
 |
 |
 |
 |
 |
Revisão
|
|
| Funcionalização de ligações C-H em estágio tardio em síntese orgânica Late-Stage Functionalization OF C-H Bonds In Organic Synthesis |
|
Eduardo C. S. Rocha; Yasmin N. Salmazo; Marcio Hayashi; César A. D. Zaragoza; Emilio C. de Lucca Júnior*
Instituto de Química, Universidade Estadual de Campinas, 13083-970 Campinas - SP, Brasil Recebido em 30/05/2022 *e-mail: eluccajr@unicamp.br The development of new strategies for the functionalization of the historically inert C-H bond arises as an excellent way to create new carbon-carbon and carbon- heteroatom bonds. With increasingly chemo- and site-selective methods that enables the late-stage functionalization of C-H bonds, the modification of specific sites in natural products and pharmaceuticals without altering their scaffold emerges as a powerful means for the diversification of complex molecules. In this review, we will introduce concepts of late-stage modifications, the use of C-H bond functionalization to forge new carbon-carbon and carbon-heteroatom bonds using metal catalysis and photochemistry in simple examples and their applications in natural products and pharmaceuticals. The aim of the review is to update and display to the reader the contributions and implications of this methodology to organic synthesis. INTRODUÇÃO Produtos naturais e seus derivados têm sido utilizados no tratamento de doenças e, mais recentemente, no design de novos fármacos, além de servirem como inspiração para o desenvolvimento de novas metodologias sintéticas. Em muitos casos, as propriedades biológicas do produto natural isolado não são boas o suficiente para que os mesmos sejam utilizados como medicamento e a sua diversificação estrutural se torna uma opção.1 Uma estratégia capaz de rapidamente expandir bibliotecas de derivados de produtos naturais é a funcionalização de estágio tardio (do inglês, late-stage functionalization). Tobias Ritter define essa estratégia como: "A transformação quimiosseletiva desejada em uma molécula complexa para fornecer ao menos um análogo em quantidade suficiente e pureza para um dado objetivo sem a necessidade de instalar um grupo funcional que sirva exclusivamente para uma dita transformação".2,3 Para que a terminologia "funcionalização em estágio tardio" possa ser utilizada, a molécula complexa a ser funcionalizada pode ser tanto um produto natural como também um intermediário avançado em uma síntese, na qual desafios de quimiosseletividade sejam impostos. No geral a funcionalização em estágio tardio ocorre em locais específicos em uma molécula, em que condições reacionais distintas possam fornecer produtos funcionalizados em diferentes sítios. Por exemplo, o exemplo geral mostra uma molécula complexa que, dependendo da reação aplicada, pode funcionalizar na posição mais à esquerda (quadrado), região mais à direita (triângulo) ou na região central (hexágono), sem que ocorra a alteração do esqueleto (Esquema 1).
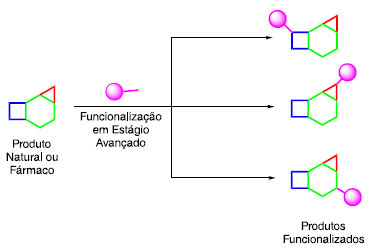 Esquema 1. Esquema geral de funcionalização em estágio avançado
A seletividade de uma reação química pode ser classificada em: quimiosseletividade, regiosseletividade e estereosseletividade.4 A quimiosseletividade, segundo a IUPAC, é "uma reação preferencial de um reagente químico com um de dois ou mais diferentes grupos funcionais" (Esquema 2a).4,5 Para a quimiosseletividade, a partir do cinamato de metila (1), o tratamento com DIBAL-H em CH2Cl2, seguido do tratamento com o sal de Rochelle forneceu o álcool 2, em que a redução ocorreu no grupo éster, enquanto a hidrogenação com Pd/C em EtOAc a temperatura ambiente forneceu o éster 3, sendo a reação seletiva para a olefina.
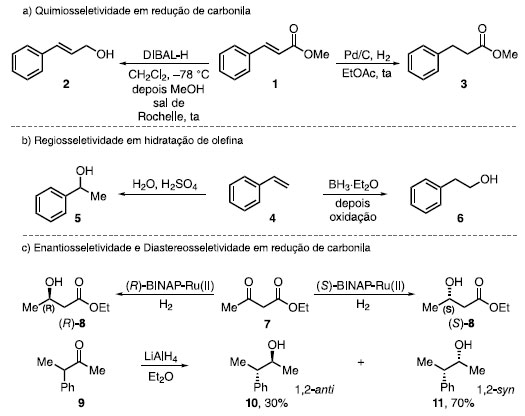 Esquema 2. Exemplos de: (a) Quimiosseletividade; (b) Regiosseletividade; (c) Enantiosseletividade e Diastereosseletividade
A regiosseletividade é "a que uma direção da formação da ligação ou a quebra ocorre preferencialmente sobre todas as outras direções possíveis" (Esquema 2b).4 A partir do estireno (4), o tratamento com H2O e H2SO4 forneceu o álcool terciário 5, com a adição da água via Markovnikov, enquanto o uso de BH3*Et2O seguido de oxidação forneceu o álcool primário 6, com a formação do produto via anti-Markovnikov. A estereosseletividade é "a formação preferencial de um este-reoisômero sobre outro em uma reação química", e a diastereosse-letividade é a formação preferencial de um diastereoisômero frente a outro (Esquema 2c).4 O tratamento de acetoacetato de etila (7) em condições de hidrogenação assimétrica catalisada por complexo de rutênio e ligante (Ã)-BINAP produziu o álcool (Ã)-8, enquanto o uso do enantiômero (S)-BINAP forneceu o correspondente (S)-8. A redução da cetona 9, como uma mistura racêmica, por LiAlH4, levou à formação de uma mistura diastereomérica 10 e 11, com uma relação 1,2-anti e 1,2-syn, respectivamente. As reações de funcionalização C-H se encaixam na mesma descrição que a "quimiosseletividade". No entanto, a IUPAC ainda não considera a ligação C-H como um grupo funcional, pelo fato dessa ligação ser tradicionalmente inerte frente aos outros grupos funcionais. A funcionalização dela se aproxima da definição de quimiosseletividade, pois um reagente consegue diferenciar uma ligação C-H frente a outras ligações C-H. Da mesma forma é relacionado com a regiosseletividade, pois funcionaliza uma ligação C-H em uma posição frente a outras posições.6,7,8 Nos últimos anos, há o aumento do uso do termo "sitiosseleti-vidade" para explicar a seletividade em reações de funcionalização da ligação C-H, apesar desse termo não estar definido na IUPAC. A sitiosseletividade, segundo Guangbin Dong, é "a capacidade de diferenciar a reatividade de um mesmo tipo de um grupo funcional presentes em ambientes diferentes na molécula."9 Conseguir a sitiosseletividade de uma reação é uma tarefa formidável, pois como são os mesmos tipos de grupos funcionais, a tendência é que possuam reatividades próximas, fornecendo o mesmo produto em uma condição reacional. Dessa forma, a diferença de energia de ativação entre as reações contendo tais grupos é pequena.9 Uma reação categorizada como sitiosseletiva é a funcionalização de ligações C-H, como, por exemplo, a oxidação C-H da artemisina (12), realizada por White e colaboradores, utilizando catalisadores de ferro non-heme (S,S)-Fe(PDP) e (5,,S)-Fe(CF3-PDP) para oxidar ligações C-H diferentes, dependendo do catalisador utilizado. Ao utilizar o catalisador (S,S)-Fe(PDP), a oxigenação ocorre na ligação C-H terciária, sendo mais seletivo do que em relação à ligação C-H de metileno, de tal forma que o produto 13 seja obtido em 54% de rendimento. Ao utilizar um catalisador mais eletrofílico (S,S)-Fe(CF3-PDP), a ligação C-H de metileno, presente na posição C10, pôde ser funcionalizada, fornecendo a cetona 14 com uma maior seletividade frente ao outro catalisador (Esquema 3).10
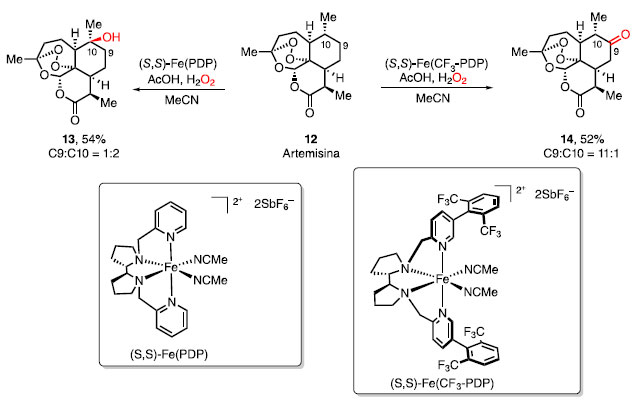 Esquema 3. Oxidação C-H de artemisina (12)
Ser capaz de modificar uma molécula bioativa de maneira precisa e eficiente, sem que altere o esqueleto, é um desafio na química orgânica altamente desejável, e a estratégia capaz de contornar esse desafio é a funcionalização da ligação C-H.2,6,11 Historicamente, a ligação C-H sempre foi tratada como inerte, com poucos trabalhos envolvendo a funcionalização C-H. Em 1970, Breslow e colaboradores reportaram a oxidação remota do colestanol (15) para obter a cetona 16, apesar do baixo rendimento, mas com uma boa seletividade, frente às outras ligações C-H (Esquema 4a).12 Para isso, a partir do éster de colestanol foi realizada a ciclização radicalar seletiva no C14 para obter um álcool terciário, seguida da desidratação para formar uma olefina. Foi realizada a ozonólise desta olefina, seguida da hidrólise para obter a cetona 16. Esse exemplo foi o pioneiro em oxidação C-H em molécula mais complexa seletivamente.
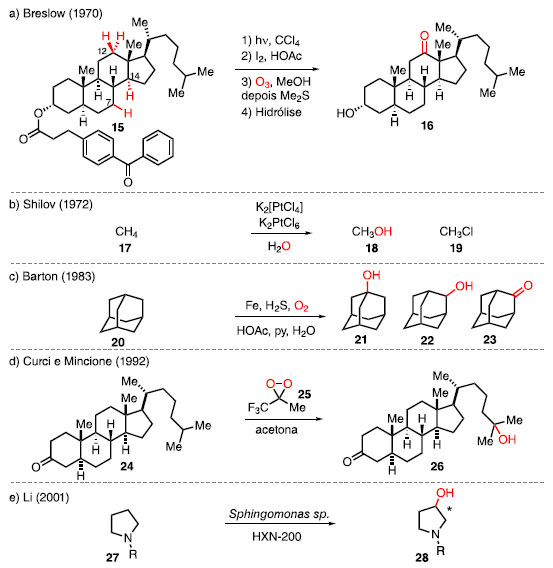 Esquema 4. Exemplos históricos de funcionalização C-H por: (a) Breslow; (b) Shilov; (c) Barton; (d) Curci e Mincione; (e) Li
Em 1972, Shilov e colaboradores desenvolveram uma reação de ativação da ligação C-H de metano (17) mediada por complexo de platina, este que passa por intermediários de platina (IV), de alta valência, para obter a mistura de metanol (18) e cloreto de metila (19). Este exemplo se tornou o primeiro a utilizar complexos metálicos para a funcionalização de metano (Esquema 4b).13 Em 1983, Barton e colaboradores reportaram a oxidação C-H do adamantano (20) utilizando catálise de ferro, obtendo majoritariamente álcool secundário 22 e cetona 23 na frente do álcool terciário 21.14 Nos primeiros exemplos, foi empregado o uso de ferro metálico, sulfeto de hidrogênio, piridina e mistura de água e ácido acético, com o oxigênio molecular como fonte para a inserção de oxigênio na molécula. Esse tipo de reação ficou conhecida como reações de Gif, em homenagem à cidade de Gif-sur-Ivette, local onde os primeiros exemplos dessa reação foram realizados (Esquema 4c). Em 1992, Curci e colaboradores introduziram a oxidação do esteroide 24 utilizando trifluorometildioxirano (TFDO) 25, um dioxirano com alta reatividade, fornecendo o produto 26 (Esquema 4d).15 Esse reagente, além de oxidar a ligação C-H mais distante de um grupo retirador de elétrons, consegue realizar a funcionalização C-H de maneira concertada, sem que ocorra a formação de radical ou de carbocátion. Além disso, em 2001, Li e colaboradores utilizaram a catálise enzimática para hidroxilar derivados de pirrolidina 27 para o produto 28 (Esquema 4e).16 As células envolvidas, além da utilização após o crescimento, foram também utilizadas após meses de congelamento, sem que houvesse perda da atividade. Após os anos 2000, o número de estudos envolvendo reações de funcionalização de ligações C-H aumentou significativamente, trazendo com isso o desenvolvimento de novas metodologias sintéticas para a funcionalização quimiosseletiva dessas ligações.7,8 Esses avanços tiveram um grande impacto tanto na área de síntese total,17 quanto na área de funcionalização em estágio avançado, em que produtos naturais foram funcionalizados para obter derivados com melhor atividade biológica. Também foram reportadas derivatizações de candidatos de fármacos, diodos orgânicos emissores de luz (da sigla, OLEDs), estruturas metalorgânicas (da sigla, MOFs) e polímeros, mostrando o alcance que essa estratégia pode ter.3,7,8,18 No geral, existem duas estratégias na lógica de funcionalização C-H para favorecer a formação de um produto de forma seletiva: a "dirigida" e a "inata".19 A funcionalização C-H dirigida utiliza um segundo grupo funcional na molécula, o qual é chamado de grupo diretor ou grupo dirigente, para coordenar a um complexo metálico, e direcionar reatividade a uma ligação C-H mais próxima. Um exemplo deste tipo de funcionalização é o uso da 8-amino quinolina como grupo diretor para a arilação C-H, introduzida por Daugulis e colaboradores. No exemplo, é utilizado o substrato 29 que, na presença do iodeto de arila 30, Pd(OAc)2 e AgOAc, sofre a reação de arilação para fornecer o produto 31 (Esquema 5a).19,20,21
 Esquema 5. Exemplos de funcionalização C-H: (a) dirigida; (b) inata
Já a funcionalização inata é a funcionalização da ligação C-H baseada na reatividade natural do substrato frente a um reagente, sem o uso de grupos direcionadores.9,19 A reação de cloração radicalar de esclareolídeo (32), desenvolvida por Alexian, Vanderwal e colaboradores, na presença da cloroamida 33, Cs2CO3 em benzeno, e sob luz visível, fornece o produto clorado 34 (Esquema 5b).22 O desenvolvimento dessa lógica permite com que ligações C-H, antes não possíveis de serem exploradas, possam ser funcionalizadas. Um exemplo recente desta lógica aplicada a produtos naturais é o trabalho desenvolvido por Stoltz e colaboradores, onde diversas estratégias de funcionalização de ligações C-H para a obtenção de análogos de um derivado da ciantiwigina 36 foram utilizadas (Esquema 6).23
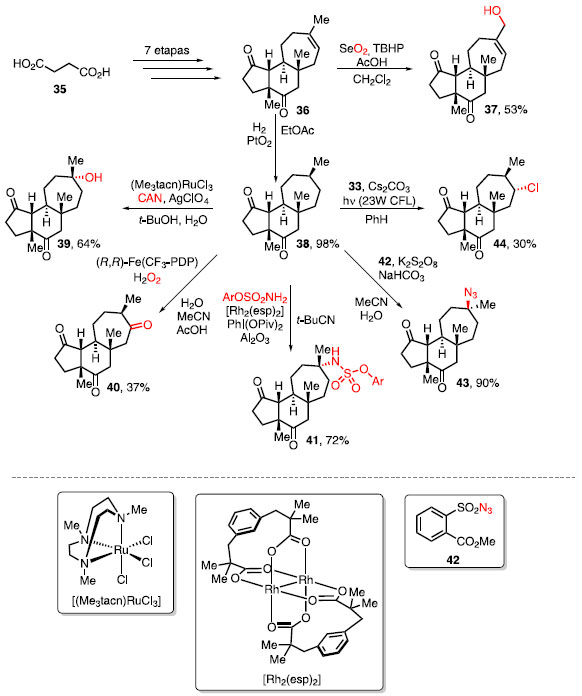 Esquema 6. Análogos de ciantiwigina via funcionalização C-H inata
O composto 36 foi obtido em 7 etapas a partir do ácido succínico (35), e a partir dele, foi realizada uma oxidação de Riley para acessar o produto 37 e uma hidrogenação para acessar ao intermediário 38. A partir do composto 38, foram realizadas reações de oxidação catalisadas por rutênio [(Me3tacn)RuCl3] e ferro (Ã,Ã)-Fe(CF3-PDP) para obter o álcool e a cetona 39 e 40, respectivamente. Do mesmo intermediário 38, foram realizadas uma aminação calatisada por ródio [Rh2(esp)2] e uma azidação envolvendo a molécula 42, obtendo os produtos 41 e 43, respectivamente. Por fim, foi realizada a cloração sitiosseletiva utilizando o reagente 33, cujo produto 44 foi obtido.23 Herzon e colaboradores aplicaram a mesma lógica de funcio-nalização C-H para obter derivados da pleuromutilina (51), um diterpeno utilizado como antibiótico, capaz de inibir a síntese de proteínas.24 Nesse caso, reações de sililação C-H catalisadas por irídio seguidas por uma oxidação de Tamao-Fleming para formar 1,3- e 1,4- dióis foram utilizadas (Esquema 7a).25 Diferente da metodologia descrita por Breslow, em que ocorre a ozonólise de olefina para a formação de dicetonas, a oxidação de Tamao-Fleming faz a conversão de uma ligação C-Si para o álcool correspondente, com o uso de H2O2 como oxidante.
