
 |
 |
 |
 |
 |
Revisão
|
|
| Uma nova era da incorporação de átomo de flúor em compostos orgânicos: métodos modernos e uso da espectroscopia de ressonância magnética de 19F A new era of incorporation of fluorine atom on organic compounds: modern methods and use of 19F magnetic resonance spectroscopy |
|
Cassia Chiari; Cláudio F. Tormena*
Departamento de Química Orgânica, Instituto de Química, Universidade Estadual de Campinas, 13083-970 Campinas - SP, Brasil Recebido em: 29/06/2023 *e-mail: tormena@unicamp.br During the last decades, the incorporation of the fluorine atom in organic molecules suffered an increase, mainly in the pharmaceutical and agrochemical industries, due to the unique properties of the fluorine atom and the desired behaviour of the molecules resulting from fluorination processes. We present the rise and historical evolution of both fluorination agents, nucleophilic and electrophilic agents, and the three fluorination conditions, nucleophilic, electrophilic and neutral (radical) conditions. The advantages and disadvantages of each type of fluorination agent and condition are highlighted employing modern methods of fluorination, along with the presentation of the author's mechanistic proposals and discussion of the scope of the reaction. Green chemistry, more sustainable procedures and the use of alternative methods are gaining more attention and are the challenges of the fluorination of organic compounds. 19F NMR technique is also highlighted as a powerful tool for structural elucidation, intermediates identification and reaction monitoring. 1D and 2D NMR experiments involving fluorine are widely employed in biological areas and need to be more widespread in organic synthesis to help face the challenges of this area. INTRODUÇÃO O átomo de flúor é o 13º elemento mais abundante na superfície terrestre, o 24º no universo e o 1º entre a família dos halogênios, no entanto, está exclusivamente presente na natureza na forma de sais, sendo majoritariamente depositado na terra na forma de fluoreto de cálcio (CaF2), também conhecido como fluorita. Curiosamente, compostos naturais orgânicos contendo esse átomo são raros e isto é devido ao fato da fluorita apresentar baixa solubilidade em água e o íon hidratado de flúor ser pouco nucleofílico, com uma energia livre de solvatação em água estimada em −117,5 kcal mol−1.1,2,3 Átomos de flúor podem ser incorporados em moléculas orgânicas através de processos de fluoração ou fluoroalquilação, sendo o último responsável por introduzir grupos trifluorometila (CF3), difluorometila (CHF2) ou monofluorometila (CH2F).4 Embora a forma natural de flúor encontrada na terra seja o CaF2, o emprego da fluorita na química orgânica tende a ser quase inexistente devido à sua baixa reatividade, tornando necessário o uso de ácidos fortes como o ácido sulfúrico para gerar ácido fluorídrico, o qual por sua vez é mais reativo que a fluorita. Infelizmente, o ácido fluorídrico é tóxico, pois reage com o cálcio do corpo de seres vivos, pode penetrar a pele causando queimaduras graves e, além do seu caráter corrosivo, possui ainda a capacidade de reagir com silício, dissolvendo vidros e consequentemente dificultando sua manipulação em laboratório.2,5 Meramente pelas informações até agora apresentadas sobre o flúor pressupõe-se que esse átomo não seria um grande alvo de estudos, contudo ele é o segundo heteroelemento mais empregado em pesquisas em ciências da vida, estando somente atrás do nitrogênio,5 e à vista disso pode-se inferir que outros métodos foram elaborados para sintetizar compostos fluorados. Grande parte do interesse pelo flúor reside nas indústrias agroquímica6,7,8 e farmacêutica,9 em especial a farmacêutica em virtude das alterações que a incorporação do átomo de flúor gera em moléculas orgânicas, tais como alterações nas propriedades físicas e químicas, na lipofilicidade, basicidade, capacidade de permear a membrana celular e biodisponibilidade,2,10,11,12,13,14 decorrentes de alterações na estabilidade e no equilíbrio conformacional.15 Drogas farmacologicamente ativas podem ser fluoradas através de estratégias de late-stage functionalization, estratégia definida como sendo uma transformação quimiosseletiva de uma molécula complexa sem a necessidade de instalar um outro grupo funcional que auxilie nessa transformação.16 Um caminho para realizar late-stage functionalization é substituir átomos de hidrogênio por átomos de flúor, o que não leva a diferenças estéricas significativas, porém pode mudar a preferência confomacional da molécula devido a interação eletrostática entre o átomo de flúor inserido e outros grupos funcionais presentes.12 A estratégia de late-stage impulsionou a química envolvendo 18F por promover um caminho de incorporar esse átomo em moléculas complexas, uma vez que o 18F tem um tempo de meia-vida curto de aproximadamente 110 min, impossibilitando sua incorporação em etapas iniciais de síntese totais de compostos orgânicos.12 Compostos marcados com 18F são aplicados comumente em tomografia de emissão de positron (positron emission tomography, PET), uma técnica de imagem altamente sensível empregada na caracterização de processos bioquímicos.17 Historicamente, a química de organofluorados teve início em 1835 com a síntese do fluorometanol a partir de dimetil sulfato e fluoreto de potássio, como apresentada na linha temporal na Figura 1. Durante os 125 anos seguintes o processo de fluoração foi alcançado mediante reagentes como HF, F2, SbF3 e CoF3, os quais são considerados corrosivos e com capacidade de serem explosivos, e reações, tais como de Swarts, Balz-Schiemann e Halex, além do processo de fluoração eletroquímica de Simons e processos de fluoração direta.5,18
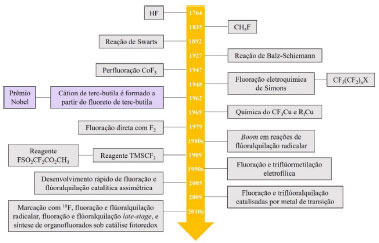 Figura 1. Desenvolvimento histórico da química de organofluorados (fonte: adapatado de Hu e colaboradores)5
Em 1964 foi reportada a síntese do primeiro reagente de fluoração contendo a ligação N-F, o perfluoro-N-fluoropiperidina, por fluoração eletroquímica de piridina ou 2-fluoropiridina (Esquema 1a). Em razão do baixo rendimento, esse reagente não se tornou popular, porém abriu as portas para o desenvolvimento de diversos reagentes de fluoração N-F.19 Dois exemplos de apliçação desse primeiro reagente de fluoração estão ilustrados no Esquema 1b.
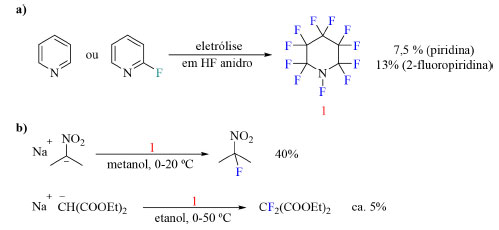 Esquema 1. Síntese do perfluoro-N-fluoropiperidina (a) e exemplos de fluoração (b) (fonte: adaptado de Hammond e colaboradores)19
Após a década de 1960, reagentes mais brandos emergiram: Selectfluor, N-fluorobenzenosulfonamida (NFSI) e PyFluor são exemplos notáveis. O desenvolvimento de reagentes de fluoração e fluoroalquilação nucleofílicos, eletrofílicos e de mecanismo radicalar contribuíram para impulsionar a química de organofluorados.5 Reagentes de fluoração são preparados a partir de substratos apropriados e reagentes como F2, KF e Et3N3HF, que por sua vez foram preparados a partir de ácido fluorídrico anidro (Figura 2). Em destaque, F2, considerado a espécie oxidante mais forte conhecida, é produzido por processo de eletrólise envolvendo KF e HF.
