
 |
 |
 |
 |
 |
Artigo
| A computational study of the antioxidant power of eugenol compared to vitamin C |
|
Hezha O. RasulI; Bakhtyar K. AzizII; Guillermo Salgado MoránIII; Luis Humberto Mendoza-HuizarIV,*; Assia BelhassanV; Lorena Gerli CandiaVI; Wilson Cardona VilladaVII; Kandasamy SadasivamVIII
I. Department of Pharmaceutical Chemistry, College of Medicals and Applied Sciences, Charmo University, 46023 Sulaimani, Iraq Recebido em 28/01/2023 *e-mail: hhuizar@uaeh.edu.mx The antioxidant power of eugenol and vitamin C was examined by analyzing the ability of these ligands to bind to the NADPH oxidase protein target and evaluating their bond interactions with critical residues. The results confirm that docked ligands are more stable in the specified active region of 2CDU during a MD simulation of 100 ns and 2CDU protein-ligand interactions with docked ligands showed significant hydrogen bond, hydrophobic, and water bridge formation. Eugenol exhibits hydrogen bond interactions with critical residues in the selective pocket in comparison to vitamin C. Also, eugenol had a similar binding orientation and very considerable stability in the selective pocket of 2CDU with a high binding energy with lipophilic energy. The electrostatic potential maps indicate that for eugenol, the -OH and -OCH3 sites, while that the -OH and -CO functional groups in vitamin C are responsible of the antioxidant activities of these compounds. HAT and SET mechanisms suggest that eugenol may become a better antioxidant than vitamin C. INTRODUCTION The role of free radicals and reactive oxygen species in the development of several chronic diseases has motivated an intense research for simple or complex compounds that have the power to scavenge or neutralize them.1,2 These compounds, called antioxidants, may neutralize free radicals by donating some of their electrons. They work as a natural free radical kill switch that interrupts chain reactions involving other molecules. In this sense, vitamin C is one of the preferred antioxidants, because, in addition to other biological vital roles,3 it is able to donate electrons. Once vitamin C loses an electron, it is converted to a free radical, ascorbic semihydroxy acid or ascorbyl radical, which is relatively stable with a half-life of 10 ⫺ 5 seconds and is pretty unreactive. Thus, ascorbate can interact with a reactive and potentially dangerous free radical. The amount of the reactive free radical is decreased, and a less reactive ascorbyl radical is produced in its place.4-6 Alternatively, some compounds with phenolic groups have long been recognized to possess antioxidant capabilities,7,8 quenching free radical species through the loss of an atom of hydrogen and the antioxidant phenolic group is converted into a radical species. Normally the resulting phenoxyl radical is less harmful and does not react with many substrate molecules. Moreover, the ease with which the phenolic group can lose an electron or electrons in order to scavenge a radical determines whether it has antioxidant properties.9 In this regard, eugenol, one of many phenolic chemicals and a component of many plants, is a strong antioxidant.10 Also, eugenol has therapeutic, nonmutagenic and noncarcinogenic properties and is generally regarded as safe,11-13 with an tolerable daily intake of up to 2.5 mg kg-1 body weight in humans.14 In addition, eugenol exhibits an exceptional reducing and it is able to donates phenolic hydroxyl group that can react with free radicals.15 Furthermore, the antioxidant power of eugenol is superior to that of most known or standard antioxidants such as Trolox.14 Interestingly here, eugenol has both antioxidant and oxidative effects, where the latter contribute to its cytotoxicity.16 Thus, at low concentrations, eugenol behaves as an antioxidant minimizing ROS-mediated oxidative stress, whereas at high concentrations it acts as a prooxidant and it increases ROS production.13,17-19 The above mentioned suggest that it is important to understand the action mechanism of eugenol, at molecular level, to understand its antioxidant power. In this sense, molecular docking has aided in the elucidation of the antioxidant activity of many compounds by analyzing the binding free energy and the interactions of ligands with the receptor. Thus, in this work we performed an in silico study of eugenol analyzing the ability of eugenol to bind to the NADPH oxidase protein target and we compare these results with those exhibited by vitamin C.