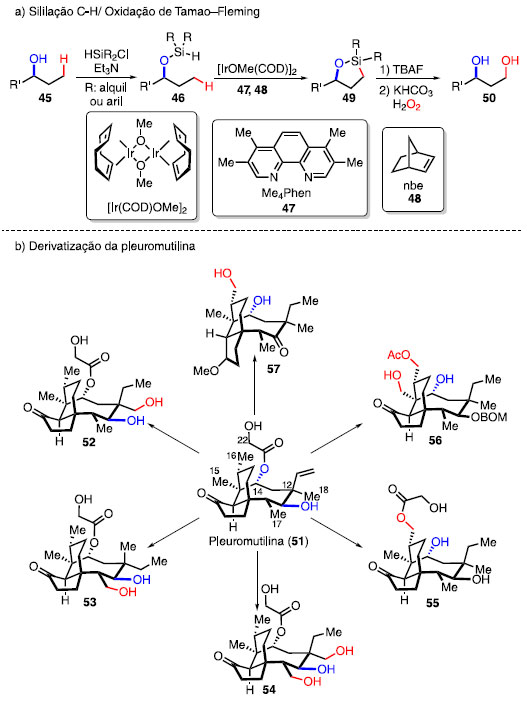 Esquema 7. Análogos de pleuromutilina (51) via funcionalização C-H dirigida
A hidroxila no C11 da pleuromutilina foi utilizada como um grupo diretor para funcionalizar as metilas nas posições C17 e C18, obtendo os produtos 52, 53 e 54 (Esquema 7b). Para obter o produto 53, a pleuromutilina foi tratada com dietilzinco para epimerizar o C12, e favorecer a oxidação no C17 da metila. Proteções das hidroxilas de C22 e C18 do intermediário 52, seguida da sequência sililação C-H e oxidação de Tamao-Fleming forneceu o produto 54. A hidroxila do C14 da pleuromutilina também foi utilizada como grupo dirigente para funcionalizar a ligação C-H das metilas das posições C15 e C16 do 51, para acessar aos compostos 55, 56 e 57. O tratamento de 51 com BOMCl, saponificação, redução de olefina, sililação C-H, oxidação de Tamao-Fleming, esterificação seletiva e hidrogenólise forneceu o produto 55, via isomerização do éster de O14 para O16. O álcool no C16 obtido foi acetilado, seguido da sililação C-H e oxidação de Tamao-Fleming, fornecendo o produto 56. O tratamento da pleuromutilina com trimetilortoformato e ácido sulfúrico forneceu o 4-epi-pleuromutilina, que foi submetida à saponificação, hidrogenação, sililação C-H e oxidação de Tamao-Fleming, formando o produto 57.26 Neste artigo de revisão abordaremos o desenvolvimento de metodologias para a funcionalização de ligações C-H que foram utilizados na modificação em estágio tardio de produtos naturais e fármacos. Os exemplos aqui apresentados envolvem catálise metálica e fotoquímica na formação de ligações C-C, C-O, C-N, C-S, C-Si, C-Halogênio. Funcionalizações de ligações C-H por meios eletroquímicos, organocatalisadas ou biocatalisadas não serão cobertos por este trabalho. Funcionalização de ligações C-H ativadas como, por exemplo, em posições alílicas ou benzílicas também não serão cobertas.
FORMAÇÃO DE LIGAÇÕES C-C A formação de ligações C-C é de importância central para a síntese orgânica ao possibilitar a obtenção de moléculas mais complexas a partir de outras mais simples e disponíveis comercialmente.27 O uso de metais de transição como catalisadores para a formação de novas ligações C-C tem se mostrado uma estratégia sintética promissora devido à sua seletividade, rendendo, por exemplo, o prêmio Nobel de Química de 2005 para Yves Chauvin, Robert H. Grubbs e Richard R. Schrock "pelo desenvolvimento do método de metátese de olefinas na síntese orgânica".28 Além disso, em 2010, Richard F. Heck, Ei-ichi Negishi e Akira Suzuki, foram laureados por reações de "acoplamento cruzado catalisadas por paládio na síntese orgânica".29 Nas últimas décadas, a alquilação e a arilação de ligações C-H têm se mostrado um campo com grande potencial permitindo sínteses mais eficientes e econômicas. A funcionalização em estado avançado de ligações C-H para a formação de novas ligações C-C através da utilização de organometálicos permitiu o desenvolvimento de novos fármacos, como por exemplo, o inibidor SPT, e produtos naturais, como o ácido litospérmico, ambos com etapas-chaves a funcionalização C-H.7,8,30 Em 2015, Shi, Xia e colaboradores desenvolveram uma metodologia capaz de arilar ligação C-H de maneira dirigida em carbonos b a éteres de oxima 58 através da utilização do sal de diaril iodônio não-simétrico 59 como agente arilante. Além disso, foram envolvidos AgNTf2 e PivOH como aditivos, e o complexo sanduíche de irídio (III) [(Cp*IrCl2)2] como catalisador, fornecendo os produtos arilados 60 (Esquema 8).31
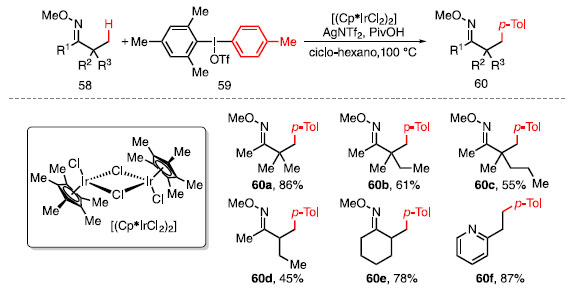 Esquema 8. Escopo de produtos obtidos a partir da arilação C-H de oximas
O aumento do volume estéreo causado pelos grupos adjacentes reduz gradualmente o rendimento, como observado em 60a-60c. Há seletividade para a funcionalização de ligações C-H na posição b primárias frente a secundárias, como observado na formação exclusiva de 60d. Substratos com hidrogênios na posição a se mostraram compatíveis com a metodologia, assim como composto cíclico 60e. Piridinas podem atuar como grupos dirigentes além de éteres de oxima, sendo capazes de fornecer produtos monoarilados com excelentes rendimentos, como visto em 60f. Após estudos computacionais, o mecanismo reacional proposto inicia-se com a coordenação da oxima 58 ao catalisador de irídio levando à formação do intermediário 61 (Esquema 9). Com o auxílio da base, a abstração do hidrogênio na posição b, formando o metalaciclo 62, ocorre via metalação-desprotonação concertada (da sigla em inglês, CMD), liberando ácido tríflico. Em seguida, a adição oxidativa do sal de diaril iodônio 59 no intermediário 62, na qual o átomo de irídio passa do estado de oxidação III para V, fornece o intermediário 63. Por fim, após uma etapa de eliminação redutiva, a oxima 60 é liberada e o ciclo catalítico é restaurado.31
 Esquema 9. Mecanismo de reação para a arilação C-H de oximas
Neste ciclo catalítico, é importante ressaltar que há a formação in situ de 59, com NTf2 como base, tornando o processo da adição oxidativa mais favorável, quando comparada com o sal de diariliodônio contendo o OTf como base. Esta metodologia foi aplicada na arilação em b de um derivado do ácido oleanóico 64, um triterpeno não tóxico encontrado nas plantas da espécie Phytolacca americana, conhecido por apresentar propriedades antivirais e antitumorais.32 Ao utilizar o sal de diaril iodônio simétrico 65, AgNTf2, PivOH e [(Cp*IrCl2)2] obteve-se unicamente o diasteroisômero 66 em 38% de rendimento (Esquema 10).31
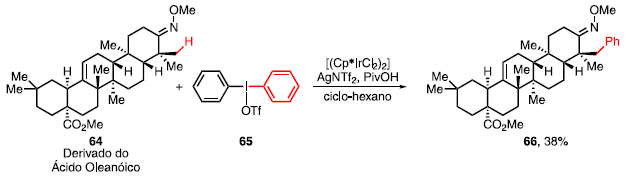 Esquema 10. Arilação do derivado do ácido oleanólico
Uma estratégia semelhante foi explorada por Chen e colaboradores, ao realizarem a arilação em b de éteres de oxima com o auxílio de sais de diaril iodônio simétricos, utilizando Pd(OAc)2 como catalisador.33 Em relação ao trabalho do Xia, Shi e colaboradores, que utilizava catalisador de irídio, a metodologia desenvolvida pelo grupo de pesquisa de Chen utilizou catalisador de paládio, mais barato do que o correspondente acima, e pôde funcionalizar ligações C-H de metilenos, expandindo o escopo da reação desenvolvida. Para a oxima 67, a arilação utilizando o sal de diaril iodônio 68, catalisador de paládio, seguida da hidrólise ácida, forneceu a cetona 69 arilada na posição b em 42% de rendimento para duas etapas. Para a oxima 70, que contém o substituinte adamantano, a arilação nas mesmas condições reacionais, e com o sal de diaril iodônio 71, produziu a oxima 72 arilada em 47% de rendimento (Esquema 11).
 Esquema 11. Arilação C-H de metileno catalisada por paládio
O derivado da metasterona 73, um esteroide anabolizante utilizado para aumentar o rendimento físico de atletas, considerada uma substância proibida em 2006 pela World Anti-Doping Agency (WADA),34 forneceu o produto monoarilado 74, em 81% de rendimento. Foi utilizado o sal de diaril iodônio 69, PivOH, Ag2CO3 e Pd(OAc)2, seguido pela hidrólise do éter de oxima em meio ácido. Essa condição reacional mostrou tolerância à presença de uma hidroxila terciária. Já o composto 75, um derivado do ácido glicirretínico, conhecido comercialmente como enoxolona, um triterpeno encontrado nas raízes das plantas da espécie Glycyrrhiza glabra (alcaçuz), que apresenta propriedades anti-inflamatórias, antivirais e antitumorais,35 forneceu um produto diarilado 76 em 69% de rendimento (Esquema 12).
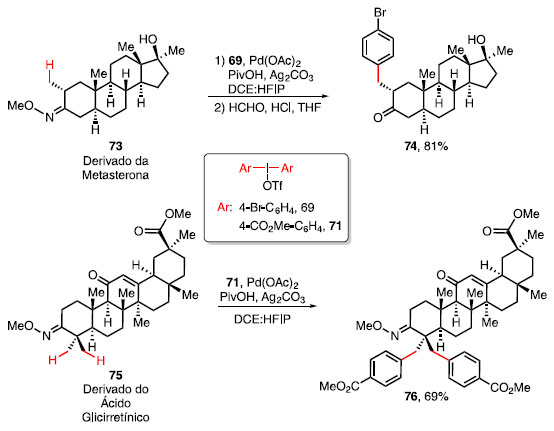 Esquema 12. β-arilação em moléculas complexas derivadas de produtos naturais
Em 2016, Gaunt e colaboradores desenvolveram uma metodologia de carbonilação C-H de aminas alifáticas 77 em posição b catalisadas por paládio para obter b-lactamas 83 (Esquema 13).36 Para isso, além dos catalisadores de paládio (II), foram utilizados o gás CO para a carbonilação, ácido adamantano-1-carboxílico (AdCO2H, 78) ou ácido 2,4,6-trimetilbenzóico (MesCO2H; 79) como ácidos impedidos estericamente para o ataque da amina ao paládio durante o ciclo catalítico.
 Esquema 13. Escopo de carbonilação de ligações C-H alifáticas em aminas
O acréscimo da benzoquinona (80) facilita a etapa de eliminação redutiva, enquanto o uso de ligantes nitrogenados, sendo uma quinolina 81 ou quinuclidina 82, permitiu que o catalisador e outros aditivos pudessem ser utilizados em uma menor quantidade. Por fim, o Cu(OAc)2 oxida o paládio, regenerando o ciclo catalítico. O aumento do volume estéreo observado em 83a-83d resulta em um menor rendimento da reação; a reação com a fenil isopropilamina 83e não teve sucesso nessas condições reacionais, por conta do efeito de ressonância entre o nitrogênio e o anel aromático nesse tipo de substrato. Uma variedade de grupos funcionais se mostrou tolerante como, por exemplo, a sulfona 83f, éster 83g e a pirimidina 83h.36 O mecanismo de reação proposto inicia-se com a troca de ligantes do pré-catalisador Pd(OAc)2 para o catalisador Pd(O2CR)2 (Esquema 14). Após coordenação da amina alifática 77 ao átomo de paládio, fornecendo o intermediário 84, ocorre a coordenação de CO para gerar o intermediário 85, com a formação do anidrido 86.
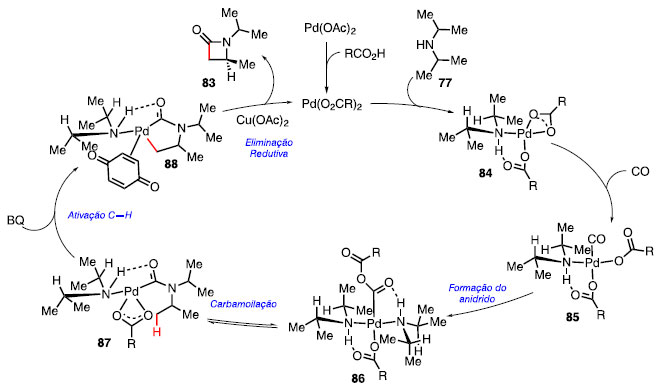 Esquema 14. Mecanismo proposto para a carbonilação C-H de aminas alifáticas
Com base nos estudos computacionais e efeito isotópico, a etapa de carbamoilação ocorre com o ataque da amina ao anidrido, obtendo a amida 87 e, com o auxílio de benzoquinona (80), ocorre a ativação da ligação C-H, formando o metalaciclo de 5 membros 88. A etapa da eliminação redutiva, juntamente com a regeneração do catalisador via oxidação por Cu(OAc)2, forneceu a b-lactama 83.36 O derivado do salbutamol 89, um fármaco utilizado no tratamento da asma por promover a dilatação dos brônquios, forneceu a b-lactama 90 com rendimento de 40%, quando utilizado a condição a (Esquema 15).37 Ao se utilizar a condição b para a reação de carbonilação C-H em um derivado da dobutamina 91, um fármaco desenvolvido nos anos 70 e usado no combate da insuficiência cardíaca por suas propriedades vascular e arritmogênica, o composto 92 foi obtido em 72% de rendimento.38
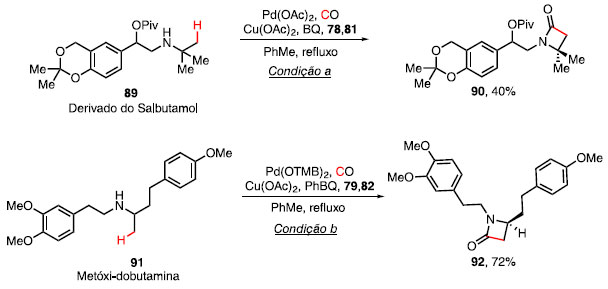 Esquema 15. Síntese de lactamas em derivados farmacêuticos
Yu e colaboradores desenvolveram uma metodologia catalisada por Pd(OAc)2 capaz de realizar reações de carbonilação e olefinação utilizando CO e etileno, respectivamente, em ligações C-H em posições ô ou g de éteres benzílicos 93, utilizando C6F5CO2H e AgOAc como aditivos em HFIP (Esquema 16).39 A utilização de um grupo diretor bidentado, aliado ao oxigênio do éter benzílico, controla a regiosseletividade da reação, obtendo ésteres 94.
