; J. Mater. Chem. A 2013, 1, 5790. 8. Liu, M.; Wang, Z.; Zong, S.; Chen, H.; Zhu, D.; Wu, L.; Hu, G.; Cui, Y.; ACS Appl. Mater. Interfaces 2014, 6, 7371. 9. Grasseschi, D.; Zamarion, V. M.; Araki, K.; Toma, H. E.; Anal. Chem. 2010, 82, 9146. 10. Stewart, M. E.; Anderton, C. R.; Thompson, L. B.; Maria, J.; Gray, S. K.; Rogers, J. A.; Nuzzo, R. G.; Chem. Rev. 2008, 108, 494. 11. Noginov, M. A.; Zhu, G.; Belgrave, A. M.; Bakker, R.; Shalaev, V. M.; Narimanov, E. E.; Stout, S.; Herz, E.; Suteewong, T.; Wiesner, U.; Nature 2009, 460, 1110. 12. Topics in Applied Physics; Lee, C.-C., ed.; Springer Netherlands: Dordrecht, 2015; Vol. 129. 13. Su, Y.; Zhou, G.; Li, F.; Wang, T.; Front. Optoelectron. Chin. 2011, 4, 264. 14. Deepa, K. G.; Lekha, P.; Sindhu, S.; Sol. Energy 2012, 86, 326. 15. Qi, J.; Dang, X.; Hammond, P. T.; Belcher, A. M.; ACS Nano 2011, 5, 7108. 16. Hagglund, C.; Zach, M.; Kasemo, B.; Hägglund, C.; Zäch, M.; Kasemo, B.; Appl. Phys. Lett. 2008, 92, 013113. 17. Toma, H. E.; Zamarion, V. M.; Toma, S. H.; Araki, K.; J. Braz. Chem. Soc. 2010, 21, 1158. 18. Grasseschi, D.; Toma, H. E.; Coord. Chem. Rev. 2017, 333, 108. 19. Morton, S. M.; Silverstein, D. W.; Jensen, L.; Chem. Rev. 2011, 111, 3962. 20. Luo, J.; Wang, P.; Fang, Y.; Vib. Spectrosc. 2014, 70, 162. 21. Zhao, X.; Chen, M.; J. Raman Spectrosc. 2014, 45, 62. 22. Beloglazkina, E. K.; Majouga, A. G.; Romashkina, R. B.; Zyk, N. V; Zefirov, N. S.; Russ. Chem. Rev. 2012, 81, 65. 23. Grasseschi, D.; Ando, R. A.; Toma, H. E.; Zamarion, V. M.; RSC Adv. 2015, 5, 5716. 24. Jensen, L.; Aikens, C. M.; Schatz, G. C.; Chem. Soc. Rev. 2008, 37, 1061. 25. Sanchez, L. A.; Birke, R. L.; Lombardi, J. R.; J. Phys. Chem. 1984, 88, 1762. 26. Lombardi, J. R.; Birke, R. L.; Sanchez, L. A.; Bernard, I.; Sun, S. C.; Chem. Phys. Lett. 1984, 104, 240. 27. Lombardi, J. R.; Birke, R. L.; Lu, T.; Xu, J.; J. Chem. Phys. 1986, 84, 4174. 28. Albrecht, A. C.; J. Chem. Phys. 1961, 34, 1476. 29. Long, D. A.; The Raman Effect: A Unified Treatment of the Theory of Raman Scattering by Molecules; John Wiley & Sons Ltd: Bradford, UK, 2002. 30. Kiyonaga, T.; Fujii, M.; Akita, T.; Kobayashi, H.; Tada, H.; Phys. Chem. Chem. Phys. 2008, 10, 6553. 31. Adams, D. M.; Brus, L.; Chidsey, C. E. D.; Creager, S.; Creutz, C.; Kagan, C. R.; Kamat, P. V.; Lieberman, M.; Lindsay, S.; Marcus, R. A.; Metzger, R. M.; Michel-Beyerle, M. E.; Miller, J. R.; Newton, M. D.; Rolison, D. R.; Sankey, O.; Schanze, K. S.; Yardley, J.; Zhu, X.; J. Phys. Chem. B 2003, 107, 6668. 32. Hosten, C. M.; Birke, R. L.; Lombardi, J. R.; J. Phys. Chem. 1992, 96, 6585. 33. Shi, C.; Zhang, W.; Birke, R. L.; Lombardi, J. R.; J. Phys. Chem. 1990, 94, 4766. 34. Shi, C.; Zhang, W.; Birke, R. L.; Lombardi, J. R.; J. Phys. Chem. 1990, 94, 4766. 35. Zhao; Jensen, L.; Schatz, G. C.; J. Am. Chem. Soc. 2006, 128, 2911. 