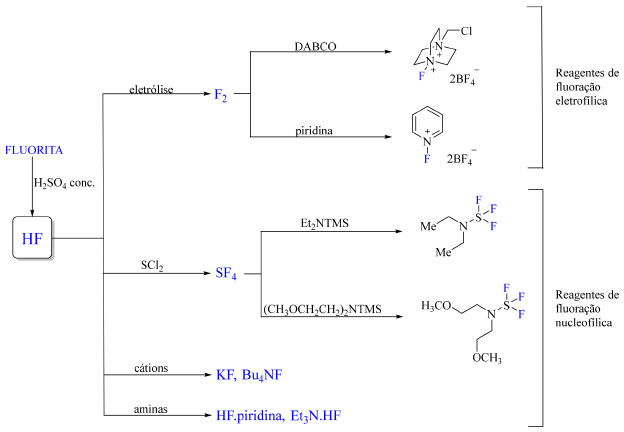 Figura 2. Preparo dos reagentes de fluoração mais comuns em química de organofluorados (fonte: adaptado de Harsanyi e Sandford)20
Sendo assim, o objetivo principal desse texto é de apresentar os reagentes de fluoração mais comumente empregados e abordar estudos de casos que os empregam em três diferentes condições: eletrofílica, nucleofílica e neutra.
MÉTODOS DE INSERÇÃO DE ÁTOMOS DE FLÚOR SOB CONDIÇÕES ELETROFÍLICAS Há duas classes em que os reagentes de fluoração podem ser inseridos: agentes nucleofílicos e agentes eletrofílicos. Nesta sessão serão abordados os agentes eletrofílicos, caracterizados por introduzirem átomos de flúor em centros ricos em elétrons e por conferir a presença de "F+". Em uma fluoração sob condição eletrofílica o substrato é uma espécie que pode assumir a forma de carbânion, ser insaturado rico em elétrons ou apresentar uma ligação lábil e nucleofílica (por exemplo C-Si, C-Sn e C-B), ou seja, o substrato necessariamente deve apresentar caráter nucleofílico.21 Em um primeiro momento foram estudados e desenvolvidos agentes de fluoração eletrofílicos que exibiam ligação C-O, como por exemplo CF3OF, ClO3F, CF3COOF, CH3COOF e CsSO4F, reagentes considerados muito reativos, não seletivos, difíceis de preparar e altamente oxidantes. O fluoreto de xenônio (XeF2) é uma alternativa mais estável, no entanto, é também altamente oxidante, intolerante a grupos funcionais sensíveis a condições oxidantes no substrato e indisponível comercialmente.4,21,22 As adversidades com os reagentes contendo ligação C-O e Xe-F foram contornadas com o desenvolvimento de reagentes de fluoração estáveis contendo ligação N-F. Esse tipo de ligação é polarizada em direção ao flúor com uma carga parcial negativa nesse átomo e o orbital σ*N-F (Figura 3a) é estericamente inacessível no átomo de nitrogênio para ataque nucleofílico, isto é, reações do tipo SN2 (substituição nucleofílica bimolecular) são viáveis de ocorrer, porém o ataque nucleofílico é no átomo de flúor, como ilustrado na Figura 3b. Outra característica pertinente do orbital a*N-F é seu pequeno coeficiente orbitalar no flúor e o baixo nível de energia, o que conferem outros possíveis mecanismos além da SN2, sendo eles mecanismos radicalares como transferência de um elétron (single-electron transfer, SET).4,22
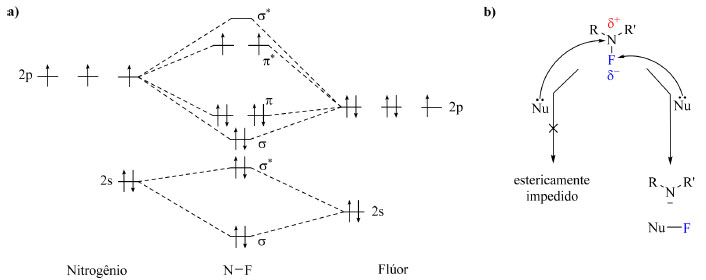 Figura 3. Diagrama de orbitais moleculares da molécula N-F (a) e representação de um ataque nucleofílico a uma molécula contendo uma ligação N-F (b)
Muitos dos reagentes de fluoração N-F estão disponíveis comercialmente e estão divididos em dois grupos, neutros e de amônio quaternário, sendo o último grupo considerado como tendo caráter mais eletrofílico.22 Os mais comercializados atualmente são os sais de N-fluoropiridínio ou NFPy (2-7),23,24 NFSI (8),25 Selectfluor (9)26 e Accufluor (10),27 e os desenvolvidos mais recentemente são derivados do NFSI (11-12),28,29 derivados quirais do Selectfluor (13-14)30,31 e reagentes baseados em N-fluoro-N-arilsulfonamidas (15),32 de acordo com a Figura 4.
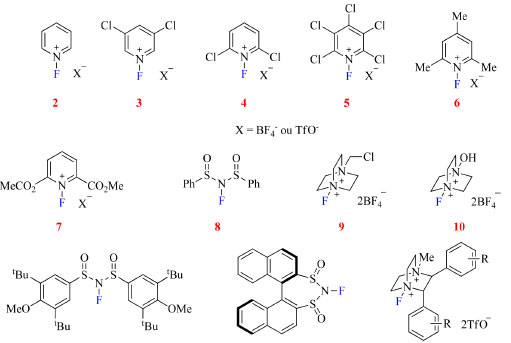 Figura 4. Exemplos de reagentes de fluoração eletrofílica da classe N-F (fonte: adaptado de Rozatian e Hodgson)22
Os sais de N-fluoropiridínio (2-7) foram reportados por Umemoto e colaboradores19 em 1986 como novos agentes de fluoração catiônicos estáveis e são caracterizados por conter um núcleo de piridínio com substituintes doadores ou retiradores de densidade eletrônica. São reportados quatro métodos distintos para síntese desses sais: no método A (Esquema 2), há a formação de um complexo piridina∙F2 seguida da adição de um ânion não nucleofílico de sal, ácido ou derivado de silício; no método B (Esquema 2), a fluoração e adição do ânion de um sal não nucleofílico ocorrem em uma única etapa; no método C (Esquema 2), o substrato é misturado com um ácido ou derivado de silício previamente à fluoração; e por fim o último método, o método D (Esquema 2), envolve a mistura do substrato com um ácido de Lewis seguida da etapa de fluoração com F2/N2.