EXPERIMENTAL PART Lipinski's rule and ADMET prediction Lipinski's rule and ADMET parameters,20 of vitamin C and eugenol, were calculated using SwissADMET,21 and pkCSM,22 web servers, respectively. Lipinski's rule including; number hydrogen bonds donor, number of hydrogen bonds acceptor, number of rotatable bonds, molecular weight and logP were determinate. Molecules violating more than one of these parameters may have problems with bioavailability and a high probability of failure to display drug-likeness.23 We used preADMET server to predict the absorption, distribution, metabolism and excretion (ADMET) properties of the studied molecules. Protein preparation The RCSB Protein Databank (https://www.rcsb.org/) was used to obtain the three-dimensional crystallographic structure of water-forming NADPH oxidase protein.24 [2CDU (resolution: 1.80 Å)].25 The protein crystal structures were created by eliminating all solvent molecules as well as receptor-binding chemicals (FAD: flavin adenine dinucleotide and ADP: adenosine 5-diphosphate) from the solution. It was then stored in PDBQT format, the preferred input format for AutoDock Vina.26 Ligand preparation Following data cleaning, a spatial data file of eugenol ligand was created. The ligand was discovered using PubChem (https://pubchem.ncbi.nlm.nih.gov/).27 As a control ligand, the reference ligand vitamin C (Vit C) was employed. The Open Babel program,28 was used to convert the files to PDB format. Using the MMFF94 force field and a 0.1 RMS gradient, ChemDraw Ultra v19.1 decreased the ligands' energy to the lowest possible value. The structures produced were saved in the PDBQT format. Docking studies AutoDock Vina was used to screening the chemical produced as a ligand against prepared protein receptors.26 The grid box size for the targets at x, y, and z coordinates was set to 80 × 80 × 80 Å, and the detailed grid centers for x, y, and z were 10.03, -1.81, and 0.608, respectively. To finalize the receptor's development, AutoDock 4.2 was employed. Eliminating solvent molecules and receptor-associated binding chemicals from each protein chain enhanced the composition of each. After the receptor had been updated to incorporate polar hydrogens and charges, it was exported in PDBQT.29,30 PyMOL was used to visualize and predict the structure of the active pockets.31 To test the docking approach's performance, the root-mean-square deviation was calculated by re-docking the crystalline ligand into the assigned protein. The compounds were subsequently subjected to a molecular docking analysis. Molecular docking was carried out in triplicate using AutoDock Vina on the Windows 10 platform (64-bit) with a Lenovo YOGA C930 Personal Computer (Intel Core Intel(R) Core (TM) i7-8550U CPU Processor 1.99 GHz, 16 GB memory). Molecular dynamics (MD) simulation To investigate receptor stability and ligand interaction, the Schrodinger program, Desmond 2019-4 package,32 was used to run Molecular dynamics (MD) simulations on the two docked complexes at 100 nanosecond time intervals. For each system, the simulation used three identically parameterized simulations. During the 100 ns MD simulation, the TIP3P water model was utilized to fill the system, which was centered in a cubic box. All of the water molecules injected into the complicated system by eugenol and Vit C were: 14685 and 14682 water molecules, respectively. For orthorhombic simulation, a buffer distance of 10 Å was built between the edge of the box and the complex atoms. After introducing the required counter-ion, the isosmotic condition of the simulation box could be maintained with 0.15 mol L-1 NaCl. Following reducing the system's energy usage, the complex underwent a development run in the NPT ensemble class. All runs were performed at a temperature of 300 K and a pressure of 1,013 bar, with a total of 1000 frames taken and saved for the trajectory. A simulation interaction diagram (SID) tool from the Desmond Package was also used to investigate the precise interactions between ligands and proteins. The RMSD, RMSF, and protein-ligand interaction patterns were shown to represent the ligand's stability for both the protein and the ligand-bound protein. Molecular mechanics/generalized Born surface area (MM-GBSA) calculation The molecular mechanics generalized Born surface area (MM/GBSA) computation was used to calculate the relative binding free energy for the NADPH oxidase protein-eugenol combination and the reference complex. This robust quantum mechanics computation aids in the reduction of false-positive molecular docking outcomes. The Prime MM/GBSA module from the Schrödinger suite was used in this study to investigate the effect of interactions on ligand-protein stability. Schrödinger's thermal mmgbsa.py python script was used to run the MM-GBSA computations. It takes a Desmond trajectory file, separates it into individual snapshots, and executes it on each frame, producing the average and standard deviation of the binding energy estimated over a time period of 100 ns.
RESULTS AND DISCUSSION Lipinski's rule and ADME prediction The Lipinski's rule including molecular weight, number of rotatable bonds, number of hydrogen bonds acceptor, number hydrogen bonds donor and logP were shown in Table 1.
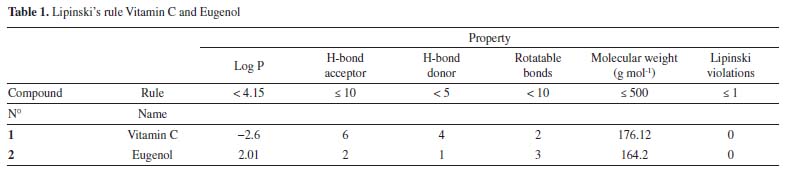
Vitamin C and eugenol respect the conditions mentioned in Lipinski's rule, without any Lipinski violations. The ADMET prediction was used in this study to calculate the pharmacokinetics parameters of vitamin C and eugenol that respect the conditions mentioned in Lipinski's rule (Table 2).