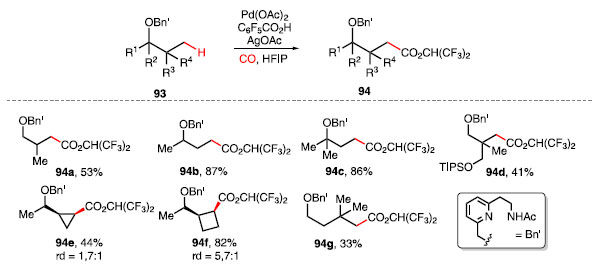 Esquema 16. Esquema de produtos obtidos a partir da carbonilação em éteres
A análise das condições reacionais revelou que os produtos 94a-94c, provenientes de álcoois primários, secundários e terciários, respectivamente, são compatíveis com o sistema desenvolvido, fornecendo produtos com rendimentos moderados a altos, assim como substratos que possuem outro oxigênio coordenante como, por exemplo, 94d. Ligações C-H metilênicas em anéis como ciclopropano 94e e ciclobutano 94f também são seletivamente funcionalizadas. A carbonilação de uma ligação ô C-H foi possível como visto no produto 94g.39 A olefinação da ligação C-H do éter 95 ocorreu através de um mecanismo similar à reação de carbonilação, mas, nesse caso, com o uso de Pd(OAc)2, AgTFA, Ag2CO3 e Cu(OAc)2 para a inserção de etileno, um gás barato e abundante com enorme potencial sintético. Foram obtidos dessa metodologia os produtos olefinados 96a e 96b como isômeros, em 14% e 38%, respectivamente (Esquema 17).40
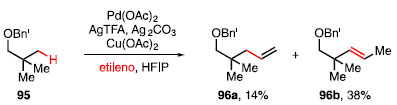 Esquema 17. Olefinação de ligações C-H quando há utilização de gás etileno
O derivado do estradiol (97), esteroide de importância para o sistema vascular e sexual feminino, se mostrou um substrato eficaz para expor o potencial de ambas as transformações de carbonilação e olefinação sob as condições mencionadas, obtendo o éster 98 em 54% e os isômeros 99a e 99b em 11% e 27%, respectivamente (Esquema 18).40
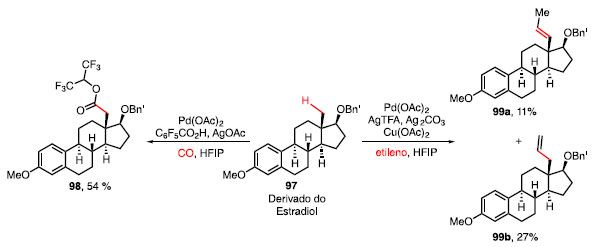 Esquema 18. Carboxilação e olefinação em derivado obtido a partir do estradiol
Em 2016, Rovis e colaboradores desenvolveram uma reação de acoplamento entre uma amida 100 e um alceno 101 disponível comercialmente, catalisada por complexo de irídio e sob irradiação de luz azul, fornecendo o produto 102 (Esquema 19).41 Para isso, além da amida e o alceno, foram utilizados o fotocatalisador [Ir{dF(CF3) ppy}2(dtbbpy)]PF6, K3PO4 como base para abstrair o próton da ligação N-H, em PhCF3.
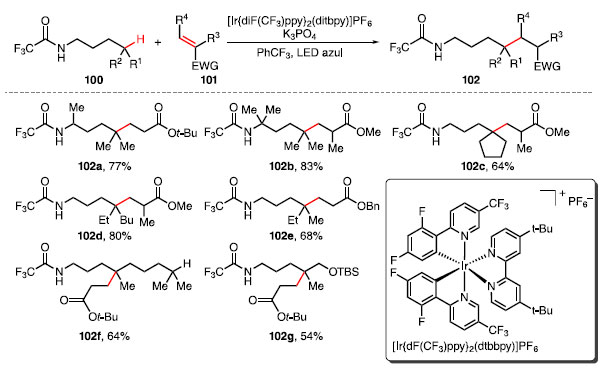 Esquema 19. Escopo dos produtos obtidos a partir da ativação da ligação C-H e funcionalização via 1,5-HAT
A presença do grupo trifluoroacetamida como grupo diretor facilita a transferência 1,5 do átomo de hidrogênio (1,5-HAT). Estudos revelaram que a reação ocorre em substratos que possuem substituintes a ao nitrogênio, fornecendo o produto 102a em bom rendimento. A variação do ambiente estéreo dos grupos substituintes ao redor da ligação C-H inerte tem pouca influência no rendimento das reações, fornecendo 102b-102e em rendimentos de moderados a bons. Há uma preferência para a funcionalização da ligação C-H através do 1,5-HAT frente a 1,6-HAT ou outra C-H terciária, como visto em 102f e 102g. O ciclo fotocatalítico proposto para essa reação consiste na irradiação de luz azul sobre o complexo de irídio, que passa para o estado excitado Ir(III)* que, com auxílio de uma base, abstrai o próton da ligação N-H da amida 100, formando o radical 103, e o complexo Ir(II) com a base protonada [B-H]+ (Esquema 20). Esse processo de transferência de um elétron entre duas espécies (do inglês, single-electron transfer, SET), é uma etapa importante para a formação de intermediário contendo radical, e para reações que passam por mecanismos radicalares.
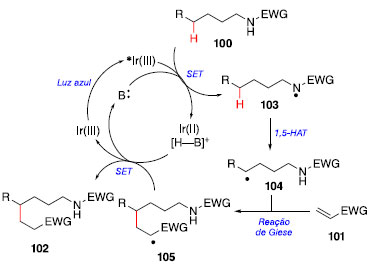 Esquema 20. Ciclo catalítico para a formação de ligação C-C via 1,5-HAT
Após a formação do radical 103, ocorre a 1,5-HAT, fornecendo o intermediário radicalar 104, que realiza a adição conjugada com o alceno 101, via reação de Giese, gerando o intermediário 105. Esse intermediário, em conjunto com [B-H]+ e Ir(II), realizam um segundo SET, fornecendo o produto 102 e regenerando o ciclo fotocatalítico.41 O derivado da pregabalina 106, um fármaco utilizado no tratamento da ansiedade e dor periférica por conta de seus efeitos analgésico, ansiolítico e anticonvulsivante também foi funcionalizado fornecendo 108 em 70% de rendimento (Esquema 21).42 Já a funcionalização do derivado do ácido litocólico 109 possibilitou a obtenção de uma mistura diastereoisomérica de 111 com rendimento de 70%.
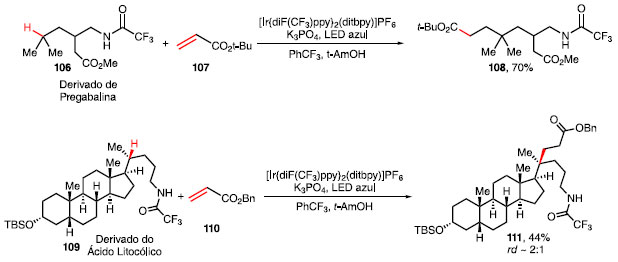 Esquema 21. Funcionalização de ligações C-H em esteróide e fármaco
Davies e colaboradores exploraram a alquilação de ligações C-H terciárias catalisada pelo complexo quiral de dirródio Rh2(S-TCPTAD)4 utilizando diazocompostos 112 como fonte de carbeno e três equivalentes de substrato 113, fornecendo o produto alquilado 114 em bom rendimento e enantiosseletividade (Esquema 22).43
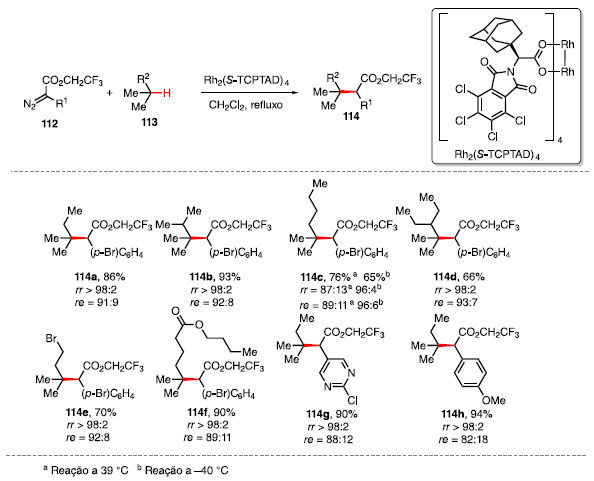 Esquema 22. Escopo de produtos obtidos através da inserção de carbenóide
O escopo da reação revelou que o sistema é sensível ao ambiente estéreo próximo ao sítio terciário, sendo que quando esse se encontra impedido a reação ocorre em um sítio secundário ou terciário menos impedido 114a-114d. A diminuição da temperatura aumenta a regiosseletividade e enantiosseletividade em detrimento do rendimento, como indicado no produto 114c. A reação ocorre de maneira satisfatória com substratos que contêm outros grupos funcionais como, por exemplo, bromo 114e, éster 114f, e grupos nucleofílicos, como pirimidinas 114g.43 O ciclo catalítico para essa reação consiste na reação do complexo de dirródio com o composto diazo 112, formando o carbenóide 115, com a liberação de N2. O complexo 115, mais reativo, irá realizar a inserção C-H de alcano, fornecendo o produto 114, e regenerando o ciclo catalítico (Esquema 23).44
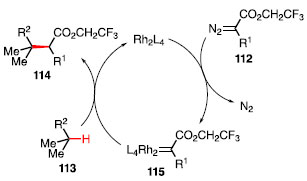 Esquema 23. Ciclo catalítico de inserção C-H via carbenóide
Esta metodologia mostrou-se compatível com acetato de vitamina E (116) e acetato de colesterol (119), utilizando o diazo composto 117 (Esquema 24). A alquilação ocorreu exclusivamente na ligação C-H terciária representada, mesmo na presença de outros sítios terciários e secundários nesses compostos, fornecendo os produtos 118 e 119 com razões regioisoméricas superiores a 98:2.43 A mudança do catalisador Rh2(S-TCPTAD)4 para o enantiômero correspondente levou à um aumento do rendimento para ambos os casos.
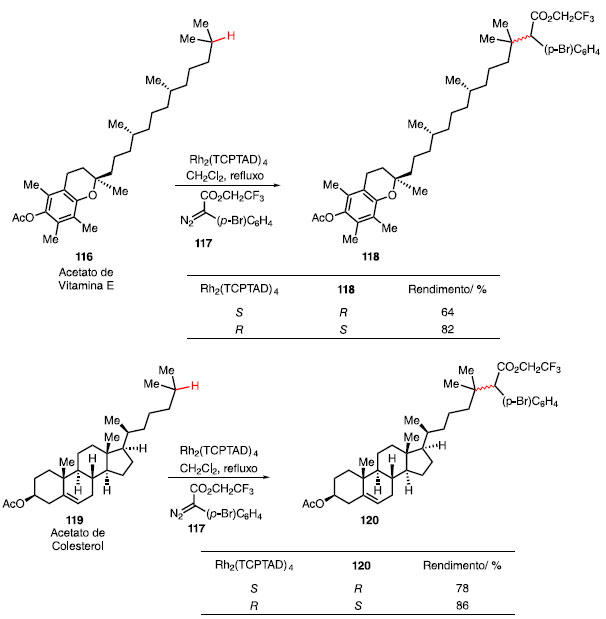 Esquema 24. Funcionalização C-H em acetatos de colesterol e vitamina E
MacMillan e colaboradores desenvolveram uma metodologia capaz de realizar a arilação direta de ligações C-H alifáticas de substratos 121 com haletos de arila 122 (Esquema 25).
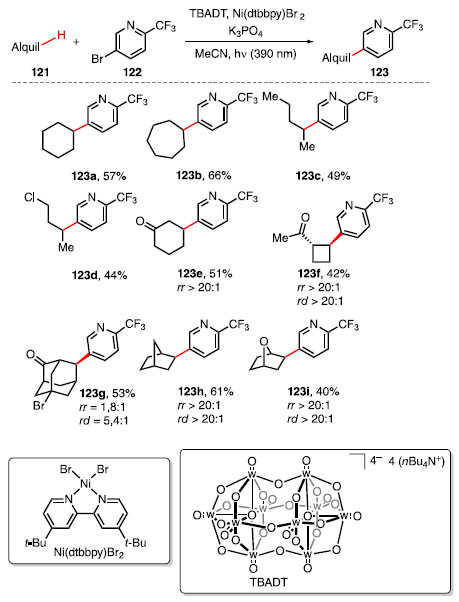 Esquema 25. Escopo de produtos obtidos a partir da arilação de ligações C-H
Nesse estudo, utilizou-se da catálise metalo fotoredox por Ni(dtbbpy)Br2, decatungstato de tetrabutilamônio (TBADT), K3PO4 em MeCN promovida por radiação próxima a região do UV, obtendo produtos 123 com uma ligação C-C entre um grupo alquil e um grupo aril.45 No escopo investigado, nota-se que cicloalcanos, variando de seis a sete carbonos, fornecem produtos com rendimentos moderados, como observado em 123a e 123b. Sistemas lineares alifáticos apresentam estatisticamente maior preferência de arilação na posição 2 menos impedida estericamente, entre todos os regioisômeros formados, mesmo quando apresentam grupos retiradores de densidade eletrônica, fornecendo os produtos 123c e 123d com rendimentos moderados. A presença de um grupo funcional retirador de elétrons como cetonas é tolerável, sendo que a arilação ocorre em posições distantes a ela, como nota-se em 123e-123g. Alcanos bicíciclos e aqueles contendo heteroátomo, especialmente derivados do adamantano são funcionalizados nas posições metilênicas, fornecendo produtos como 123h-123j, com rendimentos moderados.45 O ciclo catalítico proposto para a reação começa com o ciclo de decatungstato, aonde ocorre a fotoexcitação de [W10O32]4-para *[W10O32]4-, seguido de processo HAT com o substrato 121, permitindo a formação do radical 124 e do catalisador reduzido [W10O32]5-H+. O desproporcinamento do catalisador reduzido via SET regenera o ciclo catalítico do decatungstato e forma a espécie [W10O32]6- 2H+. O catalisador LnNi0 captura o radical 124, obtendo o intermediário de NiI 125, que ao sofrer a adição oxidativa do haleto de arila 122, produz a espécie de Nim 126. Após a etapa da eliminação redutiva, o produto 123 é proporcionado, e a espécie do catalisador de NiI é regenerada via SET, liberando HBr, [W10O32]5- H+ e o catalisador de Ni0, fechando os três ciclos catalíticos (Esquema 26).45
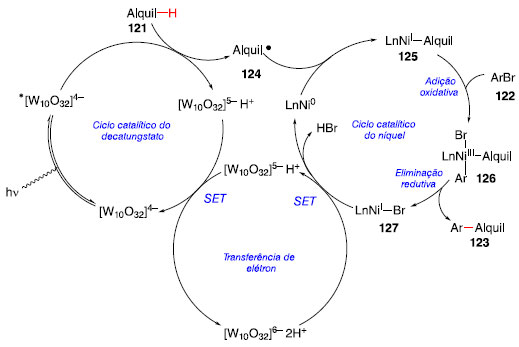 Esquema 26. Ciclo catalítico para a arilação C-H fotocatalisada por TBADT e níquel
A funcionalização de ligações C-H no esclareolídeo (32), canfeno (102) e fenchona (104), produtos naturais da classe dos terpenos que apresentam propriedades antifúngicas, utilizando a condição padrão mostrou-se eficaz (Esquema 27).46 Eles se mostraram parceiros de acoplamento competentes, fornecendo os produtos arilados 101, 103 e 105 em 41%, 70% e 38% de rendimento, respectivamente, com razões regioisoméricas e diasteroisoméricas acima de 1,4:1 e 20:1.