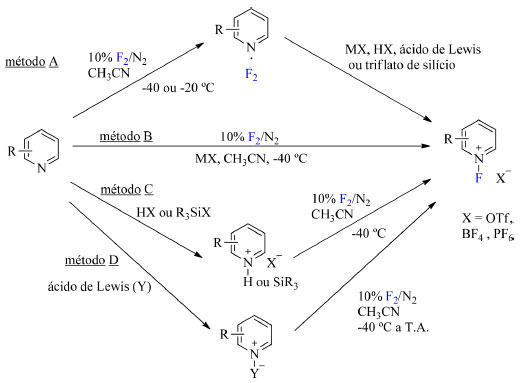 Esquema 2. Métodos para sintetizar sais de N-fluoropiridínio (fonte: adaptado de Umemoto e colaboradores)19
A reatividade desses reagentes é consequência da densidade eletrônica no átomo de nitrogênio, que por sua vez é fruto da natureza eletrônica dos substituintes no anel piridínio, isto é, quanto menor o valor do pKa da piridina correspondente maior o poder de fluoração do sal de N-fluoropiridínio (Figura 5). Além disso, sais triflato costumam ser mais efetivos que os tetrafluoroboratos devido a sua maior solubilidade em solventes halogenados.19
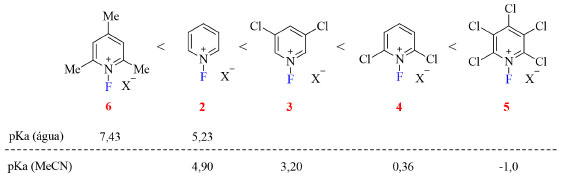 Figura 5. Poder de fluoração dos sais de N-fluoropiridínio (fonte: adaptado de Umemoto e colaboradores).19 Valores de pKa das piridinas correspondentes em água33,34 e em acetonitrila35
Já o NFSI (N-fluorobenzenosulfonamida, (8) foi sintetizado por Differding e Ofner em 1991 a partir da benzenosulfonamida na presença de F2/N2 em acetonitrila (Esquema 3). Esse reagente é capaz de fluorar nucleófilos tais como éteres enólicos de trimetilsilano (TMS), ânions enólicos de cetonas e ésteres, aril- e vinil-lítios. Entretanto, anéis aromáticos como anisol e tolueno, menos nucleofílicos, necessitam de temperaturas mais elevadas e condições de reação anidra (Esquema 4).19
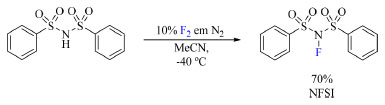 Esquema 3. Síntese da N-fluorobenzenosulfonamida (NFSI) (fonte: adaptado de Differding e Ofner)25
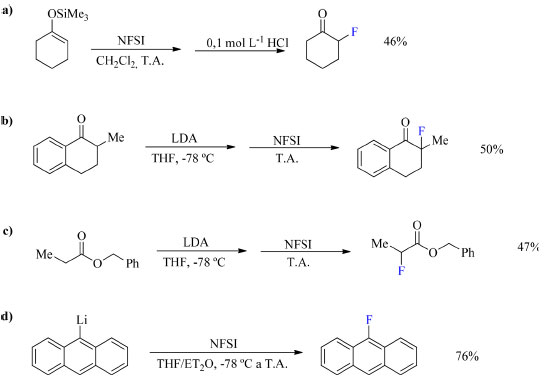 Esquema 4. Substratos passíveis de fluoração com NFSI (fonte: adaptado de Umemoto et al.)19
O último exemplo de preparo de reagentes de fluoração N-F a ser abordado é o 1-clorometil-4-fluorodiazoniabiciclo[2.2.2]octano bis(tetrafluoroborato), conhecido pelo nome de Selectfluor (9), um dos reagentes de fluoração mais empregado nos dias de hoje e que foi desenvolvido para sanar a procura por um reagente de fluoração eletrofílico que fosse seguro, estável, reativo e compatível com produção industrial.36 Seu preparo consiste na alquilação da trietilenodiamina (TEDA ou DABCO) seguida da substituição do contra íon por BF4- e, por fim, fluoração com F2, como descrito no Esquema 5.
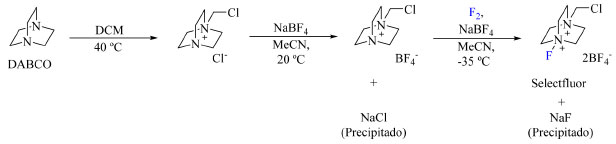 Esquema 5. Rota de síntese empregada na produção do Selectfluor (fonte: adaptado de Nyffeler e colaboradores)36
Derivados do Selecfluor podem ser sintetizados simplesmente alterando o grupo alquila presente no material de partida, o DABCO (Figura 6). Os derivados menos reativos são aqueles com substituintes metila, etila e octila, ao passo que o derivado f é o mais reativo e o Selectfluor segundo mais reativo, isto é, substituintes com caráter mais retirador de densidade eletrônica acentuam a eletrofilicidade do grupo fluorado. Apesar de não ser o mais reativo dentre seus derivados, Selectfluor é do ponto de vista econômico o reagente ideal devido a sua produção simples e eficiente.36
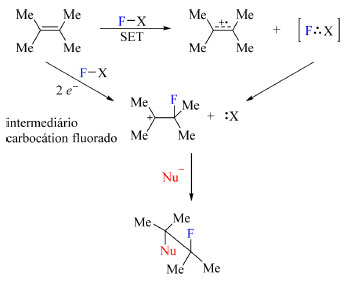 Figura 6. Derivados do Selectfluor (fonte: adaptado de Nyffeler e colaboradores)36
O contra íon também influencia na reatividade observada, por exemplo o uso de triflato, em vez de tetrafluoroborato, leva a diminuição de subprodutos bisfluorados e consequentemente ao aumento do rendimento do produto desejado. Além disso sais de triflato de Selectfluor apresentam maior solubilidade em nitrometano, solvente geralmente empregado em fluorações com esse reagente, que os sais de tetrafluoroborato.36 Compilando as informações até agora expostas sobre os mais conhecidos reagentes de fluoração eletrofílicos tem-se um comparativo da reatividade de cada um deles tendo como referência as médias das constantes de velocidades relativas ao Selecfluor determinadas através de estudos cinéticos (Figura 7). Nesse estudo, foi observado que o sal de NFPy 5 é quase duas unidades mais reativo que Selectfluor, enquanto NFPy 6 é quase 6 unidades menos reativo. Adicionalmente foi constatado que a diferença na reatividade dos reagentes comparando os dois diferentes possíveis contra íons, triflato e tetrafluoroborato, não é significativa (~ 0,3 unidades nos reagentes estudados).22
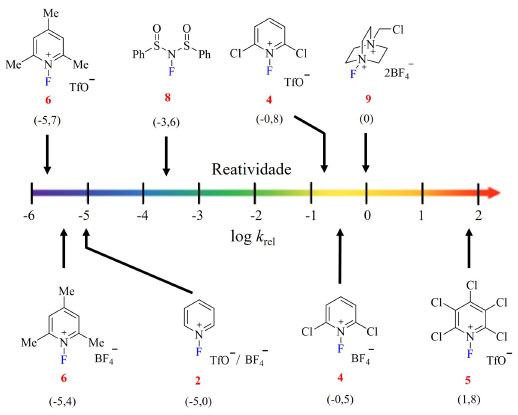 Figura 7. Escala de reatividade relativa ao Selecfluor de reagentes N-F de fluoração eletrofílica (fonte: adaptado de Rozatian e Hodgson)22
Reagentes de fluoração N-F podem apresentar dois possíveis mecanismos que levam ao mesmo intermediário, um carbocátion fluorado (Esquema 6), e esses possíveis mecanismos são substituição nucleofílica do tipo SN2 ou transferência de um elétron,36 esse último também conhecido pela sigla SET (single-electron transfer). Muitos autores já buscaram desvendar esses dois mecanismos,37,38,39,40 entretanto, cada caso deve ser estudado minunciosamente a fim de concluir sobre qual mecanismo está em ação.