36. Aikens, C. M.; Schatz, G. C.; J. Phys. Chem. A 2006, 110, 13317. 37. Sun, M.; Liu, S.; Li, Z.; Duan, J.; Chen, M.; Xu, H.; J. Raman Spectrosc. 2009, 40, 1172. 38. Rao, C. N. R.; Rao, C. N. R. In The Chemistry of Nanomaterials; Rao, C. N. R., Müller, A., Cheetham, A. K., eds.; 1st ed., Wiley: Bielefeld, Germany, 2004. 39. Daniel, M.-C.; Astruc, D.; Chem. Rev. 2004, 104, 293. 40. Pérez-Juste, J.; Pastoriza-Santos, I.; Liz-Marzán, L.; Mulvaney, P.; Coord. Chem. Rev. 2005, 249, 1870. 41. Nalbant Esenturk, E.; Hight Walker, A. R.; J. Raman Spectrosc. 2009, 40, 86. 42. Azcárate, J. C.; Corthey, G.; Pensa, E.; Vericat, C.; Fonticelli, M. H.; Salvarezza, R. C.; Carro, P.; J. Phys. Chem. Lett. 2013, 4, 3127. 43. Leite, E. R.; Ribeiro, C.; Crystallization and Growth of Colloidal Nanocrystals; SpringerBriefs in Materials; Springer New York: New York, NY, 2012. 44. Pearson, R. G.; J. Am. Chem. Soc. 1963, 85, 3533. 45. Pearson, R. G.; Proc. Natl. Acad. Sci. U. S. A. 1986, 83, 8440. 46. Jensen, L.; Zhao, L. L.; Schatz, G. C.; J. Phys. Chem. C 2007, 111, 4756. 47. Moore, J. E.; Morton, S. M.; Jensen, L.; J. Phys. Chem. Lett. 2012, 3, 2470. 48. Grasseschi, D.; Zamarion, V. M.; Toma, H. E.; J. Raman Spectrosc. 2018, 49, 1478. 49. Grasseschi, D.; Tese de Doutorado, Universidade de São Paulo, Brasil, 2015. 50. Liu, S.; Zhao, X.; Li, Y.; Chen, M.; Sun, M.; Spectrochim. Acta. A. Mol. Biomol. Spectrosc. 2009, 73, 382. 51. Pan, W.-X.; Lai, Y.-C.; Wang, R.-X.; Zhang, D.-J.; Zhan, J.-H.; J. Raman Spectrosc. 2014, 45, 54. 52. Mullin, J. M.; Autschbach, J.; Schatz, G. C.; Comput. Theor. Chem. 2012, 987, 32. 53. Zhao, L. L.; Jensen, L.; Schatz, G. C.; Nano Lett. 2006, 6, 1229. 54. Yao, J.; Yang, M.; Duan, Y.; Chem. Rev. 2014, 114, 6130. 55. Mirkin, C. A.; Letsinger, R. L.; Elghanian, R.; Mucic, R. C.; Storhoff, J. J.; Abstr. Pap. Am. Chem. Soc. 1997, 214, 17. 56. Crudden, C. M.; Horton, J. H.; Ebralidze, I. I.; Zenkina, O. V; McLean, A. B.; Drevniok, B.; She, Z.; Kraatz, H.-B.; Mosey, N. J.; Seki, T.; Keske, E. C.; Leake, J. D.; Rousina-Webb, A.; Wu, G.; Nat. Chem. 2014, 6, 409. 57. Rocha, J. C.; Mattioni, J. V.; Sihn, L. M.; Shinohara, J. S.; Melo, F. M.; Franco, M. P.; Braga, A. A. C.; Toma, H. E.; J. Raman Spectrosc. 2020, jrs. 5872. 58. Grasseschi, D.; de O. Pereira, M. L. M. L. M. L.; Shinohara, J. S. J. S. J. S.; Toma, H. E. H. E.; J. Nanopart. Res. 2018, 20, 35. 59. Lombardi, J. R.; Birke, R. L.; Acc. Chem. Res. 2009, 42, 734. 60. Maier, S. A.; Plasmonics: Fundamentals and Applications, Springer: New York, 2007. 61. Aroca, R.; Llorente, S. R.; J. Mol. Struct. 1997, 408, 17. 62. Li, M.; Cushing, S. K.; Wu, N.; Analyst 2015, 140, 386. 63. Liu, Y.; Ling, J.; Huang, C. Z.; Chem. Commun. 2011, 47, 8121. 64. Raschke, G.; Kowarik, S.; Franzl, T.; So, C.; Nano Lett. 2003, 3, 935. 