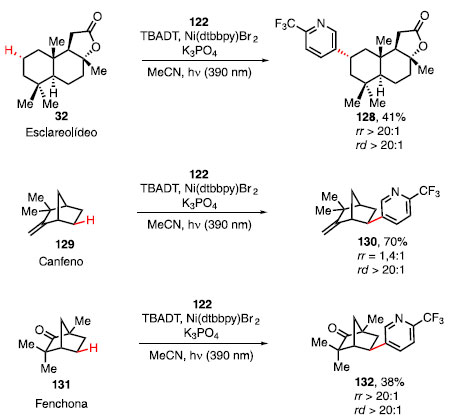 Esquema 27. Arilação direta em ligações C-H em produtos naturais
FORMAÇÃO DE LIGAÇÕES C-O A funcionalização de ligação C-H em estágio avançado para formação de ligação C-O é muito utilizada como um caminho para chegar em outras ligações.2,3,5,22,47 A capacidade de derivatizar um composto contendo a ligação C-O pode ser exemplificado pelo esquema 28, onde ocorre a conversão de um análogo de esteroide 133 em outros compostos através de reações de substituição nucleofílica alifática. A substituição por um tiol (reação a), substituição por bromo (reação b), substituição por iodo (reação c), substituição por azida (reação d), e, bem como substituição por PPh2 (reação e), fornece os respectivos produtos 134-138.47
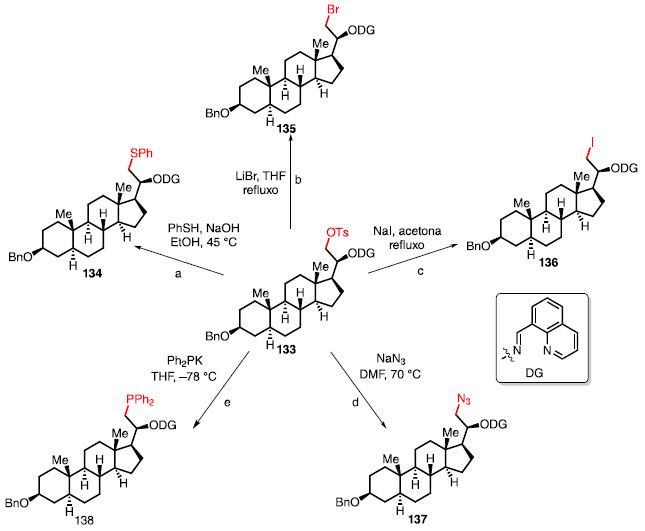 Esquema 28. Ligação C-O possibilitando formação de outras ligações
Entre 2007 e 2013, White e colaboradores desenvolveram os catalisadores não-heme Fe(PDP) e Fe(CF3-PDP) e métodos de oxidação de ligação C-H com o uso de peróxido de hidrogênio como oxidante, ácido acético como aditivo e acetonitrila como solvente foram reportados (Esquema 29).10,48 Nessas reações não há a necessidade de cuidado com oxigênio e umidade.
 Esquema 29. Reações de oxidação com catalisador a base de ferro e escopo das reações
Enquanto o catalisador Fe(PDP) é utilizado preferencialmente na conversão de ligação C-H de metinos 139 aos correspondentes 140, o catalisador Fe(CF3-PDP) é utilizado na conversão de ligação C-H de metilenos 141 a cetonas 142. O composto 143 mostra que a hidroxilação ocorre preferencialmente no carbono terciário mais rico em elétrons, ou seja, mais longe do grupo retirador de elétrons com 56% de rendimento, e com a relação de produto de oxidação distante:próximo maior que 99:1.48 Já a partir da formação do álcool 144 é possível observar que, em ligações C-H com o mesmo ambiente eletrônico, o catalisador vai atuar preferencialmente em um local estericamente mais acessível, pois a outra ligação C-H terciária está orientada mais próxima ao acetato, tornando-se menos acessível estericamente. Efeitos estereoeletrônicos como, por exemplo, a hiperconjugação também influenciam na seletividade e o composto 145, proveniente de uma oxidação da posição carbinólica, é formado em 41% de rendimento. Ácidos carboxílicos apresentam um efeito benéfico na atividade catalítica desses sistemas e são amplamente utilizados como aditivos.49 Na ausência desses aditivos, moléculas contendo ácidos carboxílicos podem dirigir a reação como, por exemplo, na síntese da lactona 146 em 63% de rendimento. Há substratos onde fatores estéreos e eletrônicos divergem fortemente para favorecer locais distintos e, nesses casos, Fe(PDP) acaba sendo pouco seletivo. Nesses substratos, Fe(CF3-PDP) forneceu sitiosseletividade útil com base em sua capacidade de superar influências eletrônicas no substrato em favor de fatores estéreos como pode ser observado para o aminoácido 147. Essa abordagem com catalisadores também funcionou com moléculas complexas (Esquema 30). O (-)-Triacetóxi Tricalisiolídeo (148) é um metabólito putativo derivado do cafestol, que é um diterpeno extraído do café. A oxidação de 148 utilizando o (R,R)-Fe(CF3-PDP) forneceu o álcool 149 seletivamente no C6 do diterpeno com 61% de rendimento. O esclareolídeo (32) possibilitou a reação com (R,R)-Fe(PDP), obtendo o 2-oxo-esclareolídeo 150 com 46% de rendimento.10,48,50
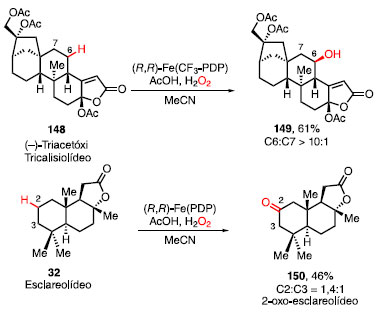 Esquema 30. Oxidação C-H do (-)-triacetóxi tricalisiolídeo e do esclareolídeo
Em geral, reações de oxidação em moléculas nitrogenadas levam à oxidação direta do átomo de nitrogênio, obtendo-se compostos N-oxo. Para contornar esse obstáculo, White e colaboradores desenvolveram entre 2015 e 2017 estratégias para funcionalizar a ligações C-H em moléculas contendo aminas, piridinas e amidas.51 Enquanto o uso de HBF4 permitiu a complexação inicial entre esse ácido e o átomo de nitrogênio básico (aminas e piridinas), MeOTf foi utilizado como um agente alquilante reversível para amidas. Em ambos os casos, oxidações deletérias foram desativadas e a oxidação remota de ligações C-H foi favorecida. O derivado do dextrometorfano (151), um fármaco utilizado para prevenir tosse, foi tratado inicialmente com HBF4 e, após oxidação utilizando o catalisador (S,S)-Fe(CF3-PDP) forneceu a cetona 152 e o álcool 153 em 45% de rendimento e uma seletividade de 2,5:1 cetona/álcool (Esquema 31).
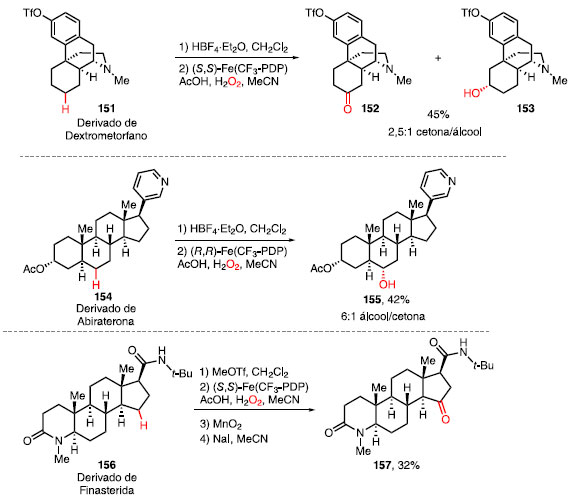 Esquema 31. Oxidação com catalisadores de ferro em moléculas contendo átomos de nitrogênio
O derivado da abiraterona (154), um fármaco recentemente aprovado pela Agência Europeia de Medicamentos (da sigla EMA) para o tratamento de câncer de próstata, forneceu o álcool 155 em 42% de rendimento como único diastereoisômero após o tratamento com HBF4 seguido da oxidação com (R,R)-Fe(CF3-PDP). O derivado de finasterida (156), utilizado para prevenir a queda de cabelo e no tratamento de hiperplasia protástica benigna e câncer de próstata, foi convertido na cetona 157 em 32% de rendimento após o tratamento com MeOTf seguido da oxidação com (S,S)-Fe(CF3-PDP). Para moléculas contendo anéis aromáticos, uma estratégia complementar foi desenvolvida. Em 2019 e 2020, White e colaboradores reportaram o uso dos catalisadores de manganês Mn(CF3-PDP) e Mn(PDP) e ácido cloroacético como aditivo para a oxidação quimiosseletiva de ligações C-H de metilenos e metinos, respectivamente.52 O derivado de efavirenz (158), um fármaco utilizado para inibir o vírus HIV da AIDS, forneceu a metilcetona 159 em 58% de rendimento utilizando o sistema Mn(CF3-PDP)/ácido cloroacético. A oxidação do análogo da ciclo-heximida (160), um antibiótico utilizado para inibir a atividade de mofos, leveduras e fungos, com Mn(PDP) levou à formação do álcool 161 em 63% de rendimento (Esquema 32).
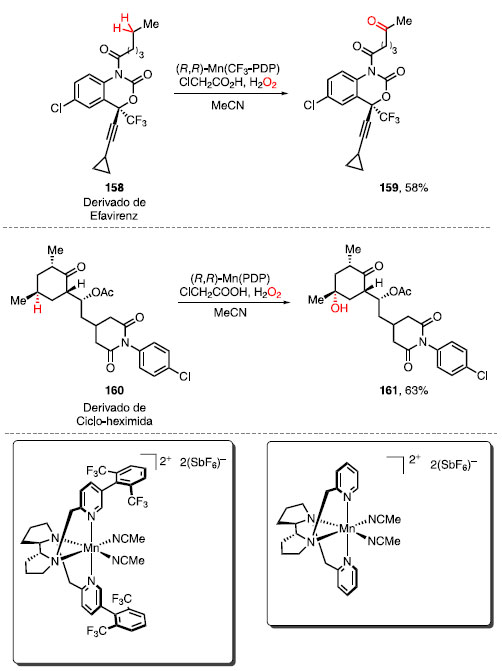 Esquema 32. Oxidação com catalisadores de manganês em moléculas contendo anel aromático
O ciclo catalítico para a transformação foi proposta por Chen, Wang e colaboradores, a partir de cálculos computacionais (Esquema 33).53 O ciclo inicia-se com a dissociação do pré-catalisador [Mn] (NCMe)2 para [Mn], que durante a coordenação com H2O2, forma o complexo [Mn]-OH, com a liberação do radical «OH. Em seguida, ocorre a troca de ligante com o ácido cloroacético, liberando água e formando o complexo 162.
 Esquema 33. Ciclo catalítico proposto para a oxidação C-H utilizando manganês
Uma nova troca de ligantes ocorre com a adição de H2O2, formando o complexo 163 e liberando o ácido cloroacético, que retorna ao ciclo catalítico, fornecendo o catalisador ativo 164 e liberando água. O cálculos indicaram que o intermediário 164 pode ser tanto uma mistura do complexo como tripleto ou quinteto, isto é, se a ligação Mn-oxo estará como uma ligação simples (tripleto) ou como uma ligação dupla (quinteto). Com o catalisador ativo 164, ocorre a oxidação C-H do substrato 165 via formação do radical mais distante do grupo retirador de elétrons, fornecendo o álcool 166 e o complexo 162, completando o ciclo catalítico para o manganês. O álcool 166 sofre uma nova oxidação pelo catalisador ativo 164, produzindo a cetona 167, juntamente com os complexos [Mn]-OH ou 162, uma vez que os cálculos mostraram que ambos os caminhos para obter os dois complexos são favoráveis. Em 2017, Bietti, Costas e colaboradores utilizaram o catalisador a base de manganês, (S,S)-Mn-(dMMpdp) para a oxidação de ligações C-H. Essa abordagem foi possível para a conversão de uma molécula bioativa derivada da pregabalina (168) e possibilitando a formação a lactona 169 em rendimento de 60% (Esquema 34).54
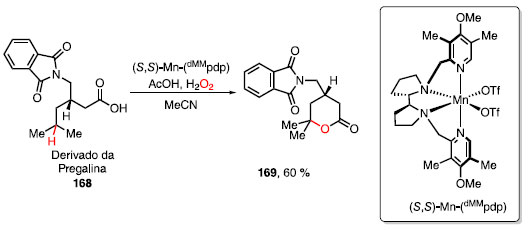 Esquema 34. Oxidação de molécula bioativa com catalisador de manganês
Além de catalisadores não-heme de ferro e manganês, Yang e colaboradores, em 2018, reportaram a oxidação C-H de amidas catalisada por cobre (Esquema 35).55 Eles utilizaram o grupo diretor benzoato para direcionar a funcionalização da molécula 170, e formar a ligação C-O do produto 171. Para isso, foi o CuI como catalisador, PhI(OTFA)2 como oxidante, e reagente de transferência do grupo -OTFA, TBAB e Br2 como aditivos, em CH2Cl2.
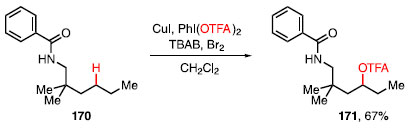 Esquema 35. Reação de oxidação C-H catalisada por cobre
Essa estratégia foi passível de ser utilizada na trifluoroacetoxilação do derivado do ácido litocólico (172) e o composto 173 foi obtido seletivamente em 63% de rendimento (Esquema 36).55
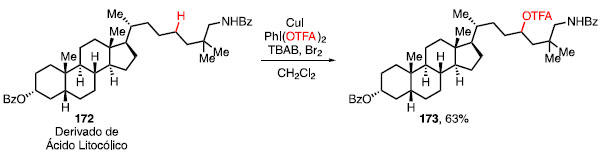 Esquema 36. Trifluoroacetoxilação de um derivado do Ácido Litocólico
FORMAÇÃO DE LIGAÇÕES C-N A formação de ligações C-N apresenta grande relevância sintética tendo em vista sua grande presença em diversos produtos naturais e fármacos. Um exemplo disso é o fato de que 59% das pequenas drogas aprovadas pelo Food and Drug Administration (FDA) dos EUA apresentam pelo menos um heterocíclo nitrogenado.56 Nos últimos anos, reações de formação de ligações C-N a partir de ligações C-H em estágio avançado de moléculas complexas têm surgido como alternativa interessante às reações clássicas, que usualmente necessitam de outros grupos funcionais prévios como, por exemplo, álcoois ou carbonilas, para a instalação de funcionalidades nitrogenadas.57 Hartwig e colaboradores desenvolveram uma metodologia para realizar reações de azidação de ligações C-H em compostos como 174 catalisadas por Fe(OAc)2 na presença do ligante de bisoxazolina quiral (5,5)-iPr-pybox e do composto 175, como fonte de azida (Esquema 37).58
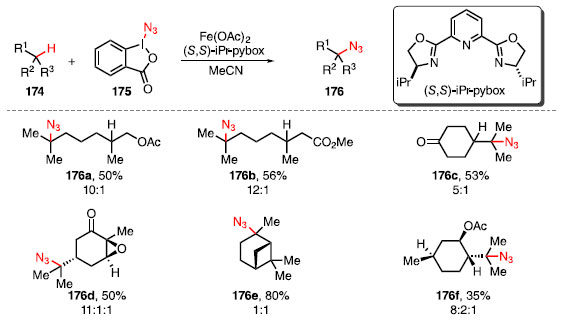 Esquema 37. Produtos de azidação da ligação C-H catalisadas por ferro
A reação de azidação foi seletiva para ligação C-H terciária mais rica em elétrons, resultando em rendimento moderado para o isômero isolado, como em 176a e 176b. Para ambos os exemplos a seletividade favoreceu a ligação C-H mais distante do retirador de elétrons. Tal reatividade também foi observada em compostos cíclicos 176c e a oxidação ocorreu na ligação C-H terciária mais rica eletronicamente, e com uma seletividade maior para a ligação C-H mais distante da cetona, fornecendo o produto em 53% de rendimento. Esse método também se mostrou tolerante à presença de epóxidos e anéis de quatro membros 176d e 176e, também com rendimentos de moderados a bons. Para o produto 176d, a reação foi bastante seletiva, funcionalizando a ligação C-H do grupo isopropila. Para o produto 176e, mesmo com o alto rendimento, não foi seletiva a ponto de o produto não apresentar excesso diastereoisomérico. Em casos em que as ligações C-H eram eletronicamente similares, como 176f, apesar de ser menos acessível estericamente, a flexibilidade conformacional do grupo isopropila promove a azidação seletiva da ligação C-H da mesma.58 O mecanismo da reação, proposto pelo grupo de pesquisas de Hartwig em uma publicação recente,59 mostra que ocorre primeiramente a quebra homolítica da ligação I-N do composto 175, formando os radicais 177 e »N3, e ambos são responsáveis pela abstração do átomo de hidrogênio (do inglês hydrogem atom abstraction, HAA), formando o ácido carboxílico 178, HN3 e o intermediário radicalar 179. No ciclo catalítico do ferro, o [Fen] reage com o 175, podendo formar diretamente o complexo [Fe111]-N3 ou o complexo 180, nesse caso ocorrendo a troca de ligante com HN3, convergindo ao mesmo complexo [FeIn]-N3. Este complexo é o responsável por reagir com o radical 179, fornecendo o produto 176 e o complexo [Fe11], completando o ciclo catalítico (Esquema 38).