Esquema 6. Origem do carbocátion fluorado obtido através de dois mecanismos, SN2 ou SET (fonte: adaptado de Nyffeler e colaboradores)36
Exemplos modernos de aplicação de fluoração sob condições eletrofílicas Em 2022, Peters e colaboradores41 relataram a fluoração catalítica assimétrica enantiosseletiva de isoxazolinonas formando um centro estereogênico α-carbonílico contendo átomo de flúor, um tipo de catálise muito rara. A motivação desse trabalho foi de obter precursores de γ-aminoálcoois, β-aminoácidos e β-lactamas de interesse medicinal. N-fluorobenzenosulfonamida (NFSI) foi adotado como reagente de fluoração, paladaciclos quirais contendo ferroceno foram empregados como catalisadores, diferentes sais de prata foram responsáveis pela ativação do catalisador, ácido acético ou NaOAc foram empregados como aditivo e clorobenzeno ou tolueno atuaram como solvente da reação. As condições otimizadas da reação estão apresentadas no Esquema 7.
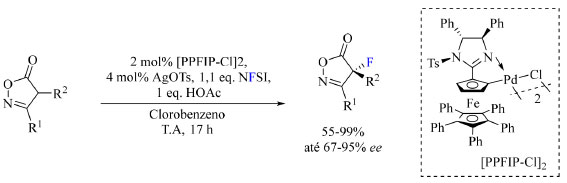 Esquema 7. Condições de reação otimizadas para fluoração assimétrica de isoxazolinonas (fonte: adaptado de Peters e colaboradores)41
O mecanismo dessa reação consiste na coordenação do átomo de nitrogênio do substrato isoxazolínico com o Pd(II) na posição trans ao átomo de nitrogênio do catalisador. Após a coordenação ao paládio, há a enolização do substrato e duas possíveis conformações são esperadas: A, que resulta no produto principal, e B, que apresenta repulsão estérica entre o substituinte do substrato e o ferroceno e entre um dos oxigênios com o ligante cloro do Pd (Figura 8). Por fim, o reagente de fluoração reage com a face do enol mais distante do ferroceno, menos impedida, levando à formação do centro estereogênico com a configuração observada pelos autores.
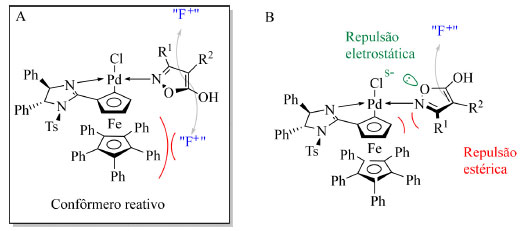 Figura 8. Possíveis conformações do complexo organometálico após etapa de enolização: à esquerda a conformação mais reativa e à direita a menos favorecida (fonte: adaptado de Peters e colaboradores)41
Como apresentado no Esquema 8, os autores demonstraram que esse método é compatível com substituintes R2 benzil, nos quais o anel aromático contém átomos e grupos funcionais como haletos, ciano, nitro, alcóxi e alquil, ligados ao substrato. Quando R2 são grupos alquil, etiloxicarbonilmetil, alil, propargil e fenil, foi constatada diminuição da enantiosseletividade. Além disso, grupos σ-aceptores e π-doadores como R1 são compatíveis com o método, enquanto grupos π-aceptores levam a diminuição do rendimento e da enantiosseletividade da reação.
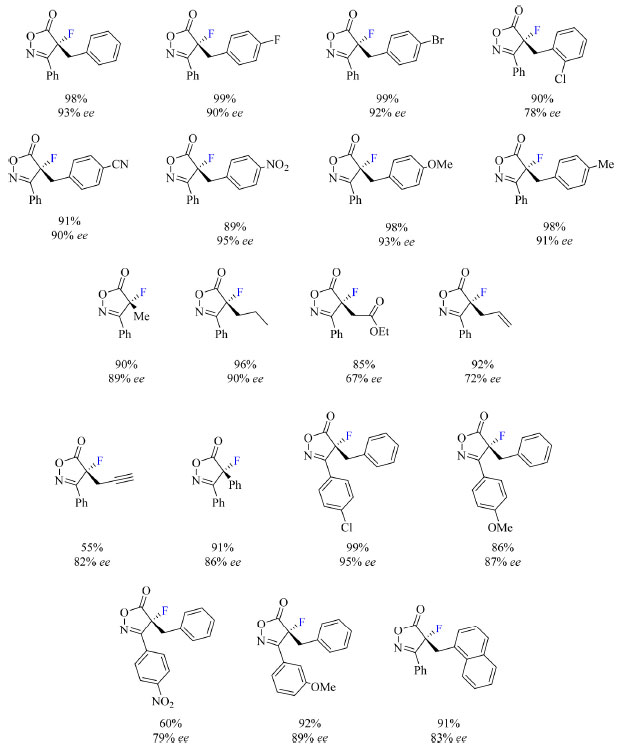 Esquema 8. Escopo do protocolo de fluoração assimétrica de isoxazolinonas com rendimento dos produtos isolados (fonte: adaptado de Peters e colaboradores)41
Nesses casos em que há diminuição do rendimento seria interessante mudar o reagente de fluoração para um que fosse mais reativo, como por exemplo NFPy 5 ou Selectfluor, e observar se há mudanças significativas. Os autores não deixaram clara a razão pela preferência por NFSI, portanto, seria válido testar outros reagentes, a fim de ampliar o escopo do trabalho. Outra sugestão seria, se possível, isolar o intermediário enolizado (Figura 8) e caracterizá-lo por espectroscopia de ressonância magnética nuclear e/ou difração de raios X (se for um sólido monocristalino). Deste modo seria possível se certificar que há mais de uma conformação através do experimento NOESY (nuclear overhauser enhancement spectroscopy), pois seria observado NOE (nuclear overhauser effect) entre os hidrogênios do substituinte do substrato e do ferroceno no confôrmero B, comprovando sua existência no meio reacional. ESI-MS (electrospray ionization-mass spectroscopy) de injeção direta também é uma técnica que poderia ser empregada na caracterização dos intermediários. Reagentes de fluoração eletrofílico podem atuar em dois mecanismos diferentes e os autores não deixaram claro como foi determinado que o mecanismo é SN2, uma vez que na Figura 8 está representada a espécie "F+", e não o SET. Tanto essa quanto a sugestão anterior seriam beneficiadas com um estudo teórico do trabalho. Em um trabalho sobre arilação enantiosseletiva catalisada por Pd(II) usando grupo diretor transiente (TDG, transient director group, do inglês) quiral desenvolvido por Park e colaboradores,42 foi questionado se a seletividade da eliminação redutiva poderia ser modificada alterando o ligante do intermediário Pd(IV)-F de modo a obter o produto fluorado, uma vez que espécies "F+" podem favorecer também a eliminação redutiva de outras ligações entre C(sp3) e heteroátomos (Esquema 9).