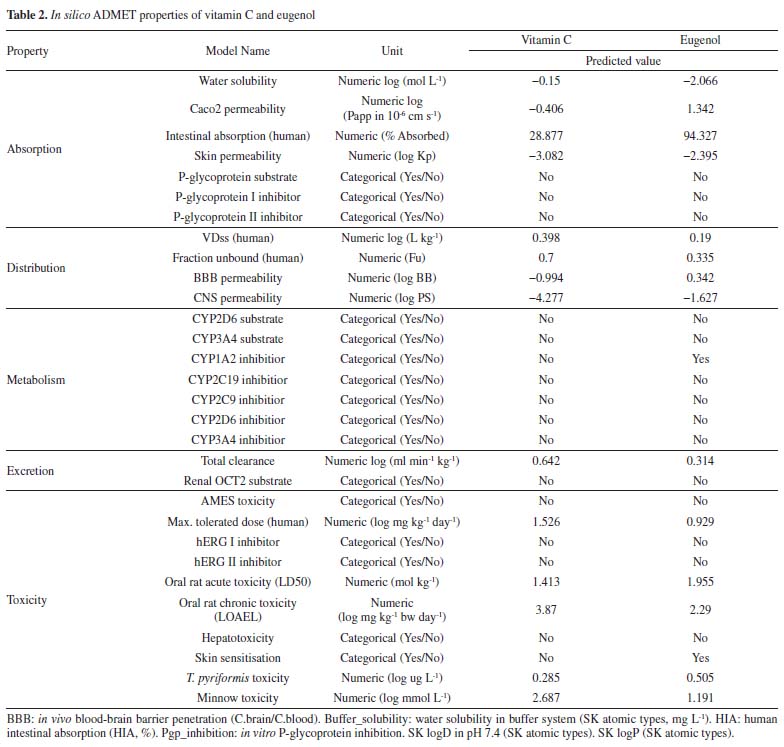
Vitamin C and eugenol have low BBB. Eugenol is completely absorbed (94.327%); whereas vitamin C has acceptable absorption percentage 28.877%. For metabolism, the both molecules could not be inhibitors for all cytochromes except eugenol that could inhibit CYP1A2. Additionally, the other pharmacokinetics parameters such as human intestinal absorption (HIA) and water solubility (log mol L-1) are all acceptable. Docking studies The current study utilized a crystalline structure of the water-forming NADPH oxidase protein co-crystallized with FAD and ADP (PDB ID: 2CDU) to understand better how the selected chemicals interact with NADPH oxidase protein. In graphic representations of protein-ligand interactions, hydrogen bonds are formed between co-crystallized ligand ADP (through GLY 158, TYR 159, Ile 160, GLY 180, HIS 181, TYR 188, and Val 214 amino acid residues), co-crystallized ligand FAD (through HIS 10, ALA 11, MET 33, Val 81, SER 115, ASP 288, PRO 298, ALA 300, THR 301 and PHE 425 amino acid residues) and the active pocket of NADPH oxidase protein (Figure 1).
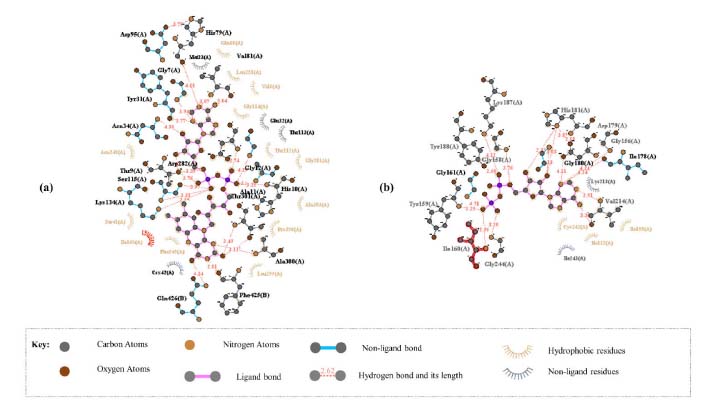 Figure 1. Protein-ligand interactions for 2CDU with co-crystallized ligand: (a) FAD, (b) ADP
Eugenol was examined for its ability to bind to an NADPH oxidase protein target. The results were compared with vitamin C. By introducing the selected ligands into the NADPH oxidase without the initial inhibitor, and molecular docking was achieved. Binding affinity energies were assigned a numerical value. Figure 2 shows the docking results of the compounds and vitamin C. The binding affinity energies of eugenol were measured for the NADPH oxidase target, with a binding affinity of -6.2 kcal mol-1. Vit C displayed comparable docking confirmation, with a binding affinity of -6.5 kcal mol-1.