 Esquema 38. Mecanismo proposto para a azidação C-H
A seletividade observada por esse sistema permitiu com que a metodologia fosse aplicada na funcionalização em estágio tardio de produtos naturais. A azidação da ciclo-heximida, uma molécula de alta toxicidade, permitiu a obtenção exclusiva de 181 em 40 % de rendimento (Figura 1). O análogo do ácido betulínico, um triterpenóide contendo um sistema trans-decalina A/B, forneceu o produto 182 em um bom rendimento, considerando a complexidade da molécula.
 Figura 1. Produtos de azidação da ligação C-H de produtos naturais
A azidação do derivado de digoxigenina, um esteroide contendo o sistema cis-decalina A/B, resultou no produto 183 em 68% como um sistema trans-decalina A/B. Isso ocorre pois durante a etapa da abstração do átomo de hidrogênio, há a formação de um intermediário radicalar centrado no carbono terciário, e a inserção do grupo azida irá gerar o produto mais estável, que é o sistema trans-decalina A/B. Para o di-hidrodipterocarpol, a abstração da ligação C-H não ocorreu no sistema trans-decalina A/B, e sim na ligação C-H terciária mais disponível estericamente, fornecendo o produto 184. Para um derivado do ácido artemisínico, que está representado como uma lactona, houve a formação de uma mistura entre três produtos de azidação 185a-185c, em que a funcionalização ocorreu nas ligações C-H mais acessíveis estericamente, com a preferência para a maior formação de 185a por ser uma ligação C-H mais rica frente a outros. Por fim, a reação de azidação envolvendo o óxido de di-hidrocariofileno ocorreu para a formação de 186 em 44% de rendimento, sem que ocorresse a abertura do ciclobutano e do epóxido.58 Em 2015, Groves e colaboradores reportaram uma metodologia para a formação de ligações C-N a partir de ligações C-H como na conversão dos compostos 187 em 188 (Esquema 39). Foram utilizados na reação catalisadores a base de manganês, azida de sódio, como fonte de nitrogênio, iodosobenzeno para oxidar o complexo de manganês, e acetato de etila ou metila como solvente.60
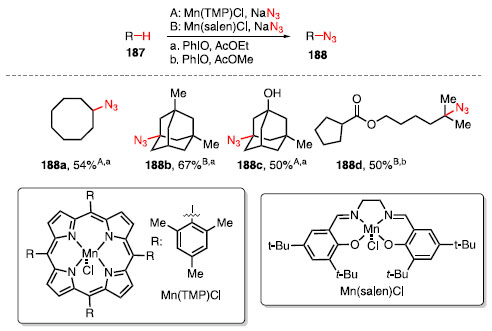 Esquema 39. Azidação C-H utilizando catalisadores de manganês
Os catalisadores de manganês utilizados foram o Mn(TMP)Cl, com o ligante porfirínico, e o Mn(salen)Cl, com ligante sendo uma base de Schiff. O escopo reacional mostra que é possível realizar a reação de azidação de ligações C-H de metilenos, como o caso de 188a, no entanto, como mostrado pelos produtos 188b-188d, a funcionalização de ligação C-H terciária, por ser mais rica em elétrons, é favorecida. Esta metodologia também se mostrou tolerável a outros grupos funcionais como, por exemplo, álcool e éster, para os produtos 188c e 188d. O mecanismo de reação proposto para a reação consiste primeiramente na troca de ligantes, partindo de [Mn111]-L para [Mn111]-N3, seguido da oxidação com PhIO para [MnV](O)-N3, sendo este complexo mais reativo, e realizando o processo de HAA sobre o substrato 187, formando o radical R» e o complexo HO- [MnIV]-N3. Esse complexo reage com o radical formado, fornecendo o produto da azidação 188, e liberando o [Mn111]-L, restaurando o ciclo catalítico (Esquema 40).60
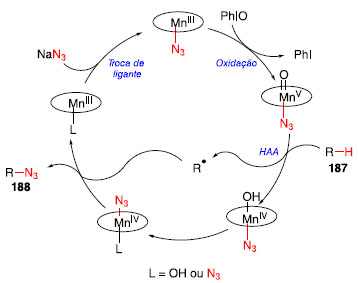 Esquema 40. Mecanismo proposto para a azidação C-H catalisada por manganês
Essa metodologia pôde ser aplicada na modificação de produtos naturais e a reação de azidação catalisada por manganês levou à formação do composto 189, um produto de funcionalização na posição C2 do esclareolídeo (32), com 57% de rendimento e uma razão a:fi de 7,5:1. Esse método também foi eficiente para moléculas frágeis como a artemisinina (12), com a azidação exclusiva na posição C10 como produto 190 em 20% de rendimento (Esquema 41).61
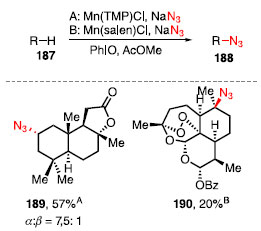 Esquema 41. Reação de azidação aplicada na funcionalização de produtos naturais
Em 2015, White e colaboradores reportaram uma metodologia de formação de ligações C-N de maneira intramolecular a partir de ligações C-H. Nesse trabalho foram utilizados o catalisador a base de manganês, o [Mn(t-BuPc)], um sal de aril iodônio levando à conversão de ésteres sulfâmicos 191 nos produtos 192 (Esquema 42).62
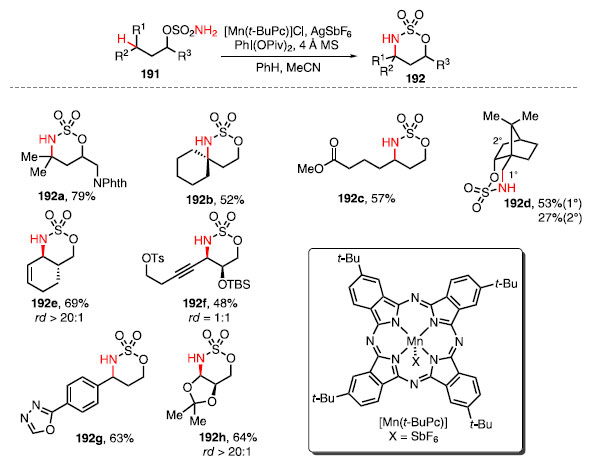 Esquema 42. Escopo de Aminação C-H Intramolecular catalisadas por manganês
A condição reacional desenvolvida possibilitou a funcionalização de ligações C-H primárias, secundárias e terciárias, além de tolerar diversos grupos funcionais, como ftalimidas 192a. Essa técnica mostrou-se efetiva para a formação de azaespirociclo 192b. O catalisador é sensível a efeitos eletrônicos dos substratos, e o composto 192c pôde ser obtido em 57% de rendimento. Grupos retiradores de elétrons mais próximo ao éster sulfâmico reduzem a reatividade. Observou-se também a funcionalização preferencial de posições g formando um anel de seis membros frente a posições b que formam anéis de cinco membros, como observado no composto 192d. Essa metodologia mostrou-se altamente diasteroseletiva para o ciclo-hexeno 192e. O composto 192f foi formado com rendimentos moderados, mostrando que a técnica é tolerante à presença de alcinos. A obtenção do composto 192g mostra que essa metodologia também é tolerante a heterociclos aromáticos como o oxadiazol. Já o composto 192h foi obtido com bom rendimento e seletividade. O mecanismo da reação proposto consiste na formação de imi-noiodinano 193 a partir do éster sulfâmico 191, e o intermediário formado reage com o complexo [MnLn] para a formação do meta-lonitreno 194. O nitreno formado é responsável pela clivagem da ligação C-H, formando o radical 195, que rapidamente se recom-bina, fornecendo o produto 192 e o catalisador [MnLn], restaurando o ciclo catalítico (Esquema 43).62 A etapa da recombinação é rápida a ponto de que essa transformação apresenta estereorretenção, isto é, sem que ocorra a racemização/epimerização do produto, como normalmente ocorre em transformações clássicas envolvendo formação de radical.
 Esquema 43. Mecanismo proposto para a aminação C-H intramolecular
Para os compostos mais complexos percebe-se que a tendência dos substratos mais simples se manteve (Esquema 44). O composto 196, um derivado da picrotoxinina, foi obtido com 57% de rendimento. O derivado do estigmasterol produziu o composto 197 em 66%, com um único disateroisômero.
 Esquema 44. Aminação C-H intramolecular em estágio tardio
O derivado da pleuromutilina forneceu o composto 198 em 84% de rendimento como um único isômero, frente a outros sítios possíveis de funcionalização. O derivado do isoesteviol 199 forneceu o composto 200 em 92% de rendimento formando apenas o diasteroisômero syn. O produto 200 pôde ser facilmente diversificado para formar os compostos 201a e 201b.62 Em 2019, Dauban e colaboradores utilizaram catalisadores a base de ródio [Rh2(CF3CONH)4] e [Rh2(esp)4] para realizar reações de aminação em estágio avançado de ligações C-H utilizando também ésteres sulfâmicos e reagente de iodo hipervalente como oxidante (Esquema 45).63 Algo que chama a atenção é a estrutura ao redor do centro bimetálico do catalisador, lembrando uma "roda de pá" (do inglês paddlewhell).
 Esquema 45. Aminação C-H intramolecular utilizando catalisadores de dirródio
Aplicando a derivados de produtos naturais contendo esqueletos abietanos, a aminação intramolecular dos compostos 202, 204 e 206 resultaram na obtenção dos produtos 203, 205 e 207, respectivamente, como um único estereoisômero. É notório a tolerância dessa metodologia frente a outros grupos funcionais, como uma olefina conjugada ao anel aromático, em 204, e de anel mais rico em elétrons, como em 206.63
FORMAÇÃO DE LIGAÇÃO C-S A ligação C-S apresenta versatilidade para a química orgânica, uma vez que compostos contendo enxofre podem servir de trampolim para a instalação de outras funcionalidades.64,65 Essa versatilidade é evidenciada pela conversão de um único ditiocarbamato derivado da nicotinamida 208 em outros compostos através de reações bem estabelecidas como, por exemplo, eliminação tipo-Chugaev, transferência de azida, remoção do grupo ditiocarbamato, reações radicalares de alilação e deuteração, para obter os respectivos produtos 209-213 (Esquema 46).64
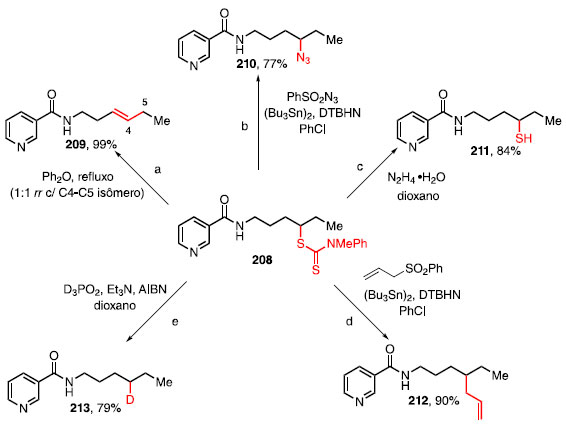 Esquema 46. Diversificação de um derivado da nicotinamida
Algumas das reações para formação de ligação C-S a partir de ligações C-H acontecem via transferência intramolecular de átomos de hidrogênio que é inspirado na reação de Hofmann-Loffler-Freytag (HLF), uma reação radicalar.65,66 Uma abordagem para a formação de ligações C-S a partir da funcionalização de ligações C-H envolve transferência de átomo de hidrogênio de A-ditiocarbamatos, descrita por Alexanian e colaboradores em 2018 (Esquema 47).65
 Esquema 47. Reação geral de ditiocarbamilação e produtos simples de ditiocarbamilação
A formação inicial do ditiocarbamato acontece através do acoplamento do ácido carboxílico com uma amina (via formação do cloreto de ácido) obtendo a amida 214 que, então sofre irradiação de luz azul LED em PhCF3 para a funcionalização da ligação C-H, fornecendo o produto ditiocarbamilado 215.65 A ditiocarbamilação não se limitou à inserção em ligações C-H metilênicas como em 215a e 215b, e a funcionalização em ligações C-H metílicas também foi bem sucedida como demonstrado pela obtenção de 215c e 215d. Por fim, substratos cíclicos também se mostraram compatíveis com essa transformação e o composto 215e foi obtido em 81% de rendimento.64 Produtos naturais e fármacos também foram funcionalizados a partir dessa abordagem (Figura 2). A rimantadina, um agente antiviral que inibe especificamente a replicação do vírus influenza tipo A,67 forneceu o produto 216 com 79% de rendimento. O gemfibrozil, utilizado no tratamento de hiperlipidemias, que são alterações metabólicas que ocorrem quando níveis de lipídeos estão altos na corrente sanguínea,68 possibilitou a realização do produto 217 com 57% de rendimento.
 Figura 2. Derivados de produtos naturais e fármacos com a ditiocarbamilação
O ácido nicotínico, uma vitamina com propriedades hipolipemian-tes, que reduz os níveis de triglicérides e LDL colesterol (lipoproteína de baixa densidade), ambas gorduras armazenadas no corpo, e aumenta o nível de HDL colesterol, uma lipoproteína de alta densidade,69 gerou o ditiocarbamato 218 com 81% de rendimento. Já a progesterona, que é um hormônio esteroide natural, produzido pelo corpo lúteo ovariano após a ovulação e pela placenta durante a gestação,70 foi funcionalizada e o produto 219 foi obtido em 46% de rendimento. O ácido tranexâmico, um fármaco antifibrinolítico, que previne a dissolução do coágulo, levando à redução da perda sanguínea,71 forneceu o produto 220 em bons rendimentos. O ácido linóico, um ácido graxo essencial na dieta humana, sendo o componente principal de muitas gorduras presentes nos alimentos incluindo os óleos extraídos de sementes de soja, milho e girassol,72 foi funcionalizado em 53% de rendimento gerando o composto 221. Uma outra abordagem para a instalação de ligações C-S a partir de ligações C-H via HLF foi descrita por Yang e colaboradores em 2020 (Esquema 48).66 A reação entre N-fluorotosil amida 222 e arildissulfeto 223, catalisada por cobre, 10-phen como ligante, Na2HPO4 como base e índio como aditivo, sob irradiação de luz LED azul, forneceu os compostos 224.