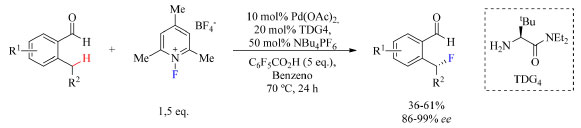 Esquema 9. Fluoração enantiosseletiva de 2-alquilbenzaldeídos substituídos como substratos catalisada por Pd(II) (fonte: adaptado de Park e colaboradores)42
Como grupos TDG os autores empregaram aminoácidos e observaram o aumento do rendimento do produto fluorado com o aumento estérico do TDG. O reagente de fluoração aplicado foi o NFPy 6 (Figura 4) e constatou-se que com aminoácidos menos volumosos esse reagente atua como oxidante auxiliar fornecendo o produto acetilado como principal, ao invés do fluorado. O uso de C6F5CO2H como aditivo da reação permitiu também obter o produto fluorado com alta enantiosseletividade e boa quimiosseletividade. A distinção entre reações de acetilação e de fluoração é que a etapa de eliminação redutiva da primeira ocorre por meio do mecanismo de SN2 enquanto a da segunda reação ocorre por esfera interna, como apresentado no Esquema 10.
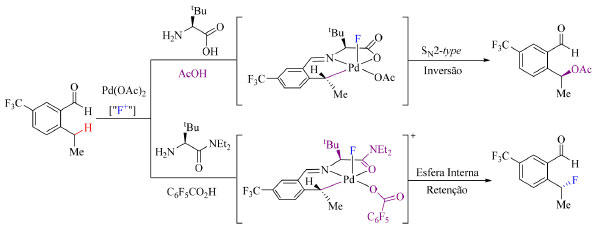 Esquema 10. Efeito do grupo diretor transiente na etapa de eliminação redutiva do Pd(IV) (fonte: adaptado de Park e colaboradores)42
A hipótese é que TDG's que apresentam o grupamento amida (NEt2) proporcionam a formação de um intermediário penta coordenado de Pd(IV) conveniente para a eliminação redutiva C(sp3)-F, isto devido à redução da energia do orbital a*(Pd-C), orbital molecular não ocupado de menor energia (LUMO), que afeta a propriedade da ligação Pd-F, o que por sua vez tem maior influência na eliminação redutiva por esfera interna. O escopo do trabalho nas condições otimizadas (Esquema 11) abrange substratos com grupos retiradores de densidade eletrônica, como CF3, nitro, carboximetil e isopropilcetona, halogenados e substratos com dois grupos, um doador (metil e metoxi) e outro retirador de elétrons em R1. Substratos contendo somente grupos doadores de elétrons são incompatíveis com a metodologia empregada. Substratos contendo flúor como substituinte R1 podem posteriormente ser submetidos a outras reações de modo a substituir a ligação C-F por ligações C-O, C-S ou C-N.
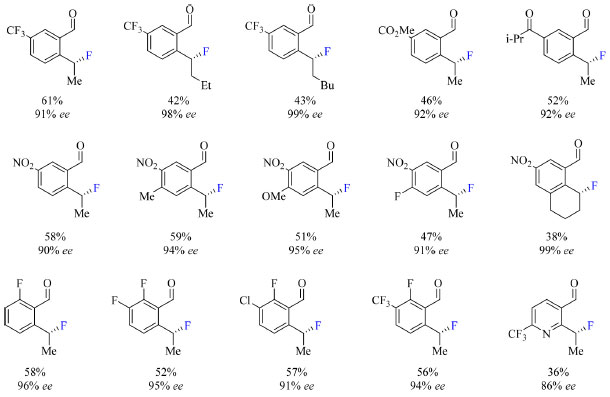 Esquema 11. Escopo do protocolo de fluoração enantiosseletiva catalisada por Pd(II) com rendimento do produto isolado ou obtido por RMN Ή (fonte: adaptado de Park e colaboradores)42
O ácido C6F5CO2H auxilia na redução da formação da ligação C(sp3)-O e no favorecimento da eliminação redutiva C(sp3)-F, no entanto, não há dados que sugerem que a quantidade de ácido foi otimizada, uma vez que os autores relataram o uso de 5 equivalentes. O uso excessivo de ácido sem necessidade pode acarretar maiores custos no tratamento do descarte e, consequentemente, a aplicação dessa reação em escala industrial não seria vantajosa. O último trabalho a ser exposto nessa sessão é de Yamamoto e colaboradores43 sobre fluoração C(sp2)-H direta aromática empregando catálise metálica com paládio como mostrado no Esquema 12. Essa abordagem não gera intermediários organometálicos envolvendo o substrato, algo que é comum em funcionalização C-H em aromáticos, em vez disso uma espécie eletrofílica de Pd reativa é gerada cataliticamente para a fluoração de arenos, no qual seus ligantes favorecem a oxidação do complexo antes de interagir com o substrato. Essa abordagem tem destaque por gerar uma espécie reativa, eletrofílica no átomo de flúor e capaz de transferir flúor para sistema arenos.
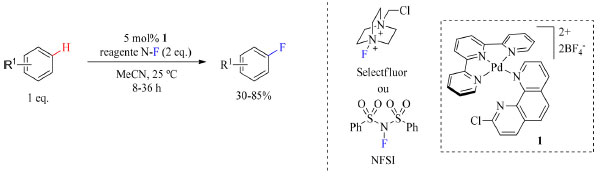 Esquema 12. Fluoração de arenos catalisada por Pd na presença de Selectfluor ou NFSI (fonte: adaptado de Yamamoto e colaboradores)43
O catalisador é um complexo de Pd(II) que é oxidado por NFSI ou Selectfluor para obter um complexo de Pd(IV)-F. O único estado de transição é caracterizado por ser diradicalar singleto e o mecanismo da reação compreende em duas transferências eletrônicas acopladas com flúor que ocorrem de modo assíncrono, como demostrado no Esquema 13.