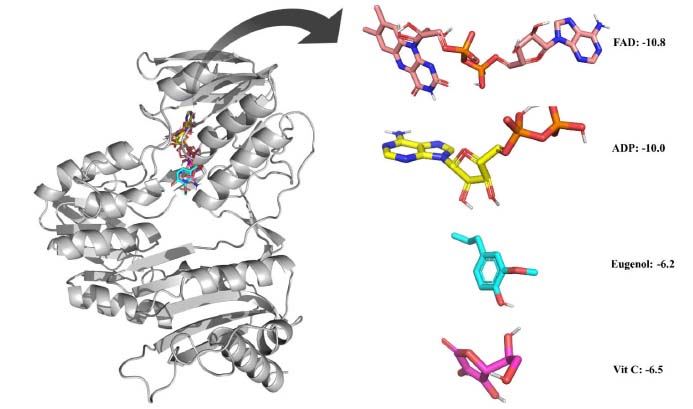 Figure 2. Ligands accommodation in water-forming NADPH oxidase active sites and the zoomed view of the ligands. The binding affinity in kcal mol-1 with 3D structures of the compounds are represented in boxes
Figure 3 depicts the molecular interactions between the selected components and the therapeutic targets. As with the eugenol compound, it forms a hydrogen bond with CSX 42, a cysteine derivative of L-cysteine carrying an S-hydroxy-substituent. Furthermore, residues like SER 41, ASP 282, and PRO 298 may form hydrogen bonds. HIS 10, ALA 11, ALA 300, and ALA 303 residues helped form a strong hydrophobic contact with the ligand. Furthermore, in Vit C compound, hydrogen interactions between the residues THR 9, HIS 10, ALA 11, GLY 12, THR 112, and ASP 282 firmly secure the ligand in its binding pocket. Thus, it is clear that the hydroxyl groups of eugenol and vitamin C are essential for the inhibitory activity on NADPH oxidase, a similar result was found during the interaction of flavonoids with the xanthine oxidase.33
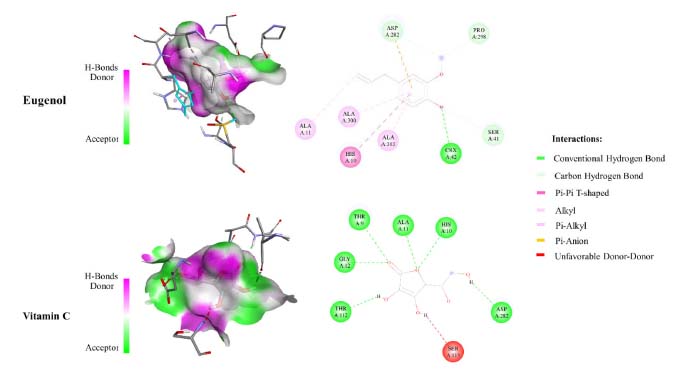 Figure 3. Binding mode visualization (3D-left and 2D-right) of eugenol and vitamin C with 2CDU
Using the re-docking technique, the current study validated the docking procedure for docked molecules obtained by AutoDock Vina. The PDB format of the co-crystal ligand (ADP) was obtained from the "Protein Data Bank Japan database" (https://pdbj.org/chemie/summary/ADP). The co-crystal ligand (ADP) was re-docked into the catalytic binding site of NADPH oxidase using the previously described technique; the observed binding energy was -10.0 kcal mol-1. When the resulting conformer was superposed in the active pocket of the NADPH oxidase, it was revealed to have a significant overlap with the co-crystal ligand. The PyMol software was used to compute the RMSD between the re-docked complex and native co-crystal ligand positions. The calibration of the docking procedure was confirmed by a low RMSD value of less than 2 Å. An RMSD of less than 2 Å is appropriate for computational validation of the ligand-protein.34,35 MD simulations The Desmond package (Schrodinger 2019-4) was used to undertake a molecular dynamics simulation to examine the stability of ligand-protein docked complexes of the NADPH oxidase receptor with ligands eugenol and vitamin C. 2CDU-eugenol, and 2CDU-vitamin C complexes were used in molecular dynamics simulations. When calculating the scalar distance between the protein (Cα backbone) and the ligand throughput trajectory, the RMSD values of the protein and ligand complexes were used to investigate the response of MD. Accordingly, the RMSD method measured protein backbone and ligand-protein complex structural stability. Figure 4 depicts the results as a function of the simulation time. During the 100 ns simulation interval, the eugenol ligand showed an acceptable variance (< 3 Å) in structural conformation until 35 ns, then fluctuated and then somewhat normalized at 95 ns for the duration of the simulation. Despite this, the control ligand (Vit C) remained very steady (< 2 Å) during the whole simulation time. The RMSD of ligands as a function of simulation time (Figure 5) reveal that the eugenol molecule and the control ligand Vit C travel approximately 0.8 Å relative to their reference location within the active site throughout the simulation period.