65. Martín-Sánchez, C.; González-Rubio, G.; Mulvaney, P.; Guerrero-Martínez, A.; Liz-Marzán, L. M.; Rodríguez, F.; J. Phys. Chem. Lett. 2019, 10, 1587. 66. Zhao, P.; Li, N.; Astruc, D.; Coord. Chem. Rev. 2013, 257, 638. 67. Toma, S. H.; Bonacin, J. a.; Araki, K.; Toma, H. E.; Eur. J. Inorg. Chem. 2007, 2007, 3356. 68. Xu, X.; Rosi, N. L.; Wang, Y.; Huo, F.; Mirkin, C. A.; J. Am. Chem. Soc. 2006, 128, 9286. 69. Liu, J.; Mazumdar, D.; Lu, Y.; Angew. Chem., Int. Ed. 2006, 45, 7955. 70. Kim, K.; Lee, J. W.; Shin, K. S.; Analyst 2013, 138, 2988. 71. Zamarion, V. M.; Timm, R. A.; Araki, K.; Toma, H. E.; Inorg. Chem. 2008, 47, 2934. 72. Pemberton, J. E.; Buck, R. P.; J. Phys. Chem. 1981, 85, 248. 73. Pemberton, J. E.; Buck, R. P.; J. Am. Chem. Soc. 1982, 104, 4076. 74. Toma, H. E.; da Silva Shinohara, J.; Grasseschi, D. In Nanoscale Imaging, Sensing, and Actuation for Biomedical Applications XII; Cartwright, A. N., Nicolau, D. V., eds.; 2015; Vol. Proc. SPIE, p. 933702. 75. Grasseschi, D.; Parussulo, A. L. A.; Zamarion, V. M.; Guimarães, R. R.; Araki, K.; Toma, H. E.; RSC Adv. 2013, 3, 24465. 76. Grasseschi, D.; Parussulo, A. L. A.; Zamarion, V. M.; Guimarães, R. R.; Araki, K.; Toma, H. E.; J. Raman Spectrosc. 2014, 45, 758. 77. Grasseschi, D.; Parussulo, A. L. A.; Zamarion, V. M.; Guimaraes, R. R.; Araki, K.; Toma, H. E.; Int. J. Nanotechnol. 2015, 12, 263. 78. Ruile, S.; Kohle, O.; Péchy, P.; Grätzel, M.; Inorg. Chim. Acta 1997, 261, 129. 79. Jang, Y. H.; Jang, Y. J.; Kochuveedu, S. T.; Byun, M.; Lin, Z.; Kim, D. H.; Nanoscale 2014, 6, 1823. 80. Brown, M. D.; Suteewong, T.; Kumar, R. S. S.; D'Innocenzo, V.; Petrozza, A.; Lee, M. M.; Wiesner, U.; Snaith, H. J.; Nano Lett. 2011, 11, 438. 81. Standridge, S. D.; Schatz, G. C.; Hupp, J. T.; J. Am. Chem. Soc. 2009, 131, 8407. 82. Gangishetty, M. K.; Lee, K. E.; Scott, R. W. J.; Kelly, T. L.; ACS Appl. Mater. Interfaces 2013, 5, 11044. 83. Theil, F.; Zedler, L.; März, A.; Xie, W.; Csáki, A.; Fritzsche, W.; Cialla, D.; Schmitt, M.; Popp, J.; Dietzek, B.; J. Phys. Chem. C 2013, 117, 1121. 84. Zedler, L.; Theil, F.; Csáki, A.; Fritzsche, W.; Rau, S.; Schmitt, M.; Popp, J.; Dietzek, B.; RSC Adv. 2012, 2, 4463. 85. Šloufová, I.; Vlčková, B.; Procházka, M.; Svoboda, J.; Vohlídal, J.; J. Raman Spectrosc. 2014, 45, 338. 86. Sheet, D.; Halder, P.; Paine, T. K.; Angew. Chem., Int. Ed. 2013, 52, 13314. 87. Holmberg, R. J.; Hutchings, A.-J.; Habib, F.; Korobkov, I.; Scaiano, J. C.; Murugesu, M.; Inorg. Chem. 2013, 52, 14411. 88. Xue, C.; Birel, O.; Li, Y.; Ma, X.; Gao, M.; Urbas, A.; Li, Q.; J. Colloid Interface Sci. 2013, 398, 1. 89. Scaiano, J. C.; Netto-Ferreira, J. C.; Alarcon, E.; Billone, P.; Alejo, C. J. B.; Crites, C.