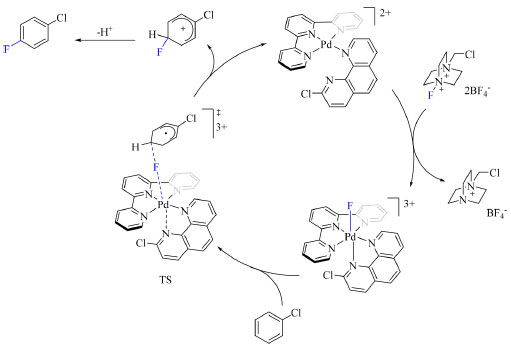 Esquema 13. Ciclo catalítico para a fluoração do clorobenzeno (fonte: adaptado de Yamamoto e colaboradores)43
O escopo do trabalho compreende grupos funcionais como nitrilas, brometos de arila, cloretos, heterociclos, sulfonamidas, cetonas, amidas, estéres, carbamatos, éteres e hidroxilas, e não é compatível com heteroarenos de cinco membros com grupos funcionais oxidativamente lábeis como aminas e tióis por serem incompatíveis com reagentes de fluoração eletrofílica como o Selectfluor e o NFSI (Esquema 14). As reações foram realizadas a temperatura ambiente ou a 50 °C em acetonitrila, como apresentado no Esquema 12.
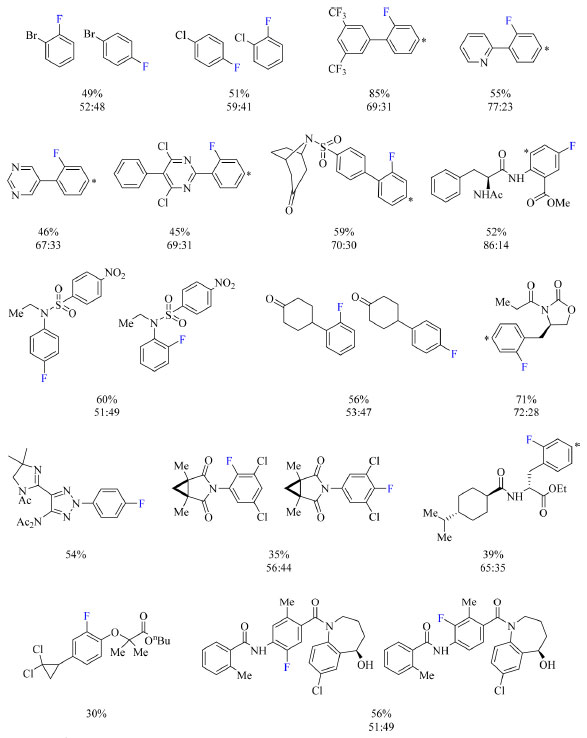 Esquema 14. Escopo do protocolo de fluoração de arenos com rendimento dos produtos obtido por RMN de 19F (fonte: adaptado de Yamamoto e colaboradores).43 O arterisco denota a posição no anel aromático de fluoração do isômero constitucional não mostrado
A inserção do átomo do flúor no substrato se dá por um mecanismo radicalar, no entanto, essa inserção é considerada como uma condição eletrofílica, pois os reagentes de fluoração transferem "F+" para o Pd nas condições de estudo. Outros trabalhos corroboram que o mecanismo dessa reação seja entendido como substituição eletrofílica aromática (SEAr).44 Além disso, o protocolo apresenta um problema de regiosseletividade, uma vez que em alguns substratos mais de uma posição, ativada pelo substituinte, foi fluorada. Os trabalhos apresentados não trazem substratos fortemente nucleofílicos e são métodos modernos de inserção de átomos de flúor. Isso demonstra que atualmente o desenvolvimento de novos métodos visa ampliar as classes de compostos que podem sofrer fluoração sob condições brandas empregando reagentes de fluoração conhecidos e simples de serem preparados.
MÉTODOS DE INSERÇÃO DE ÁTOMOS DE FLÚOR SOB CONDIÇÕES NUCLEOFÍLICAS Nas fluorações sob condição nucleofílica, o substrato atua como eletrófilo enquanto o flúor atua como nucleófilo. Levando em consideração as propriedades do átomo de flúor seria esperado que o método mais simples de sua inserção em uma molécula orgânica seria a substituição de um grupo de saída pelo flúor (via SN2). Todavia essa reação não é tão simples assim, uma vez que o ânion fluoreto tem alta energia de hidratação atuando mais como base e sendo um nucleófilo fraco.2,45 A fim de contornar essa questão, HF pode ser ligado a uma espécie básica, como trietilamina (16) ou piridina (17), no entanto, esses complexos podem afetar negativamente reações orgânicas ao reduzir a acidez do meio e/ou complexar fortemente com catalisadores de metal de transição prejudicando seu desempenho.1 Ou então podem ser empregadas outras fontes de flúor tais como fluoretos de metais alcalinos (18-20), silicatos ou estananas hipervalentes (21-22), fluoreto de tetrabutilamônio (23) e iodo hipervalente (24), como mostrado na Figura 9.
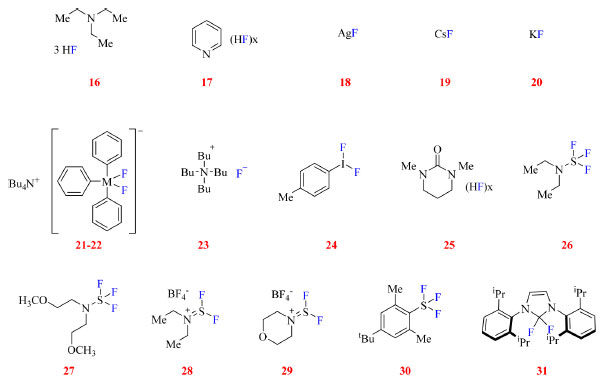 Figura 9. Reagentes de fluoração nucleofílica (fonte: adaptado de Hollingworth e Gouverneur, e Paquin e colaboradores)45,21
Os fluoretos de metais alcalinos são abundantes, no entanto, apresentam baixa solubilidade em solventes orgânicos.46 Esse problema, em alguns casos, pode ser resolvido usando álcoois volumosos para que o flúor seja transferido para a fase orgânica e possa vir a reagir como nucleófilo. O par iônico M+F− (M = K ou Cs, por exemplo) interage via ligação de hidrogênio com moléculas do álcool volumoso comprometendo a força da ligação iônica MF, o que por sua vez leva a solvatação do íon fluoreto, permitindo-o atuar como nucleófilo, como ilustrado na Figura 10.1,47 De acordo com os cálculos de DFT (density functional theory) e as simulações de dinâmica molecular conduzidas por Pliego Junior,48 nos casos nos quais o metal ligado ao flúor ionicamente é potássio e o álcool volumoso o terc-butanol, o par iônico KF é solvatado por cinco moléculas de solvente em média, sendo que o íon fluoreto participa de quatro ligações de hidrogênio com as moléculas do álcool, e o íon de potássio apresenta quatro átomos de oxigênio ao seu redor provenientes também do álcool. Em resumo, três moléculas de solvente solvantam tanto F− quanto K+, uma molécula extra interage com o fluoreto e uma outra molécula extra interage somente com o íon de potássio. Chi e colaboradores,47 pesquisadores que desenvolveram esse tipo de reação de fluoração SN2 usando álcool como solvente, também reportaram que o uso de álcoois volumosos são eficientes na supressão da formação de produtos laterais.