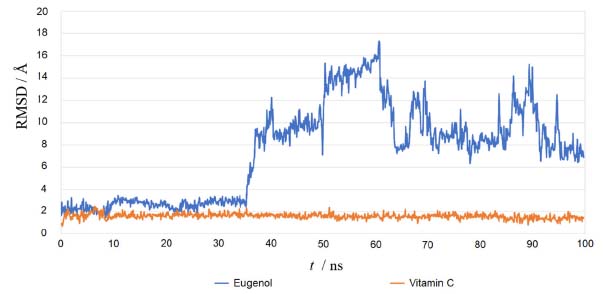 Figure 4. Root mean square deviation (RMSD) plot for Cα atoms (Å) with the ligands
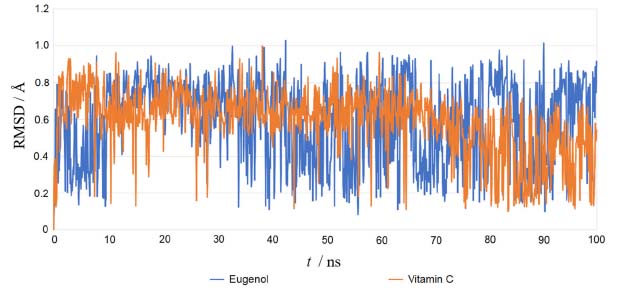 Figure 5. Root mean square deviation (RMSD) plot for ligand atoms (Å) with the ligands
Throughout 100 ns simulation trajectories, the local modifications and flexibility of the NADPH oxidase receptor and the screening ligand in their respective complexes were analyzed using RMSF (Figure 6). The RMSF is the cumulative displacement of an atom or atom group relative to the reference structure. It can be used to investigate a structure's time-dependent dynamics. It is common practice to use the RMSD to identify if a structure is stable during a simulation or drifting away from its original coordinate system.36 The N- and C-terminal tails of proteins naturally change more than any other protein part. Secondary structural components of a protein, such as alpha helices and beta strands, are often stiffer than the unstructured segment and exhibit less variation than loop regions. The output shows that during the 100 ns simulation procedure, the loop region and the C-terminal of NADPH oxidase receptor showed similar changes in the amino acid residues of the protein in the docked complexes of the screened ligands (RMSF: 1.5-3.0 Å). Consequently, the computed results confirm that docked ligands are more stable in the specified active region of 2CDU during the 100 ns MD simulation.
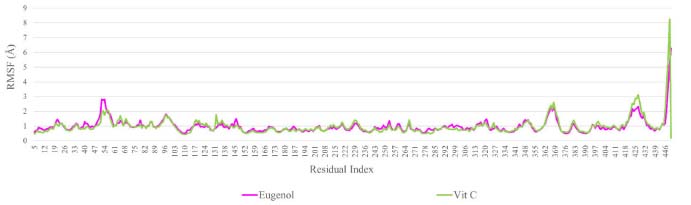 Figure 6. Root mean square fluctuation (RMSF) plot for 2CDU with the ligands
Furthermore, the stability of docked NADPH oxidase-ligand complexes was investigated for intermolecular interactions between proteins and ligands such as hydrogen bonds, hydrophobic, ionic, and water bridges (Figure 7). The docked ligands interacted well with the amino acid residues in the selective pocket of the 2CDU crystal structure throughout the 100 ns simulation run. Furthermore, 2CDU protein-ligand interactions with docked ligands showed significant hydrogen bond, hydrophobic, and water bridge formation. The selected eugenol was assessed for hydrogen bond interactions with critical residues (ALA 11) in the selective pocket of NADPH oxidase compared to the control ligand (Vit C). Compared to the control ligand, the eugenol ligands had a similar binding orientation and very considerable stability in the selective pocket of 2CDU.