 Esquema 48. Reação de tiolação de aminas alifáticas e exemplos simples
Os dissulfetos substituídos com grupos doadores de elétrons, tais como p-metil e p-metoxi levaram à formação dos produtos 224a e 224b em bons rendimentos, já a presença de grupos retiradores de elétrons como, por exemplo, o grupo nitro, levou a uma diminuição no rendimento para o 224c. Para o produto 224d, o rendimento da tiolação para ligação C-H primária foi de 50%. Cicloalcanos com vários tamanhos de anel tiveram um bom rendimento com a tiolação, como no caso do produto 224e. O grupo adamantino pôde ser funcionalizado e o produto 224f foi obtido em 72% de rendimento. Por fim, essa transformação foi aplicada com sucesso para formar um centro quaternário com um grupo tiol no composto 224g.66 Compostos biologicamente ativos também foram utilizados nessa metodologia (Esquema 49).66 Enquanto a funcionalização de um derivado de 5b-androstano (225) forneceu o composto 226 em 57% de rendimento, a tiolação do derivado de ácido desidroabiético (227), um ácido de resina diterpênica do tipo abietano que possui propriedades antibacterianas, cardiovasculares e antioxidantes, levou à formação do composto 228 em 46% de rendimento como um único diastereoisômero.
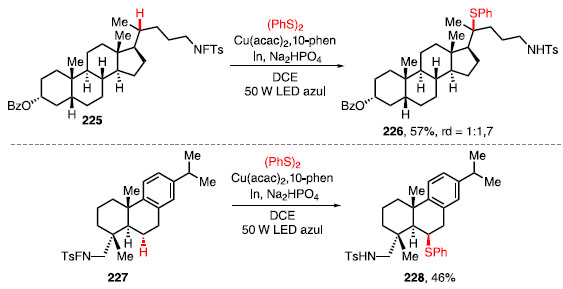 Esquema 49. Reação de tiolação de compostos biologicamente ativos
Em 2016, Alexanian e colaboradores descreveram uma abordagem para a formação de ligação C-S a partir da reação de xantilação intermolecular entre um alcano 229, N-xantilamida 230 em PhCF3, sob irradiação de luz azul LED, para fornecer xantatos de alquila 231 (Esquema 50).64
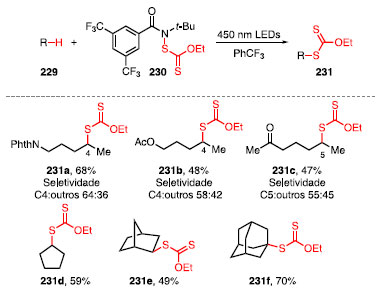 Esquema 50. Reação de xantilação para funcionalização C-H e produtos da reação
A funcionalização de substratos contendo grupos retiradores de elétrons forneceu os compostos 231a-231c em bons rendimentos, com preferência para a xantilação na posição ô em relação à posição g, indicando uma preferência para metilenos mais ricos em elétrons. Logo, para 231a, a seletividade em 64% é a obtenção do produto xantilado na posição ô, indicado em negrito, e 36% é a soma dos produtos de funcionalização de outras posições na mesma molécula. Para 231b e 231c, o mesmo raciocínio se aplica, cujo produtos apresentam uma seletividade em 58% e 55% frente aos subprodutos, respectivamente. Reações com cicloalcanos forneceram produtos com bons rendimentos, como é possível ver com o produto 231d. A xantilação de norbornano forneceu o produto 231e em 49% de rendimento como o único diastereoisômero. A funcionalização do adamantano favoreceu a ligação C-H de carbono terciário, fornecendo como único produto o xantato 231f com rendimento de 70%. Exemplos de xantilação de moléculas complexas derivadas de produtos naturais são mostrados na Figura 3. A funcionalização do esclareolídeo possibilitou a formação do produto 232 com 55% de rendimento de forma seletiva.73
 Figura 3. Xantilação C-H aplicado a produtos naturais
O acetato de androsterona e o 5a-androstanediona são derivados do núcleo dos esteroides androstano e forneceram os produtos de xantilação 233 e 234 com 56% e 44% de rendimento, respectivamente. Por fim, o 5a-colestano, um derivado do núcleo dos esteroides colesterol, foi funcionalizado levando à formação do produto 235 com 60% de rendimento.64 Em 2016, Glorius e colaboradores desenvolveram um método para a instalação de um grupo trifluorometilsulfanil em um grande número de ligações C-H. O tratamento de compostos 236 com PhthSCF3, como o reagente de inserção do grupo SCF3, um fotoca-talisador de irídio, benzoato de sódio, como aditivo, em acetonitrila, sob irradiação de luz azul, forneceu os produtos 237 (Esquema 51).74
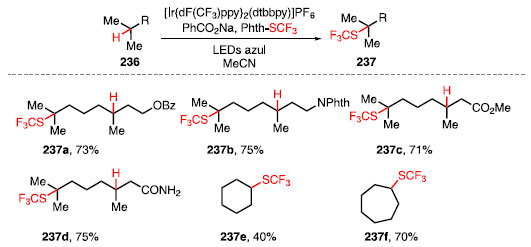 Esquema 51. Reação de trifluorometiltiolação C-H e o escopo
A formação dos compostos 237a-237d mostram que a trifuorometiltiolação ocorre preferencialmente na ligação C-H do carbono terciário mais rico em elétrons com bons rendimentos e alta seletividade, tolerando uma variedade de grupos funcionais, como benzoatos, ftalimidas, ésteres e amidas. Em geral, metilenos não reagiram bem para essa transformação, no entanto, o ciclo-hexano 237e e o ciclo-heptano 237f foram tri-fluorometiltiolados em 40% e 70% de rendimento, respectivamente.74 Esta metodologia também foi utilizada na funcionalização em estágio tardio de moléculas biologicamente ativas (Figura 4). Em todos os casos, o grupo SCF3, foi introduzido seletivamente na ligação C-H mais rica em elétrons, que normalmente é mais suscetível a alterações metabólicas de degradação.74
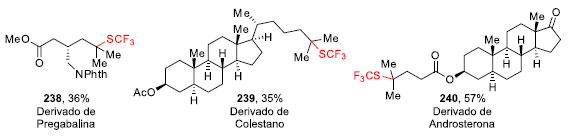 Figura 4. Trifluorometiltiolação aplicado a produtos naturais
A pregabalina é um fármaco anticonvulsivante e antiepilético, e sua funcionalização levou à formação do produto 238 em 36% de rendimento. O colestano, um triterpeno tetracíclico saturado produzido pela diagênese do colesterol e sua presença em amostras ambientais indica vida animal, gerou o produto 239 em 35% de rendimento. Por fim, a androsterona, um hormônio esteroidal, forneceu o produto 240 com 57% de rendimento.
FORMAÇÃO DE LIGAÇÕES C-SI Organossilanos são uma classe de reagentes multifuncionais e valiosos, possuindo aplicabilidade nos campos da química sintética e ciência de materiais.75 Além disso, a substituição bioisostérica de átomos de carbono por silício tem encontrado grande uso em química medicinal nos últimos anos.76 Desde a década de 2000 houve um crescente número de publicações que tratam sobre a sililação de ligações C-H através de sua ativação, catalisada por metais de transição.77 Uma característica notável da ligação C-Si é a sua versatilidade: podem ser facilmente instaladas, funcionalizadas e removidas, além de apresentar estabilidade frente a alguns grupos funcionais, sendo um ótimo material de partida para fornecer novas ligações C-C e C-heteroátomo. Embora a ativação de ligações C-H de carbonos saturados para obter ligações C-Si foi historicamente menos explorada em comparação às ligações C-H de carbonos insaturados, devido à sua menor reatividade, grupos C-SiR3 sofrem reações de oxidação, carboxilação, aminação, halogenação e acoplamento cruzado com maior facilidade.78 Hartwig e colaboradores desenvolveram a síntese de 1,3-dióis através de um protocolo em duas etapas que envolve inicialmente uma sililação intramolecular de ligações C-H secundárias em posição g a hidroxilas seguidas por uma oxidação de Tamao-Fleming (Esquema 52).
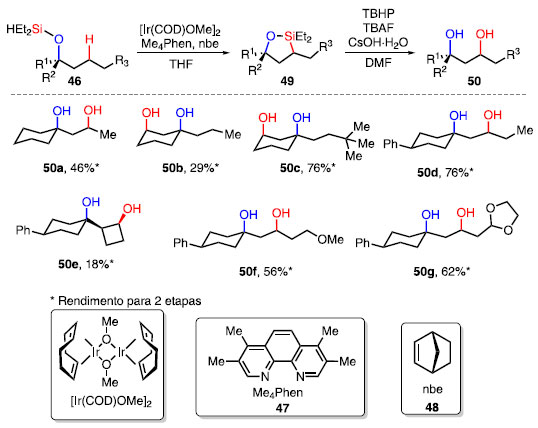 Esquema 52. 1,3-dióis obtidos a partir da sililação C-H e oxidação de Tamao-Fleming
O tratamento de sililéteres 46, com a utilização do catalisador [Ir(COD)OMe]2, Me4Phen como aditivo e norborneno (48), como aceptor de H2 em THF leva à formação do intermediário 49. Em seguida, a oxidação do composto 49 com TBHP, CsOH»H2O e TBAF em DMF, forneceu o 1,3-diol 50.26,79 Álcoois cíclicos terciários sofrem a funcionalização em ligações C-H secundárias, podendo fornecer uma mistura de isômeros como em 50a e 50b, em 46% e 29% de rendimento, respectivamente. O efeito estéreo de grupos presentes no substrato tem grande influência na sítiosseletividade da reação de sililação, e o grupo volumoso t-butil na cadeia lateral do ciclo-hexano dirige a reação para a ligação C-H axial, fornecendo 50c em 76% de rendimento como único isômero. A presença de um grupo fenila em posição equatorial em 50d faz com que a funcionalização ocorresse na cadeia lateral, fornecendo o diol em 76% como único isômero. A presença de um substituinte cíclico lateral 50e, ou de grupos funcionais oxigenados em 50f e 50g não impedem o prosseguimento da reação, gerando os produtos em 18%, 56% e 62%, respectivamente. O mecanismo da reação de sililação de ligações C-H foi proposto por Sunoj e colaboradores a partir de cálculos computacionais (Esquema 53).80 O pré-catalisador [Ir(COD)OMe]2, na presença do ligante Me4Phen e norborneno (48), é convertido no catalisador ativo 241, seguido da adição oxidativa da ligação Si-H, obtendo-se o intermediário 242, que sofre então inserção migratória do norborneno coordenado, formando o complexo 243. Com a transferência do próton ligado ao átomo de irídio para o norborneno, tem-se a formação do intermediário 244, seguido da conversão para o composto 245, em que há a interação agóstica entre a ligação C-H do norborneno e o centro metálico.
 Esquema 53. Ciclo catalítico da sililação C-H
A primeira etapa da eliminação redutiva é impulsionada por 48, fornecendo o complexo 247 e norbornano (246). A partir de 247, ocorre a adição oxidativa da ligação C-H, gerando o intermediário 248 que, após a conversão para o correspondente 249, sofre eliminação redutiva, fornecendo o produto 49 e o catalisador 241. Segundo os cálculos computacionais, a etapa de eliminação redutiva para a formação da ligação C-Si é a etapa determinante, com o maior valor de energia do estado de transição.80 O colesterol é um dos principais esteroides sintetizados pelos animais, auxiliando no funcionamento de suas células, além de ser precursor de diferentes hormônios esteróides e de vitaminas. O protocolo de sililação de ligação C-H seguida por oxidação pôde ser aplicado na funcionalização do derivado do colesterol 250, levando à formação do composto 251 em 68% de rendimento para três etapas (Esquema 54).81
 Esquema 54. Sililação de ligação C-H seguida por sua oxidação em derivado do colesterol
Hartwig e colaboradores puderam estender a metodologia para a sililação seletiva de ligações C-H em posição b a aminas alifáticas, catalisadas por irídio, utilizando a fenantrolina 253 como ligante, nas condições semelhantes às utilizadas no protocolo estabelecido no trabalho anterior (Esquema 55).82 Os substratos utilizados 252 foram sililmetilaminas, as quais formam produtos cíclicos de cinco membros 254 que posteriormente puderam ser oxidadas para formar ligações C-O.
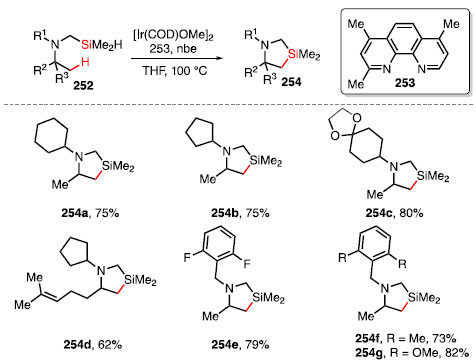 Esquema 55. Escopo de produtos obtidos a partir da sililação de ligações C-H em posição β a aminas alifáticas
A funcionalização ocorre de forma seletiva em aminas que possuem ciclos ligados diretamente ao nitrogênio, fornecendo os produtos 254a e 254b em bom rendimento. Substratos com grupos funcionais como cetais 254c, olefinas 254d, haleto de arila 254e e grupos doadores de elétrons em posição orto 254f, 254g se mostraram tolerantes às condições reacionais empregadas, apresentando bons rendimentos.82 O derivado do propranolol 255, um dos 200 fármacos mais prescritos nos Estados Unidos em 2016 por conta de seu tratamento contra a ansiedade,83 foi submetido à sililação de sua ligação C-H b ao nitrogênio, fornecendo o produto 256 em 90% de rendimento. Após oxidação nas condições de Tamao-Fleming seguida de proteção com o grupo Boc, o produto 258 foi obtido em 54% de rendimento para 2 etapas (Esquema 56).
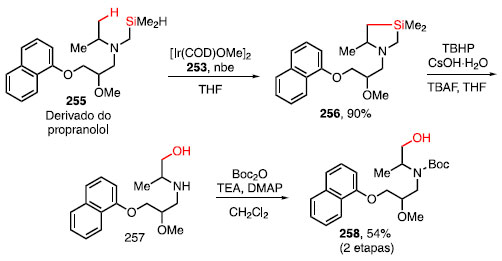 Esquema 56. Combinação entre a sililação de ligação C-H seguida por oxidação da ligação C-Si em derivado de fármaco
O grupo de pesquisas de Hartwig desenvolveu uma reação de sililação intramolecular enantiosseletiva através da funcionalização de ligações C-H metílicas de substratos 259, utilizando [Ir(COD) OMe]2, ligante quiral 260 e nbe, levando à formação dos produtos 261 (Esquema 57).84
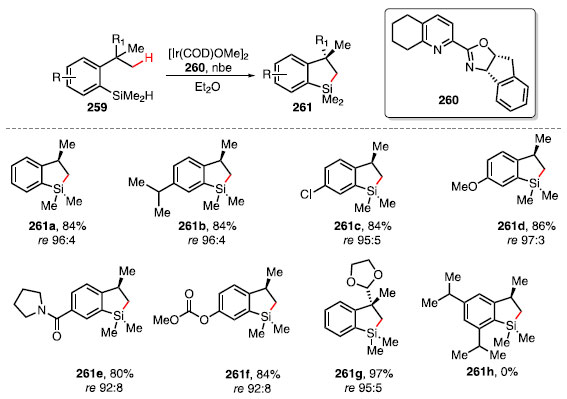 Esquema 57. Escopo de produtos obtidos a partir da sililação de ligações C-H enantiosseletiva
Essa metodologia permite a formação da ligação C-Si a partir da reação de dessimetrização enantioseletiva das metilas de um grupo isopropil para formar um centro de quiralidade em um anel de cinco membros.85 Os estudos mostraram que produtos provenientes de substratos não substituídos 261a, substituintes alquila 261b, e haletos de arila 261c são obtidos com bons rendimentos e razões enantioméricas. Substratos que possuem grupos funcionais com oxigênio ou nitrogênio, como alcóxi 261d, amida 261e, carbonato 261f e cetal 261g se monstraram toleráveis ao sistema reacional. Em geral, a metodologia é tolerante a diversos grupos funcionais, com a capacidade de gerar centros de quiralidade quarternários. No entanto, a reação não ocorreu em substratos contendo grupos volumosos em posições meta e orto, como visto em 261h.84 A metodologia foi aplicada no derivado do ácido desidroabiético 262, um diterpeno com propriedades antivirais, antitumorais e atividade citotóxica,86 levando à formação do composto 263 com 78% de rendimento e razão diastereoisomérica de 96:4 (Esquema 58).