-O. L.; Decan, M.; Fasciani, C.; González-Béjar, M.; Hallett-Tapley, G.; Grenier, M.; McGilvray, K. L.; Pacioni, N. L.; Pardoe, A.; René-Boisneuf, L.; Schwartz-Narbonne, R.; Silvero, M. J.; Stamplecoskie, K. G.; Wee, T.-L.; Pure Appl. Chem. 2011, 83, 913. 90. Novoselov, K. S.; Science (80-. ). 2004, 306, 666. 91. Chhowalla, M.; Shin, H. S.; Eda, G.; Li, L.-J.; Loh, K. P.; Zhang, H.; Nat. Chem. 2013, 5, 263. 92. Lv, R.; Robinson, J. A.; Schaak, R. E.; Sun, D.; Sun, Y.; Mallouk, T. E.; Terrones, M.; Acc. Chem. Res. 2015, 48, 56. 93. Liang, L.; Wang, J.; Lin, W.; Sumpter, B. G.; Meunier, V.; Pan, M.; Nano Lett. 2014, 14, 6400. 94. Castellanos-Gomez, A.; Vicarelli, L.; Prada, E.; Island, J. O.; Narasimha-Acharya, K. L.; Blanter, S. I.; Groenendijk, D. J.; Buscema, M.; Steele, G. A.; Alvarez, J. V.; Zandbergen, H. W.; Palacios, J. J.; Van Der Zant, H. S. J.; 2D Mater. 2014, 1, 025001. 95. Kaloni, T. P.; Schreckenbach, G.; Freund, M. S.; J. Phys. Chem. C 2014, 118, 23361. 96. Bhimanapati, G. R.; Lin, Z.; Meunier, V.; Jung, Y.; Cha, J.; Das, S.; Xiao, D.; Son, Y.; Strano, M. S.; Cooper, V. R.; Liang, L.; Louie, S. G.; Ringe, E.; Zhou, W.; Kim, S. S.; Naik, R. R.; Sumpter, B. G.; Terrones, H.; Xia, F.; Wang, Y.; Zhu, J.; Akinwande, D.; Alem, N.; Schuller, J. A.; Schaak, R. E.; Terrones, M.; Robinson, J. A.; ACS Nano 2015, 9, 11509. 97. Butler, S. Z.; Hollen, S. M.; Cao, L.; Cui, Y.; Gupta, J. A.; Gutiérrez, H. R.; Heinz, T. F.; Hong, S. S.; Huang, J.; Ismach, A. F.; Johnston-Halperin, E.; Kuno, M.; Plashnitsa, V. V.; Robinson, R. D.; Ruoff, R. S.; Salahuddin, S.; Shan, J.; Shi, L.; Spencer, M. G.; Terrones, M.; Windl, W.; Goldberger, J. E.; ACS Nano 2013, 7, 2898. 98. Falkovsky, L. a.; J. Phys. Conf. Ser. 2008, 129, 012004. 99. Castro Neto, A. H.; Guinea, F.; Peres, N. M. R.; Novoselov, K. S.; Geim, A. K.; Rev. Mod. Phys. 2009, 81, 109. 100. Li, L.; Yu, Y.; Ye, G. J.; Ge, Q.; Ou, X.; Wu, H.; Feng, D.; Chen, X. H.; Zhang, Y.; Nat. Nanotechnol. 2014, 9, 372. 101. Dai, J.; Zeng, X. C.; J. Phys. Chem. Lett. 2014, 5, 1289. 102. Lee, S.-K.; Rana, K.; Ahn, J.-H.; J. Phys. Chem. Lett. 2013, 4, 831. 103. Johll, H.; Kang, H. C.; Tok, E. S.; Phys. Rev. B 2009, 79, 245416. 104. Jing, Y.; Tang, Q.; He, P.; Zhou, Z.; Shen, P.; Nanotechnology 2015, 26, 095201. 105. Cao, C.; Wu, M.; Jiang, J.; Cheng, H.-P.; Phys. Rev. B 2010, 81, 205424. 106. Guo, B.; Fang, L.; Zhang, B.; Gong, J. R.; Insciences J. 2011, 1, 80. 107. Ding, Y.; Wang, Y.; J. Phys. Chem. C 2015, 119, 10610. 108. Lv, R.; Li, Q.; Botello-Méndez, A. R.; Hayashi, T.; Wang, B.; Berkdemir, A.; Hao, Q.; Elías, A. L.; Cruz-Silva, R.; Gutiérrez, H. R.; Kim, Y. A.; Muramatsu, H.; Zhu, J.; Endo, M.; Terrones, H.; Charlier, J.-C.; Pan, M.; Terrones, M.; Sci. Rep. 2012, 2, 586. 109. Ling, X.; Xie, L.; Fang, Y.; Xu, H.; Zhang, H.; Kong, J.; Dresselhaus, M. S.; Zhang, J.; Liu, Z.; Nano Lett. 2010, 10, 553. 