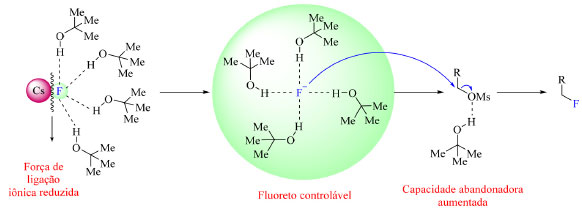 Figura 10. Efeito de interações do tipo ligação de hidrogênio intermolecular na nucleofilicidade de fluoreto de césio (fonte: adaptado de Liang e colaboradores)1
Uma outra abordagem para que o aumento da solubidade de sais de fluoreto possa ser alcançado é o uso de macrocíclicos, os quais também atuam interagindo com ambas espécies, cátions e ânions, presentes no meio reacional, solubilizando o sal e ativando o nucleófilo (íon F-).49 Alguns exemplos desenvolvidos nos últimos anos (Figura 11) incluem os trabalhos de Kim e colaboradores50,51 com os macrocíclicos nomeados de BACCA e BTC5A, os quais são destaques por serem constituídos por um éter de coroa que atua como quelante do cátion metálico e cadeias contendo álcoois que interagem via ligação de hidrogênio com o íon fluoreto, e os trabalhos de Pliego Junior e colaboradores52,53,54 que empregam éteres de coroa da classe de 18-crown-6, os quais atuam como quelantes do cátion metálico, juntamente com álcoois volumosos que interagem via ligação de hidrogênio com o íon fluoreto.
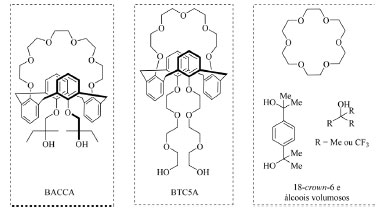 Figura 11. Macrocíclicos empregados em reações de fluoração nucleofílica (fonte: adaptado de Kim e colaboradores e de Pliego Junior e colaboradores)50,51,52,53,54
Uma terceira alternativa para o uso de sais de fluoreto, tais como fluoreto de prata e fluoreto de césio, em reações de fluoração de caráter nucleofílico é o emprego adicional de espécies catalíticas de Pd(0/II) e de Cu(I/III). Reações de acoplamento cruzado a fim de inserir um átomo de flúor em sistemas aromáticos foram reportados por Buchwald e colaboradores55 usando espécies de Pd(0/II), e por Liu e colaboradores56 usando espécies de Cu(I/III). A discussão e elucidação do mecanismo dessas transformações do ponto de vista computacional, através de cálculos de DFT, foram realizados por Pliego Junior,57,58 sendo considerada uma leitura essencial para aqueles interessados em se aprofundar no tema e obter conhecimento de cálculos teóricos de reações de fluoração. Reagentes tais como 1,3-dimetil-3,4,5,6-tetraidro-2(1H)-pirimidinona (DMPU, 25),59 trietilamina (16) e piridina (17)60 também são candidatos a interações do tipo ligação de hidrogênio intermolecular com HF (Figura 12). Em particular o DMPU, comparado a 16 e 17, exibe maior afinidade por realizar ligações de hidrogênio, menor afinidade em se coordenar a catalisadores de metal de transição, e consequentemente prejudicar o desempenho desses catalisadores, além de sua menor nucleofilicidade, o que significa que não compete com o HF em reações nucleofílicas.1
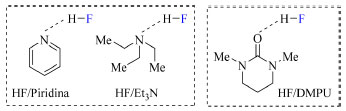 Figura 12. Interações do tipo ligação de hidrogênio intermolecular entre HF e piridina, Et3N e DMPU (fonte: adaptado de Liang e colaboradores)1
Para a realizar substituições nucleofílicas em substratos alifáticos uma alternativa encontrada foi desenvolver reagentes a partir de SF4 para preparar o fluoreto de alquila desejado através de álcoois, cetonas, aldeídos e ácidos carboxílicos (Esquema 15).2
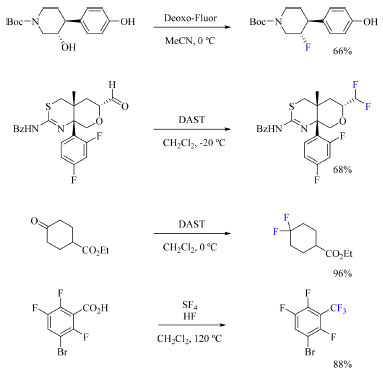 Esquema 15. Preparo de fluoretos de alquila através de reações de deoxifluoração e deoxofluoração (fonte: adaptado de Caron)2
Tetrafluoreto de enxofre (SF4) é produzido através de dicloreto de enxofre e fluoreto de sódio e é um gás termicamente instável e explosivo, o que esclarece o motivo pelo qual é pouco utilizado em reações de substituição nucleofílica.2 Na procura por reagentes mais estáveis e que não fossem gasosos, foram desenvolvidos o trifluoreto de dietilaminoenxofre (DAST, 26),61 que é líquido, e seu derivado mais estável termicamente, o trifluoreto de bis(2-metoxietil)aminoenxofre (Deoxo-Fluor, 27),62 cujas estruturas estão apresentadas na Figura 9. Tanto DAST quanto Deoxo-Fluor são preparados a partir da reação da amina de interesse com TMSCl seguida da reação com SF4 em Et2O. Em seguida, os sais de tetrafluoroborato aminodifluorosulfínio, XtalFluor-E (28) e XtalFluor-M (29) foram desenvolvidos entre 2009 e 2010,63,64 que apresentam melhoria na estabilidade térmica em relação aos reagentes anteriores. O Fluorolead (30) desenvolvido em 2010 apresenta boa estabilidade térmica e não é sensível a umidade,65 e, por fim, foi desenvolvido em 2011 o PhenoFluor (31)66 que apresenta melhora na seletividade em reações de deoxifluorações. Infelizmente, os reagentes 28, 29 e 31 necessitam de uma fonte de flúor adicional, como triidrofluoreto de trietilamina (16) e fluoreto de potássio (20) para alcançar conversões adequadas nas fluorações. Exemplos modernos de aplicação de fluoração sob condições nucleofílicas Em 2021, Zhang e colaboradores67 desenvolveram um novo protocolo de síntese de anilidas fluoradas que não dispõe do uso de metais ou de oxidantes. A motivação do trabalho foi de introduzir átomo de flúor em aminas aromáticas, uma classe de compostos que é amplamente explorada nas indústrias farmacêuticas, agroquímica e de pigmentos (Esquema 16).