 Esquema 58. Sililação C-H enantiosseletiva em derivado do ácido desidroabiético
Hartwig e colaboradores também conseguiram executar a sililação intramolecular de ligações C-H primárias em posição S a álcooisterciários 264, quando catalisadas pelo RhCl(Xantphos) e nbe em THF, obtendo os intermediários 265 (Esquema 59). Após uma oxidação de Tamao-Fleming os 1,4-dióis 266 são obtidos.87
 Esquema 59. Escopo de 1,4-dióis obtidos a partir da sililação C-H, seguido da oxidação de Tamao-Fleming
Álcoois terciários contendo o grupo isobutil ou o ciclo-hexano são seletivamente funcionalizados e a ligação C-H primária são sililadas para fornecer os produtos 266a e 266b. A metodologia mostrou-se compatível inclusive com a funcionalização de ligações C-H em posição neopentílica e o produto 266c foi obtido em 66% de rendimento. Na ausência de ramificações na cadeia lateral, a reação não forneceu quantidade considerável do produto arilado 266d. Grupos funcionais como haleto de arila 266e e um grupo metoxiarila 266f se mostraram compatíveis com a reação, fornecendo os produtos em 74% e 84% de rendimento, respectivamente.87 A transformação em estágio avançado do derivado do oxisterol (267), uma molécula que atua como sinalizador por ter importância fisiológica, como o transporte de lipídeos,88 se mostrou eficaz e o produto 268 foi obtido em 44% de rendimento após três etapas (Esquema 60).
 Esquema 60. Sililação da ligação C-H em derivado do oxisterol seguida por sua oxidação
FORMAÇÃO DE LIGAÇÕES C-HALOGÊNIO Compostos orgânicos halogenados apresentam grande interesse farmacológico e sintético, podendo atuar como intermediários na inserção de outros grupos funcionais através de reações clássicas como, por exemplo, substituição nucleofílica alifática e reações de acoplamento cruzado.89,90 A criação de uma ligação com um halogênio a partir de uma ligação C-H ainda é um grande desafio para a síntese orgânica e poucos exemplos podem ser encontrados como alternativa para os métodos clássicos. Estudos realizados por Groves e colaboradore mostraram que porfirinas de manganês podem ser utilizadas para a formação de ligações C-Cl e C-F em ligações C-H não ativadas (Esquema 61).91
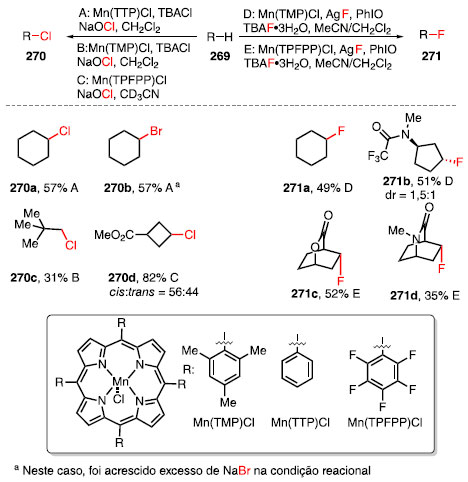 Esquema 61. Halogenações C-H utilizando catalisadores de manganês
A metodologia forneceu os produtos clorados e bromados 270a e 270b em rendimentos moderados, via procedimento A, assim como a cloração de neo-pentano, fornecendo 270c em 31%, via procedimento B. Ao utilizar o catalisador Mn(TPFPP)Cl, deficiente em elétrons, substratos contendo grupos retiradores de elétrons puderam ser funcionalizados, como o éster 270d, em 82% de rendimento via procedimento C. Utilizando o procedimento D, os compostos 271a e 271b foram obtidos em rendimentos moderados, com a relação diastereoisomérica de 1,5:1 para o produto 271b, com o produto contendo a relação dos substituintes 1,3-anti favorecida. Ao utilizar o procedimento E, biciclos contendo lactonas e lactamas puderam ser funcionalizadas, fornecendo 271c e 271d em 52% e 35% de rendimento, respectivamente, como isômeros exclusivos. Essa metodologia foi aplicada na funcionalização do esclareolídeo (32) (Esquema 62). Nesse estudo, foram realizadas tanto a cloração quanto a fluoração, fornecendo os compostos 272 e 273 em 42% de rendimento para ambos os casos, com a funcionalização preferencial na posição C2. Para a cloração C-H, o material de partida recuperado foi reagido em mais dois ciclos, fornecendo o produto 272.
 Esquema 62. Cloração e fluoração C-H do esclareolídeo
Em 2021, Hartwig e colaboradores reportaram uma reação de cloração radicalar, na presença do reagente 175 para gerar o o-iodobenzoiloxi radical, que é seletivo para a abstração do átomo de hidrogênio, e o complexo (phen)CuCl2, que entrega o átomo de cloro ao radical. Essa condição permitiu a conversão de uma ligação C-H terciária de compostos 274 ao correspondente produto clorado 275 (Esquema 63).92
 Esquema 63. Escopo de cloração C-H catalisada por cobre
Ligações C-H benzílicas e metilenos não são reativos nesse protocolo, e o produto 275a foi obtido em 75% de rendimento. A reação mostrou-se tolerante também a ésteres borônicos e ácidos carboxílicos, fornecendo os compostos 275b e 275c em 37% e 90% de rendimento, respectivamente. O benzoato de n-pentila, sem a ligação C-H terciária, não forneceu o produto esperado 275d. A reação utilizando 4-metilpentil aril éster como substrato forneceu a cloração da ligação C-H terciária em rendimentos bons a excelentes. Variando o grupo aril, a reação é tolerante a grupos doadores e retiradores de elétrons, com 100% de rendimento para 275e-g, 88% para 275h e 57% para 275i e 83% para o tiazol 275j. A reação de cloração na sulbactama, um inibidor de b-lactamase, uma enzima que destrói os antibióticos, forneceu o produto 276 em 94% de rendimento. A funcionalização da talidomida, um fármaco utilizado para tratamento de mielona, e o responsável pela má formação estrutural de fetos durante os meados do século XX, gerou o composto 277 em 98% de rendimento (Figura 5).
 Figura 5. Cloração C-H aplicado a fármacos e produtos naturais
A cloração da betulina, um triterpeno que apresenta atividade anti-cancer, levou à formação do produto 278 em 62% de rendimento, sendo seletivo a apenas uma ligação C-H terciária, de um total de sete ligações de mesma característica. Por fim, o ácido giberélico, um diterpeno pentacíclico, com uso para crescimento de plantas e fungos, forneceu o produto 279 em 71%, sendo seletivo a uma ligação terciária frente a outras quatro.
CONCLUSÕES Ligações C-H foram historicamente tratadas como inertes e sua funcionalização se dava apenas quando próximas a um grupo funcional. Nos últimos 20 anos, nossa comunidade tem visto uma mudança de paradigma e a funcionalização de ligações C-H vem ganhando espaço importante. Com o desenvolvimento de estratégias cada vez mais quimio- e sitiosseletivas, muitos dos métodos atuais são compatíveis com uma série de grupos funcionais que antes não eram tolerados, o que tem possibilitado a funcionalização de moléculas complexas como, por exemplo, produtos naturais e fármacos. O Brasil é um país com uma Química de Produtos Naturais riquíssima e uma Química de Catálise vibrante. Com esse artigo de revisão, gostaríamos de encorajar os pesquisadores dessas áreas a estabelecer colaborações e esperamos que, com isso, novos espaços químicos possam ser acessados no futuro.
AGRADECIMENTOS Agradecemos à Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) pelo apoio financeiro (Processo 2018/04837-6) e por uma bolsa de iniciação científica para E. C. S. Rocha (Processo 2021/01918-8). Nós também agradecemos ao CNPq, CAPES e FAEPEX-UNICAMP (2575/21).
REFERÊNCIAS 1. Koehn, F. E.; Carter; G. T.; Nat. Rev. Drug Discovery 2005, 4, 206 [Crossref]; Newman, D. J.; Cragg, G. M.; J. Nat. Prod. 2016, 79, 629. [Crossref] 2. Shugrue, C. R.; Miller, S. J.; Chem. Rev. 2017, 117, 11894 [Crossref]; Borgel, J.; Ritter, T.; Chem. 2020, 6, 1877 [Crossref]; Hong, B.; Luo, T.; Lei, X.; ACS Cent. Sci. 2020, 6, 622 [Crossref]; Kim, K. E.; Kim, A. N.; McCormick, C. J.; Stoltz, B. M.; J. Am. Chem. Soc. 2021, 143, 16890 [Crossref]; Zhang, L.; Ritter, T.; J. Am. Chem. Soc. 2022, 144, 2399. [Crossref] 3. Robles, O.; Romo, D.; Nat. Prod. Rep. 2014, 31, 318 [Crossref]; Blakemore, D. C.; Castro, L.; Churcher, I.; Rees, D. C.; Thomas, A. W.; Wilson, D. M.; Wood, A.; Nat. Chem. 2018, 10, 383. [Crossref] 4. IUPAC. Compendium of Chemical Terminology, 2nd ed. (the "Gold Book"). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford, 1997. Online version (2019-) created by S. J. Chalk. ISBN 0-9678550-9-8. 5. Shenvi, R. A.; O'Malley, D. P.; Baran, P. S.; Acc. Chem. Res. 2009, 42, 530. [Crossref] 6. Site-Selective Catalysis; Kawabata, T., ed.; Springer: New York, 2016. [Crossref] 7. Azambuja, F.; Correia, C. R. D.; Quim. Nova 2011, 34, 1779; Yamaguchi, J.; Yamaguchi, A. D.; Itami, K.; Angew. Chem., Int. Ed. 2012, 51, 8960 [Crossref]; Hartwig, J. E; Acc. Chem. Res. 2017, 50, 549 [Crossref]; Karimov, R. R.; Hartwig, J. F.; Angew. Chem., Int. Ed. 2018, 57, 4234 [Crossref]; White, M. C.; Zhao, J.; J. Am. Chem. Soc. 2018, 140, 13988 [Crossref]; Rogge, T.; Kaplaneris, N.; Chatani, N.; Kim, J.; Chang, S.; Punji, B.; Schafer, L. L.; Musaev, D. G.; Wencel-Delord, J.; Roberts, C. A.; Sarpong, R.; Wilson, Z. E.; Brimble, M. A.; Johansson, M. J.; Ackermann, L.; Nat. Rev. Methods Primers 2021, 1, 44 [Crossref]; Carvalho, R. L.; Dias, G. G.; Pereira, C. L. M.; Ghosh, P.; Maiti, D.; da Silva Jr., E. N.; J. Braz. Chem. Soc. 2021, 32, 917. [Crossref] 8. Wencel-Delord, J.; Glorius, F.; Nat. Chem. 2013, 5, 369 [Crossref]; Cernak, T.; Dykstra, K. D.; Tyagarajan, S.; Vachal, P.; Krska, S. W.; Chem. Soc. Rev. 2016, 45, 546 [Crossref]; Genovino, J.; Sames, D.; Hamann, L. G.; Touré, B. B.; Angew. Chem., Int. Ed. 2016, 55, 14218 [Crossref]; Moir, M.; Danon, J. J.; Reekie, T. A.; Kassiou, M.; Expert Opin. Drug Discov. 2019, 11, 1137 [Crossref]; Romero, E.; Jones, B. S.; Hogg, B. N.; Casamajo, A. R.; Hayes, M. A.; Flitsch, S. L.; Turner, N. J.; Scnepel, C.; Angew. Chem., Int. Ed. 2021, 60, 16824 [Crossref]; Guillemard, L.; Kaplaneris, N.; Ackermann, L.; Johansson, M. J.; Nat. Rev. Chem. 2021, 5, 522. [Crossref] 9. Huang, Z.; Dong, G.; Acc. Chem. Res. 2017, 50, 465. [Crossref] 10. Gormisky, P. E.; White, M. C.; J. Am. Chem. Soc. 2013, 135, 14052. [Crossref] 11. Tadross, P. M.; Jacobsen, E. N.; Nat. Chem. 2012, 4, 963. [Crossref] 12. Breslow, R.; Winnik, M. A.; J. Am. Chem. Soc. 1969, 91, 3083 [Crossref]; Breslow, R.; Baldwin, S. W.; J. Am. Chem. Soc. 1970, 92, 732 [Crossref]; Breslow, R.; Acc. Chem. Res. 1980, 13, 170. [Crossref] 13. Shilov, A. E.; Shul'pin, G. B.; Chem. Rev. 1997, 97, 2879. [Crossref] 14. Barton, D. H. R.; Gastinger, M. J.; Motherwell, W. B.; J. Chem. Soc., Chem. Commun. 1983, 41 [Crossref]; Barton, D. H. R.; Doller, D.; Acc. Chem. Res. 1992, 25, 504. [Crossref] 15. Adam, W.; Curci, R.; Edwards, J. O.; Acc. Chem. Res. 1989, 22, 205 [Crossref]; Bovicelli, P.; Lupattelli, P.; Mincione, E.; Prencipe, T.; Curci, R.; J. Org. Chem. 1992, 57, 5052 [Crossref]; Curci, R.; D'Accolti, L.; Fusco, C.; Acc. Chem. Res. 2006, 39, 1. [Crossref] 16. Li, Z.; Feiten, H. -J.; Chang, D.; Duetz, W. A.; van Beilen, J. B.; Witholt, B.; J. Org. Chem. 2001, 66, 8424. [Crossref] 17. Chen, K.; Baran, P. S.; Nature, 2009, 459, 824 [Crossref]; McMurray, L. ; O'Hara, F.; Gaunt, M. J.; Chem. Soc. Rev. 2011, 40, 1885 [Crossref]; Gutekunst, W. R.; Baran, P. S.; Chem. Soc. Rev. 2011, 40, 1976 [Crossref]; Abrams, D. J.; Provencher, P. A.; Sorensen, E. J.; Chem. Soc. Rev. 2018, 47, 8925 [Crossref]; Kanda, Y.; Nakamura, H.; Umemiya, S.; Puthukanoori, R. K.; Appala, V. R. M.; Gaddamanugu, G. K.; Paraselli, B. R.; Baran, P. S.; J. Am. Chem. Soc. 2020, 142, 10526 [Crossref]; Lam, N. Y. S.; Wu, K.; Yu, J. -Q.; Angew. Chem., Int. Ed. 2021, 60, 15767. [Crossref] 18. Para referências abordando outras aplicações, ver: Beydoun, K.; Zaarour, M. ; Williams, J. A. G.; Doucet, H.; Guerchais, V.; Chem. Commun. 