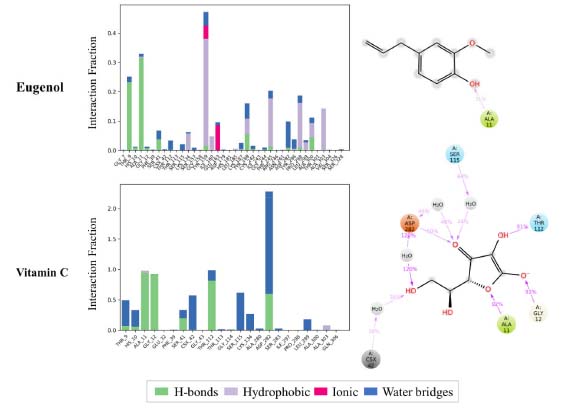 Figure 7. Protein-ligand contact plot and ligand-protein interaction residues
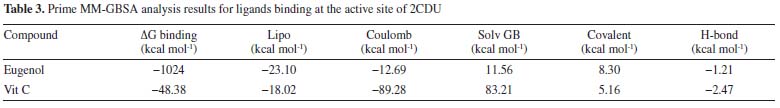
Prime MM-GBSA calculation Additional analysis was performed using 200 pictures with a 50 ps gap to establish the average binding energy for an equilibrated MD trajectory. The binding energy was calculated using the previously known formulae.37 Schrödinger's thermal mmgbsa.py python script was used to calculate the average MM-GBSA binding energy, as well as the Coulomb (Coulomb), lipophilic energy (Lipo), generalized Born electrostatic solvation energy (Solv GB), covalent binding energy (Covalent) and hydrogen-bonding energy (H-bond). The data collected is summarized in Table 3. In comparison to the control ligands, eugenol has high binding energy with lipophilic energy, according to the MM-GBSA data (Vit C). On the other hand, the eugenol ligand had a more unfavorable Coulomb energy.
Molecular electrostatic potential (MEP) A study of the charge density distribution on a molecule is useful to find the charge abundance and charge deficiency sites on it. In this sense, the determination of this charge distribution is generally possible through an analysis of the molecular electrostatic potential (MEP). Thus, MEP surfaces allow us to identify the nucleophilic and electrophilic sites on the compound. Figures 8a and 8b depicts the potential surface of eugenol and vitamin C, respectively. The MEP surfaces are ranged from -0.07862 a.u. (deepest red) to 0.07862 a.u. (deepest blue). In these Figures a red colour indicates the existence of a higher electron density site while the blue colour is related to charge deficiency while the shaded red colour indicates the presence of electron pair in that region. Thus, the deep blue colour near the hydrogen atoms around the ring indicates the presence of maximum positive charges in that region and these sites are mostly preferential for the nucleophilic attack. On the other hand, note that for eugenol and vitamin C, in all cases the electrophilic sites are located near to the oxygen atoms. Thus, in the case of eugenol the -OH and -OCH3 sites, and -OH and -CO functional groups in vitamin C can be considered as are highly abundant for the antioxidant activities.38,39 Also, from these MEP surfaces, vitamin C exhibits more active hydrogen atoms in comparison with eugenol.
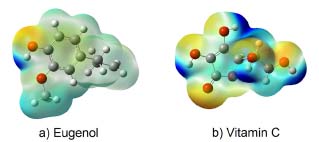 Figure 8. Molecular electrostatic potential (MEP) maps of (a) Eugenol and (b) Vitamin C plotted at a density isosurface (isovalue 0.002 e Å-3), and ranged from −0.07862 a.u. (deepest red) to 0.07862 a.u. (deepest blue)
HAT and SET mechanisms Note that from the MEP surfaces depicted in Figure 8, it is possible to qualitatively infer the ability of a molecule to be an antioxidant. However, it is also possible to evaluate the antioxidant capacity of a compound by analyzing its hydrogen atom transfer capacity (HAT) or by a single electron transfer (SET) to free radical (FR•).40,41 In this sense, the analysis of the thermochemistry in which a hydrogen bond is broken and the hydrogen is transferred to a free radical is relevant to understanding the HAT mechanism,40-43 which can be summarized by the reaction 1:44 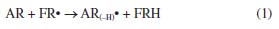 According to this reaction, a hydrogen atom is transferred from the antioxidant (AR) to the free radical (FR•) and produces a non-radical product (FRH) that is unable to propagate the chain reaction.43 Thus, a negative value of Gibbs free energy indicates that the reaction is exergonic and energetically possible. In this work, we computed the adiabatic Gibbs free energy for reaction 1, considering that AR (eugenol or vitamin C) is interacting individually with the following free radicals: CH3O•, CH3•, HOO•, OH•, and NO2•. The results are reported in Tables 1S and 2S as supplementary material. In all cases, the more negative values for Gibbs free energies are related to HAT from eugenol to FR•, suggesting a better antioxidant capacity of eugenol. Additionally, SET was evaluated by the following reaction:44,45 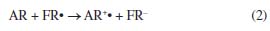 In reaction 2, a cation radical (AR+•) is formed and a single electron is transferred to the free radical (FR•). The Gibbs's free energies associated with these reactions are reported in Table 3S as supplementary material. In all cases the single electron transfers from eugenol to FR• requires less Gibbs free energy in comparison to vitamin C, which agrees with the evidence provided by the HAT mechanism.