110. Yu, X.; Cai, H.; Zhang, W.; Li, X.; Pan, N.; Luo, Y.; Wang, X.; Hou, J. G.; ACS Nano 2011, 5, 952. 111. Muehlethaler, C.; Considine, C. R.; Menon, V.; Lin, W.-C.; Lee, Y.-H.; Lombardi, J. R.; ACS Photonics 2016, 3, 1164. 112. Zhang, D.; Wu, Y.-C.; Yang, M.; Liu, X.; Coileáin, C. Ó.; Abid, M.; Abid, M.; Wang, J.-J.; Shvets, I.; Xu, H.; Chun, B. S.; Liu, H.; Wu, H.-C.; Sci. Rep. 2016, 6, 30320. 113. Ghopry, S. A.; Alamri, M. A.; Goul, R.; Sakidja, R.; Wu, J. Z.; Adv. Opt. Mater. 2019, 7, 1801249. 114. Sheng, Y.; Jiang, S.; Yang, C.; Liu, M.; Liu, A.; Zhang, C.; Li, Z.; Huo, Y.; Wang, M.; Man, B.; Chem. Phys. Lett. 2017, 682, 64. 115. Hu, C.; Rong, J.; Cui, J.; Yang, Y.; Yang, L.; Wang, Y.; Liu, Y.; Carbon N. Y. 2013, 51, 255. 116. Pan, H.; Low, S.; Weerasuriya, N.; Shon, Y.-S.; ACS Appl. Mater. Interfaces 2015, 7, 3406. 117. Wang, P.; He, H.; Jin, Y.; Small 2012, 8, 3438. 118. Hou, H.; Wang, P.; Zhang, J.; Li, C.; Jin, Y.; ACS Appl. Mater. Interfaces 2015, 7, 18038. 119. Vianna, P. G.; Grasseschi, D.; Costa, G. K. B.; Carvalho, I. C. S.; Domingues, S. H.; Fontana, J.; de Matos, C. J. S.; ACS Photonics 2016, 3, 1027. 120. Vianna, P. G.; Grasseschi, D.; Domingues, S. H.; de Matos, C. J. S.; Opt. Express 2018, 26, 22698. 121. Lermé, J.; J. Phys. Chem. C 2011, 115, 14098. 122. Lermé, J.; Baida, H.; Bonnet, C.; Broyer, M.; Cottancin, E.; Crut, A.; Maioli, P.; Del Fatti, N.; Vallée, F.; Pellarin, M.; J. Phys. Chem. Lett. 2010, 1, 2922. 123. Linic, S.; Christopher, P.; Ingram, D. B.; Nat Mater 2011, 10, 911. 124. Lemos de Souza, M.; Pereira dos Santos, D.; Corio, P.; RSC Adv. 2018, 8, 28753. 125. Hsu, L.-Y.; Ding, W.; Schatz, G. C.; J. Phys. Chem. Lett. 2017, 8, 2357. 126. Stamplecoskie, K. G.; Pacioni, N. L.; Larson, D.; Scaiano, J. C.; J. Am. Chem. Soc. 2011, 133, 9160. 127. Wee, T.-L. (Erika); Schmidt, L. C.; Scaiano, J. C.; J. Phys. Chem. C 2012, 116, 24373. 128. Liu, Z.; Hou, W.; Pavaskar, P.; Aykol, M.; Cronin, S. B.; Nano Lett. 2011, 11, 1111. 129. Lee, J.; Mubeen, S.; Ji, X.; Stucky, G. D.; Moskovits, M.; Nano Lett. 2012, 12, 5014. 130. Alvaro, M.; Aprile, C.; Ferrer, B.; Sastre, F.; Garcia, H.; Dalton Trans. 2009, 0, 7437. 131. Barbosa, E. C. M.; Fiorio, J. L.; Mou, T.; Wang, B.; Rossi, L. M.; Camargo, P. H. C.; Chem. - Eur. J. 2018, 24, 12330. 132. Yin, P. T.; Shah, S.; Chhowalla, M.; Lee, K.-B.; Chem. Rev. 2015, 115, 2483. 133. Kamat, P. V.; J. Phys. Chem. Lett. 2010, 1, 520. 134. Li, X.; Yu, J.; Wageh, S.; Al-Ghamdi, A. A.; Xie, J.; Small 2016, 12, 6640. 135. Upadhyay, R. K.; Soin, N.; Roy, S. S.; RSC Adv. 2014, 4, 3823. 136. Wu, Z.-S.; Zhou, G.; Yin, L.-C.; Ren, W.; Li, F.; Cheng, H.-M.; Nano Energy 2012, 1, 107. 137. Du, B.; Lin, L.; Liu, W.; Zu, S.; Yu, Y.; Li, Z.; Kang, Y.; Peng, H.; Zhu, X.; Fang, Z.; Laser Photon. Rev. 2017, 11, 1600148. 