2012, 48, 1260 [Crossref]; Segawa, Y.; Maekawa, T.; Itami, K.; Angew. Chem., Int. Ed. 2015, 54, 66 [Crossref]; Droge, T.; Notzon, A.; Frohlich, R.; Glorius, F.; Chem. Eur. J. 2011, 17, 11974 [Crossref]; Liu, T.; Li, D. -Q.; Wang, S. -Y.; Hu, Y. -Z.; Dong, X. -W.; Liu, X. -Y.; Che, C. -M.; Chem. Commun. 2014, 50, 13261 [Crossref]; Kondo, Y.; García-Cuadrado, D.; Hartwig, J. F.; Boaen, N. K.; Wagner, N. L.; Hillmyer, M. A.; J. Am. Chem. Soc. 2002, 124, 1164 [Crossref]; Williamson, J. B.; Lewis, S. E.; Johnson III, R. R.; Manning, I. M.; Leibfarth, F. A.; Angew. Chem., Int. Ed. 2019, 58, 8654 [Crossref]; Chen, L.; Malollari, K. G.; Uliana, A.; Sanchez, D.; Messersmith, P. B.; Hartwig, J. F.; Chem 2021, 7, 137. [Crossref] 19. Brückl, T.; Baxter, R. D.; Ishihara, Y.; Baran, P. S.; Acc. Chem. Res. 2012, 45, 826. [Crossref] 20. Zaitsev, V. G.; Shabashov, D.; Daugulis, O.; J. Am. Chem. Soc. 2005, 127, 13154 [Crossref]; Shabashov, D.; Daugulis, O.; J. Am. Chem. Soc. 2010, 132, 3965 [Crossref]; Daugulis, O.; Roane, J.; Tran, L. D.; Acc. Chem. Res. 2015, 48, 1053. [Crossref] 21. Corbet, M.; De Campo, F.; Angew. Chem., Int. Ed. 2013, 52, 9896 [Crossref]; Rouquet, G.; Chatani, N.; Angew. Chem. Int. Ed. 2013, 52, 11726 [Crossref]; Aihara, Y.; Chatani, N.; J. Am. Chem. Soc. 2014, 136, 898 [Crossref]; Sambiagio, C.; Schonbauer, D.; Blieck, R.; Huy, T. -D.; Pototschnig, G.; Schaaf, P.; Wiesinger, T.; Zia M. F. Wencel-Delord, J.; Besset, T.; Maes, B. U. W.; Schnürch, M.; Chem. Soc. Rev. 2018, 47, 6603 [Crossref]; Rej, S.; Ano, Y.; Chatani, N.; Chem. Rev. 2020, 120, 1788 [Crossref]; Murali, K.; Machado, L. A.; Carvalho, R. L.; Pedrosa, L. E; Mukherjee, R.; da Silva Jr., E. N.; Maiti, D.; Chem. Eur. J. 2021, 27, 12453. [Crossref] 22. Chen, K.; Eschenmoser, A.; Baran, P. S.; Angew. Chem., Int. Ed. 2009, 48, 9705 [Crossref]; Newhouse, T.; Baran, P. S.; Angew. Chem., Int. Ed. 2011, 50, 3362 [Crossref]; Hartwig, J. F.; Larsen, M. A.; ACS Cent. Sci. 2016, 2, 281 [Crossref]; Margrey, K. A.; Czaplyski, W. L.; Nicewicz, D. A.; Alexanian, E. J.; J. Am. Chem. Soc. 2018, 140, 4213 [Crossref]; Costas, M.; Chem. Rec. 2021, 21, 4000 [Crossref]; Fazekas, T. J.; Alty, J. W.; Neidhart, E. K.; Miller, A. S.; Leibfarth, F. A.; Alexanian, E. J.; Science, 2022, 375, 545. [Crossref] 23. Kim, K. E.; Adams, A. M.; Chiappini, N. D.; Du Bois, J.; Stoltz, B. M.; J. Org. Chem. 2018, 83, 3023. [Crossref] 24. Novak, R.; Ann. N. Y. Acad. Sci. 2011, 1241, 71. [Crossref] 25. Ma, X.; Kucera, R.; Goethe, O. F.; Murphy, S. K.; Herzon, S. B.; J. Org. Chem. 2018, 83, 6843. [Crossref] 26. Simmons, E. M.; Hartwig, J. F.; Nature 2012, 483, 70 [Crossref]; Hartwig, J. F.; Romero, E. A.; Tetrahedron 2019, 75, 4059 [Crossref]. 27. Trost, B. M.; Toste, F. T.; Pinkerton, A. B.; Chem. Rev. 2001, 101, 2067 [Crossref]; Schmidt, N. G.; Eger, E.; Kroutil, W.; ACS Catai. 2016, 6, 4286. [Crossref] 28. Chauvin, Y.; Angew. Chem., Int. Ed. 2006, 45, 3740 [Crossref]; Schrock, R. R.; Angew. Chem., Int. Ed. 2006, 45, 3748 [Crossref]; Grubbs, R. H.; Angew. Chem., Int. Ed. 2006, 45, 3760. [Crossref] 29. https://www.nobelprize.org/prizes/chemistry/2010/heck/lecture/, acessada em Maio de 2022.; Suzuki, A.; Angew. Chem., Int. Ed. 2011, 50, 6722 [Crossref]; Negishi, E.; Angew. Chem., Int. Ed. 2011, 50, 6738. [Crossref] 30. Gusevskaya, E. V.; Quim. Nova 2003, 26, 242 [Crossref]; Basu, D.; Kumar, S.; Sudhir, S.; Bandichhor, R.; J. Chem. Sci. 2018, 130, 71. [Crossref] 31. Gao, P.; Guo, W.; Xue, J.; Zhao, Y.; Yuan, Y.; Xia, Y.; Shi, Z.; J. Am. Chem. Soc. 2015, 137, 12231. [Crossref] 32. Mengogi, F.; Lichtner, M.; Battinelli, L.; Marzi, M.; Mastroianni, C. M.; Vullo, V.; Mazzanti, G.; Planta Med. 2002, 68, 111. [Crossref] 33. Peng, J.; Chen, C.; Xi, C.; Chem. Sci. 2016, 7, 1383. [Crossref] 34. https://www.wada-ama.org/sites/default/files/resources/files/WADA_ Prohibited_List_2006_EN.pdf acessada em Maio de 2022. 35. Graebin, C. S.; Verli, H.; Guimarães, J. A.; J. Braz. Chem. Soc. 2010, 21, 1595. [Crossref] 36. Willcox, D.; Chappell, B. G. N.; Hogg, K. F.; Calejja, J.; Smalley, A. P.; Gaunt, M. J.; Science 2016, 354, 851. [Crossref] 37. Apter, A. J.; Reisine, S. T.; Willard, A.; Clive, J.; Wells, M.; Metersky, M. ; McNally, D.; ZuWallack, R. L.; J. Allergy Clin. Immunol. 1996, 98, 295. [Crossref] 38. Tuttle, R. R.; Mills, J.; Circ Res. 1975, 36, 185. [Crossref] 39. Tanaka, K.; Ewing, W. R.; Yu, J.-Q.; J. Am. Chem. Soc. 2019, 141, 15494. [Crossref] 40. Saini, V.; Stokes, B. J.; Sigman, M. S.; Angew. Chem., Int. Ed. 2013, 52, 11206. [Crossref] 41. Chu, J. C. K.; Rovis, T.; Nature 2016, 539, 272. [Crossref] 42. Tassone, D. M.; Boyce, E.; Guyer, J.; Nuzum, D.; Clin. Ther. 2007, 29, 26. [Crossref] 43. Liao, K.; Pickel, T. C.; Boyarskikh, V.; Bacsa, J.; Musaev, D. G.; Davies, H. M. L.; Nature 2017, 551, 609. [Crossref] 44. Davies, H. M. L.; Antoulinakis, E. G.; J. Organomet. Chem. 2001, 617618, 47. [Crossref] 45. Perry, I. B.; Brewer, T. F.; Sarver, P. J.; Schultz, D. M.; DiRocco, D. A.; MacMillan, D. W. C.; Nature 2018, 560, 70. [Crossref] 46. Singh, S.; Gupta, P.; Gupta, J.; Curr Drug Discovery Technol 2020, 17, 619. [Crossref] 47. Xu, Y.; Yan, G.; Ren, Z.; Dong, G.; Nat. Chem. 2015, 7, 829. [Crossref] 48. Chen, M. S.; White, M. C.; Science 2007, 318, 783 [Crossref]; Chen, M. S. ; White, M. C.; Science 2010, 327, 566. [Crossref] 49. White, M. C.; Doyle, A. G.; Jacobsen, E. N.; J. Am. Chem. Soc. 2001, 123, 7194 [Crossref]; Kal, S.; Xu, S.; Que Jr., L.; Angew. Chem., Int. Ed. 2020, 59, 7332. [Crossref] 50. Bigi, M. A.; Liu, P.; Zou, L.; Houk. K. N.; White, M. C.; Synlett 2012, 23, 2768 [Crossref]; Nishimura, K.; Hitotsuyanagi, Y.; Sugeta, N.; Sakakura, K. -i.; Fujita, K.; Fukaya, H.; Aoyagi, Y.; Hasuda, T. ; Kinoshita, T.; He, D. -H.; Otsuka, H.; Takeda, Y.; Takeya, K.; Tetrahedron 2006, 62, 1512. [Crossref] 51. Howell, J. M.; Feng, K.; Clark, J. R.; Trzepkowski L. J.; White, M. C.; J. Am. Chem. Soc. 2015, 137, 14590 [Crossref]; Nanjo, T.; Lucca, E. C., Jr.; White, M. C.; J. Am. Chem. Soc. 2017, 139, 14586. [Crossref] 52. Zhao, J.; Nanjo, T.; Lucca, E. C., Jr.; White, M. C.; Nat. Chem. 2019, 11, 213 [Crossref]; Chambers, R. K.; Zhao, J.; Delaney, C. P.; White M. C.; Adv. Synth. Catal. 2020, 362, 417. [Crossref] 53. Zhao, R.; Chen, X.-Y.; Wang, Z.-X.; Org. Lett. 2021, 23, 1535. [Crossref] 54. Milan, M.; Carboni, G.; Salamone, M.; Costas, M.; Bietti, M.; ACS Catal. 2017, 7, 5903. [Crossref] 55. Tang, Y.; Qin, Y.; Meng, D.; Li, C.; Wei, J.; Yang, M.; Chem. Sci. 2018, 9, 6374. [Crossref] 56. Vitaku, E.; Smith, D. T.; Njardarson, J. T.; J. Med. Chem. 2014, 57, 10257 [Crossref]; Das, P.; Delost, M. D.; Qureshi, M. H.; Smith, D. T.; Njardarson, J. T.; J. Med. Chem. 2019, 62, 4265 [Crossref]; Scott, K. A.; Qureshi, M. H.; Cox, P. B.; Marshall, C. M.; Bellaire, B. C.; Wilcox, M.; Stuart, B. A. R.; Njardarson, J. T.; J. Med. Chem. 2020, 63, 15449. [Crossref] 57. Huang, X.; Groves, J.T.; ACS Catal. 2016, 6, 751 [Crossref]; Park, Y.; Kim, Y.; Chang, S.; Chem. Rev. 2017, 117, 9247 [Crossref]; Clark, J. R.; Feng, K.; Sookezian, A.; White, M.C.; Nat. Chem. 2018, 10, 583 [Crossref]; Ide, T.; Feng, K.; Dixon, C. F.; Teng, D.; Clark, J. R.; Han, W.; Wendell, C. I.; Koch, V.; White, M. C.; J. Am. Chem. Soc. 2021, 143, 14969. [Crossref] 58. Sharma, A.; Hartwig, J. F.; Nature 2015, 517, 600 [Crossref]; Karimov, R. R.; Sharma, A.; Hartwig, J. F.; ACS Cent. Sci. 2016, 2, 715. [Crossref] 59. Day, C.S.; Fawcett, A.; Chatterjee, R.; Hartwig, J.F.; J. Am. Chem. Soc. 2021, 143, 16184. [Crossref] 60. Huang, X.; Bergsten, T. M.; Groves, J. T.; J. Am. Chem. Soc. 2015, 137, 5300. [Crossref] 61. Miller, L. H.; Su, X.; Cell 2011, 146, 855. [Crossref'] 62. Paradine, S. M.; Griffin, J. R.; Zhao, J.; Petronico, A. L.; Miller, S. M.; White, M. C.; Nat. Chem. 2015, 7, 987. [Crossref] 63. Lapuh, M. I.; Dana, A.; di Chenna, P. H.; Darses, B.; Durán, F. J.; Dauban, P.; Org. Biomol. Chem. 2019, 17, 4736. [Crossref] 64. Czaplyski, W. L.; Na, C. G.; Alexanian, E. J.; J. Am. Chem. Soc. 2016, 138, 13854. [Crossref] 65. Na, C. G.; Alexanian, E. J.; Angew. Chem., Int. Ed. 2018, 57, 13106. [Crossref] 66. Qin, Y.; Han, Y.; Tang, Y.; Wei, J.; Yang, M.; Chem. Sci. 2020, 11, 1276. [Crossref] 67. Ferreira, S. S.; Tese de Mestrado, Universidade Estadual de Campinas, Brasil, 2013. [Crossref] 68. Schulz, I.; Arq. Bras. Endocrinol Metab. 2006, 50, 2. [Crossref] 69. Santos, R. D.; Arq. Bras. Cardiol. 2005, 85. [Crossref] 70. Vigo, F.; Lubianca, J. N.; Corleta, H. E.; Femina 2011, 39, 3. [Crossref] 71. Luz, L.; Sankarankutty, A.; Passos, E.; Rizoli, S.; Fraga, G.P.; Nascimento Jr., B.; Rev. Col. Bras. Cir. 2012, 39, 077. [Crossref] 72. Menezes, B. S.; Augusto, M. M. M.; Vetor, Rio Grande 2014, 24, 14. [Crossref] 73. Arakawa, N. S.; Tese de Doutorado, Universidade de São Paulo, Brasil, 2007. [Crossref] 74. Mukherjee, S.; Maji, B.; Tlahuext-Aca, A.; Glorius, F.; J. Am. Chem. Soc. 2016, 138, 16200. [Crossref] 75. Fleming, I.; Barbero, A.; Walter, D.; Chem. Rev. 1997, 97, 2063 [Crossref]; Mark, J. E.; Acc. Chem. Res. 2004, 37, 946. [Crossref] 76. Showell, G. A.; Mills, J. S.; Drug Discovery Today 2003, 8, 551 [Crossref]; Ma, C.; Wu, Y.; Lamb, R. A.; Pinto, L. W.; DeGrado, W. F.; J. Am. Chem. Soc. 2011, 133, 13844. [Crossref] 77. Cheng, C.; Hartwig, J. F.; Chem. Rev. 2015, 115, 8946. [Crossref] 78. Bi, L.; Dixneuf, P. H.; Chem. Soc. Rev. 2021, 50, 5062. [Crossref] 79. Simmons; E. M.; Hartwig, J. F.; J. Am. Chem. Soc. 2010, 132, 17092 [Crossref]; Li, B.; Driess, M.; Hartwig, J. F.; J. Am. Chem. Soc. 2014, 136, 6586. [Crossref] 80. Parija, A.; Sunoj, R.B.; Org. Lett. 2013, 15, 4066. [Crossref] 81. Ludke, M. C. M. M.; López, J.; Cienc. Rural 1999, 29, 181. [Crossref] 82. Su, B.; Lee, T.; Hartwig, J. F.; J. Am. Chem. Soc. 2018, 140, 18032. [Crossref] 83. Steenen, S. A.; van Wijk, A. J.; van der Heijden, G. J. M. G.; van Westrhenen, R.; de Lange, J.; de Jongh, A.; J. Psychopharmacol. 2016, 30, 128. [Crossref] 84. Su, B.; Hartwig, J. F.; J. Am. Chem. Soc. 2017, 139, 12137. [Crossref] 85. Paz, B. M.; Lucca Jr., E. C.; Pilli, R. A.; Quim. Nova 2021, 44, 1045. [Crossref] 86. González, M. A.; Guaita, D. P.; Royero, J. C.; Zapata, B.; Agudelo, L.; Arango, A. M.; Galvis, B.; Eur. J. Med. Chem. 2010, 45, 811. [Crossref] 87. Karmel, C.; Li, J.; Hartwig, J. F.; J. Am. Chem. Soc. 2018, 140, 1460. [Crossref] 88. Stappenbeck, F.; Xiao, W.; Epperson, M.; Riley, M.; Priest, A.; Huang, D.; Nguyen, K.; Jung, M. E.; Thies, R. S.; Farouz, F.; Bioorg. Med. Chem. Lett. 2012, 22, 5893. [Crossref] 89. Lu, Y.; Liu, Y.; Xu, Z.; Li, H.; Liu, H.; Zhu, W.; Expert Opin. Drug Discov. 2012, 7, 375 [Crossref]; Wilcken, R.; Zimmermann, M. O.; Lange, A.; Joerger, A. C.; Boeckler, F. M.; J. Med. Chem. 2013, 56, 1363. [Crossref] 90. Terao, J.; Kambe, N.; Acc. Chem. Res. 2008, 41, 1545 [Crossref]; Campeau, L. -C.; Hazari, N.; Organometallics, 2019, 38, 3. [Crossref] 91. Liu, W.; Groves, J. T.; J. Am. Chem. Soc. 2010, 132, 12847 [Crossref]; Liu, W.; Huang, X.; Cheng, M. -J.; Nielsen, R. J.; Goddard, W. A.; Groves, J. T.; Science 2012, 337, 1322 [Crossref]; Li, G.; Dilger, A. K.; Cheng, P. T.; Ewing, W. R.; Groves, J. T.; Angew. Chem., Int. Ed. 2018, 57, 1251. [Crossref] 92. Fawcett, A.; Keller, M. J.; Herrera, Z.; Hartwig, J. F.; Angew. Chem., Int. Ed. 2021, 60, 8276. [Crossref] |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access