CONCLUSIONS The antioxidant power of eugenol and vitamin C was evaluated by molecular docking studies, molecular dynamics and electrostatic potential maps. For both ligands, their binding capacity to the target NADPH oxidase protein was studied and their interactions with critical residues were evaluated. The results indicate that the ligands dock in the specified active region of the 2CDU and remain stable for 100 ns, also the docked ligands showed significant formation of hydrogen bonds, hydrophobic bonds and water bridges in this region. Also, eugenol had similar binding orientation and very considerable stability in the selective pocket of the 2CDU with high binding energy with lipophilic energy. The molecular electrostatic potential maps indicate that in the case of eugenol the -OH and -OCH3 sites, while that the -OH and -CO functional groups in vitamin C may be related to the antioxidant activities of these compounds. HAT and SET mechanisms suggest that eugenol may become a better antioxidant than vitamin C.
SUPPLEMENTARY MATERIAL Supplementary information is freely available at http://quimicanova.sbq.org.br, in PDF format.
ACKNOWLEDGEMENTS The authors would like to thank the Research Department of the Universidad Católica de la Santísima Concepción for its support through the DIREG 03/2020 project. L. H. M. H. thankfully acknowledges the computer resources, technical expertise and support provided by the Laboratorio Nacional de Supercómputo del Sureste de México, CONACYT member of the network of national laboratories through the project No. 202203072N and to the Universidad Autónoma del Estado de Hidalgo.
REFERENCES 1. Mahboub, R.; Memmou, F.; Nat. Prod. Res. 2015, 29, 966. [Crossref] 2. Gülçin, I.; Mshvildadze, V.; Gepdiremen, A.; Elias, R.; Phytother. Res. 2006, 20, 130. [Crossref] 3. Cerqueira, F. M.; de Medeiros, M. H. G.; Augusto, O.; Quim. Nova 2007, 30, 441. [Crossref] 4. Bielski, B. H. J.; Richter, H. W.; Chan, P. C.; Ann. N. Y. Acad. Sci. 1975, 258, 231. [Crossref] 5. Buettner, G. R.; Moseley, P. L.; Free Radical Res. Commun. 1993, 19, s89. [Crossref] 6. Padayatty, S. J.; Katz, A.; Wang, Y.; Eck, P.; Kwon, O.; Lee, J. H.; Chen, S.; Corpe, C.; Levine, M.; Dutta, A.; Dutta, S. K.; J. Am. Coll. Nutr. 2003, 22, 18. [Crossref] 7. Hidalgo, M. E.; De La Rosa, C.; Carrasco, H.; Cardona, W.; Gallardo, C.; Espinoza, L.; Quim. Nova 2009, 32, 1467. [Crossref] 8. Salgueiro, F. B.; Lira, A. F.; Rumjanek, V. M.; Castro, R. N.; Quim. Nova 2014, 37, 821. [Crossref] 9. Zhang, L. L.; Zhang, L. F.; Xu, J. G.; Hu, Q. P.; Food Nutr. Res. 2017, 61, 1. [Crossref] 10. Nagababu, E.; Rifkind, J. M.; Boindala, S.; Nakka, L.; Methods Mol. Biol. 2010, 610, 165. [Link] accessed in June 2023 11. Opdyke, D. L. J.; Food Cosmet. Toxicol. 