138. Huang, J.; Zhang, L.; Chen, B.; Ji, N.; Chen, F.; Zhang, Y.; Zhang, Z.; Nanoscale 2010, 2, 2733. 139. Wang, J.; Trindade, F. J.; de Aquino, C. B.; Pieretti, J. C.; Domingues, S. H.; Ando, R. A.; Camargo, P. H. C.; Chem. - Eur. J. 2015, 21, 9889. 140. Pereira, M. L. de O.; Grasseschi, D.; Toma, H. E.; Energy Fuels 2018, 32, 2673. 141. Fasciani, C.; Alejo, C. J. B.; Grenier, M.; Netto-Ferreira, J. C.; Scaiano, J. C.; Org. Lett. 2010, 13, 204. 142. Bakhtiari, A. B.; Hsiao, D.; Jin, G.; Gates, B. D.; Branda, N. R.; Angew. Chem., Int. Ed. Engl. 2009, 48, 4166. 143. Adleman, J. R.; Boyd, D. A.; Goodwin, D. G.; Psaltis, D.; Nano Lett. 2009, 9, 4417. 144. Rodrigues, T. S.; da Silva, A. G. M.; de Moura, A. B. L.; Freitas, I. G.; Camargo, P. H. C.; RSC Adv. 2016, 6, 62286. |
 . O terceiro termo considera o efeito RR na molécula adsorvida e sua contribuição é máxima quando o laser de excitação está em ressonância com uma transição eletrônica totalmente permitida, envolvendo o estado fundamental (eg) e o estado excitado (er) da molécula livre. Os termos Γ estão relacionados com os processos de amortecimento das vibrações moleculares, contribuindo diretamente para a forma da banda observada. Por se tratar de um produto de funções, os três termos devem ser diferentes de zero para que o efeito de intensificação seja observado, e devido a magnitude do efeito eletromagnético, este acaba intensificando os demais.
. O terceiro termo considera o efeito RR na molécula adsorvida e sua contribuição é máxima quando o laser de excitação está em ressonância com uma transição eletrônica totalmente permitida, envolvendo o estado fundamental (eg) e o estado excitado (er) da molécula livre. Os termos Γ estão relacionados com os processos de amortecimento das vibrações moleculares, contribuindo diretamente para a forma da banda observada. Por se tratar de um produto de funções, os três termos devem ser diferentes de zero para que o efeito de intensificação seja observado, e devido a magnitude do efeito eletromagnético, este acaba intensificando os demais.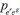 ), da transferência de carga nanopartícula-molécula (
), da transferência de carga nanopartícula-molécula (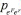 ), o termo de Herzberg-Teller,
), o termo de Herzberg-Teller, 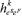 , e a integral do modo normal de vibração,
, e a integral do modo normal de vibração, 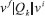 , definindo as regras de seleção para os modos vibracionais intensificados quando o ligante se encontra na superfície da nanopartícula metálica.
, definindo as regras de seleção para os modos vibracionais intensificados quando o ligante se encontra na superfície da nanopartícula metálica.On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access