1975, 13, 449. [Crossref] 12. Bendre, R. S.; Rajput, J. D.; Karandikar, P. S.; Nat. Prod. Chem. Res. 2016, 4, 212. [Crossref] 13. Jaganathan, S. K.; Supriyano, E.; Molecules 2012, 17, 6290. [Crossref] 14. Gülçin, I.; J. Med. Food 2011, 14, 975. [Crossref] 15. Rieger, A.; Sax, C.; Bauert, T.; Wäckerlin, C.; Ernst, K. H.; Chirality 2018, 30, 369. [Crossref] 16. Fujisawa, S.; Atsumi, T.; Kadoma, Y.; Sakagami, H.; Toxicology 2002, 177, 39. [Crossref] 17. Atsumi, T.; Fujisawa, S.; Tonosaki, K.; Toxicol. In Vitro 2005, 19, 1025. [Crossref] 18. Chogo, J. B.; Crank, G.; J. Nat. Prod. 1981, 44, 308. [Crossref] 19. Asha, M. K.; Prashanth, D.; Murali, B.; Padmaja, R.; Amit, A.; Fitoterapia 2001, 72, 669. [Crossref] 20. Jalkute, C. B.; Barage, S. H.; Dhanavade, M. J.; Sonawane, K. D.; Int. J. Pept. Res. Ther. 2015, 21, 107. [Crossref] 21. Daina, A.; Michielin, O.; Zoete, V.; Sci. Rep. 2017, 7, 1. [Crossref] 22. Pires, D. E. V.; Blundell, T. L.; Ascher, D. B.; J. Med. Chem. 2015, 58, 4066. [Crossref] 23. Zhao, Y. H.; Abraham, M. H.; Le, J.; Hersey, A.; Luscombe, C. N.; Beck, G.; Sherborne, B.; Cooper, I.; Pharm. Res. 2002, 19, 1446. [Crossref] 24. Berman, H. M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T. N.; Weissig, H.; Shindyalov, I. N.; Bourne, P. E.; Nucleic Acids Res. 2000, 28, 235. [Crossref] 25. Farouk, A.; Mohsen, M.; Ali, H.; Shaaban, H.; Albaridi, N.; Molecules 2021, 26, 4145. [Crossref] 26. Trott, O.; Olson, A. J.; J. Comput. Chem. 2010, 31, 455. [Crossref] 27. Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B. A.; Thiessen, P. A.; Yu, B.; Nucleic Acids Res. 2021, 49, D1388. [Crossref] 28. O'Boyle, N. M.; Banck, M.; James, C. A.; Morley, C.; Vandermeersch, T.; Hutchison, G. R.; J. Cheminf. 2011, 3, 1. [Crossref] 29. Rasul, H. O.; Aziz, B. K.; Ghafour, D. D.; Kivrak, A.; J. Mol. Model. 2022, 28, 1. [Crossref] 30. Rasul, H. O.; Aziz, B. K.; Ghafour, D. D.; Kivrak, A.; Mol. Diversity 2022, 26, 1. [Crossref] 31. DeLano, W. L.; CCP4 Newsletter on Protein Crystallography 2002, 40, 82. [Link] accessed in June 2023 32. Schrödinger Release; Desmond Molecular Dynamics System, Maestro-Desmond Interoperability Tools, New York, 2021. [Link] accessed in June 2023 33. Cos, P.; Ying, L.; Calomme, M.; Hu, J. P.; Cimanga, K.; Van Poel, B.; Pieters, L.; Vlietinck, A. J.; Vanden Berghe, D.; J. Nat. Prod. 1998, 61, 71. [Crossref] 34. Ding, Y.; Fang, Y.; Moreno, J.; Ramanujam, J.; Jarrell, M.; Brylinski, M.; Comput. Biol. Chem. 2016, 64, 403. [Crossref] 35. García-Godoy, M. J.; López-Camacho, E.; García-Nieto, J.; Nebro, A. J.; Aldana-Montes, J. F.; Molecules 2016, 21, 1575. [Crossref] 36. Martínez, L.; PLoS One 2015, 10, e0119264. [Crossref] 37. Alnajjar, R.; Mostafa, A.; Kandeil, A.; Al-Karmalawy, A. A.; Heliyon 2020, 6, e05641. [Crossref] 38. Lakshminarayanan, S.; Jeyasingh, V.; Murugesan, K.; Selvapalam, N.; Dass, G.; J. Photochem. Photobiol. 2021, 6, 100022. [Crossref] 39. Basha, A. F.; Ali Khan, F. L.; Muthu, S.; Raja, M.; Comput. Theor. Chem. 2021, 1198, 113169. [Crossref] 40. Dos Santos, R. M. B.; Simões, J. A. M.; J. Phys. Chem. Ref. Data 1998, 27, 707. [Crossref] 41. Dilabio, G. A.; Pratt, D. A.; Lofaro, A. D.; Wright, J. S.; J. Phys. Chem. A 1999, 103, 1653. [Crossref] 42. Guerra, M.; Amorati, R.; Pedulli, G. F.; J. Org. Chem. 2004, 69, 5460. [Crossref] 43. Najafi, M.; Najafi, M.; Najafi, H.; Can. J. Chem. 2012, 90, 915. [Crossref] 44. Mulder, P.; Korth, H. G.; Ingold, K. U.; Helv. Chim. Acta 2005, 88, 370. [Crossref] 45. Manzanilla, B.; Robles, J.; J. Mol. Model. 2022, 28, 1. [Crossref] |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access