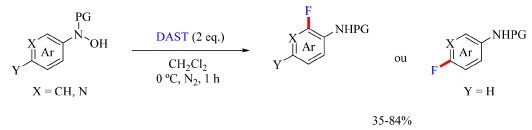 Esquema 16. Condições otimizadas de fluoração de aminas aromáticas (fonte: adaptado de Zhang e colaboradores)67
Foram testados diferentes reagentes de fluoração nucleofílica e o DAST originou os melhores resultados. Diclorometano, dimetilformamida, tolueno, dioxano, acetonitrila, acetato de etila, tetracloreto de carbono e clorofórmio foram os solventes testados e clorofórmio foi o escolhido para o desenvolvimento do escopo do trabalho. Também foi observado que o uso de aditivos destinados a promover desprotonação não teve influência no rendimento. As condições otimizadas podem ser encontradas no Esquema 16. O escopo do trabalho, como apresentado no Esquema 17, abrange grupos de proteção no nitrogênio como benzoíla, 9-fluoroenilmetoxicarbonila (Fmoc), benziloxicarbonila (Cbz), terc-butoxicarbonila (Boc), pivaloila (Piv) e acetila. Com N-naftilidroxilaminas contendo o grupo de proteção benzoíla foram testados grupos doadores e retiradores de densidade eletrônica como substituinte Y (Esquema 16) e o método foi compatível com todos estudados, como bromo, fenila, metoxila e anéis de naftil contendo metoxila e cloro. N-fenilidroxilamina substituídas com grupos OCF3, OCF2H, F, metila e etila também foram compatíveis com o método, assim como os substratos contendo o anel de piridina com bromo, metila, metoxila e fenila como exemplos de substituintes.
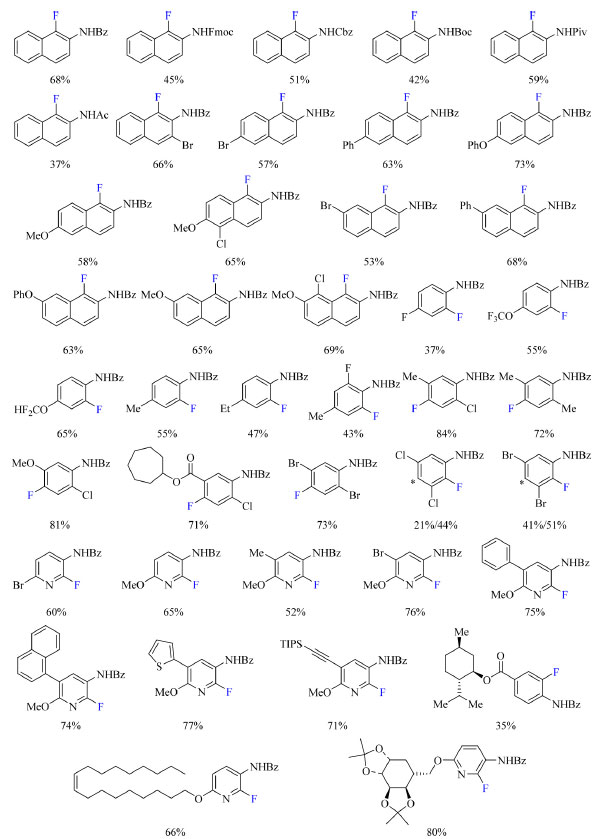 Esquema 17. Escopo do protocolo de fluoração de aminas aromáticas com rendimento dos produtos isolados (fonte: adaptado de Zhang e colaboradores)67
O mecanismo proposto inclui substituição nucleofílica de um átomo de flúor no reagente DAST pelo substrato, seguida da clivagem da ligação N-O formando uma espécie iônica. Essa espécie iônica por sua vez é transformada no carbocátion termodinamicamente mais estável de acordo com o substituinte do substrato, processo que é prontamente acompanhado do ataque do carbocátion por um íon fluoreto. O produto é obtido então após rearomatização, como demostrado no Esquema 18.
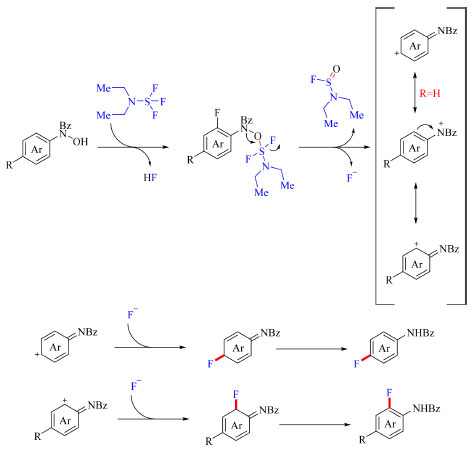 Esquema 18. Mecanismo proposto para fluoração de aminas aromáticas com DAST (fonte: adaptado de Zhang e colaboradores)67
O rendimento dos produtos isolados varia entre 35-84% e uma sugestão para aumentar esses números seria adicionar mais uma espécie no meio reacional que se ligue ao íon fluoreto liberado, de modo a aumentar sua nucleofilicidade e solubilidade no solvente da reação. Essa espécie adicionada não poderia conter grupos funcionais cetona e hidroxila para evitar uma reação lateral com DAST, e poderia interagir por meio de ligação de hidrogênio com o íon fluoreto para aumentar sua nucleofilicidade. Poderia ser usado por exemplo piperidina ou diisopropiletilamina (DIPEA) que são espécies não nucleofílicas. Gouverneur e colaboradores68 empregaram KF como reagente de fluoração e N-etil bis-ureia quiral como catalisador na síntese enantiosseletiva de β-fluoroaminas (Esquema 19). Foi descoberto que um catalisador de ureia pode interagir com íon fluoreto via ligação de hidrogênio e transportá-lo para a solução.
Esquema 19. Síntese enantiosseletiva de β-fluoroaminas 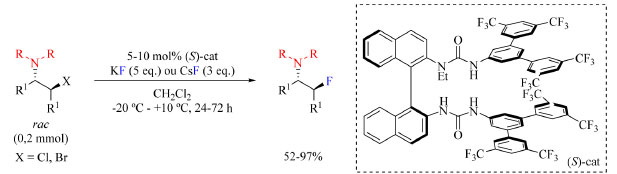 a partir de fluoreto de metal alcalino e catalisador com núcleo de ureia (fonte: adaptado de Gouverneur e colaboradores)68 a partir de fluoreto de metal alcalino e catalisador com núcleo de ureia (fonte: adaptado de Gouverneur e colaboradores)68
Para otimização do protocolo, os autores utilizaram um derivado de estilbeno β-cloro-N-dialilamina como substrato com diferentes catalisadores a base de ureia. Os catalisadores com substituintes mais volumosos na porção da amida acarretaram o aumento da enantiosseletividade da reação acompanhado da diminuição do rendimento. Tomando como mais importante a enantiosseletividade ao invés do rendimento, o catalisador mais volumoso foi escolhido para o desenvolvimento do escopo do trabalho. A temperatura da reação, assim como o solvente, também foi otimizada, empregando-se temperaturas de −15 °C, 0 °C e ambiente, e solventes como diclorometano e clorofórmio. As condições otimizadas estão apresentadas no Esquema 19. O escopo do trabalho (Esquema 20) inclui substratos contendo substituinte R como sendo N-heterociclos, R com dois diferentes N-substituintes que podem levar a formação de dois diastereoisômeros, e R1 sendo fenilas substituídas com grupos doadores de densidade eletrônica tais como Me e OMe, e halogênios. As razões enantioméricas não foram afetadas pela natureza do fluoreto, KF ou CsF.
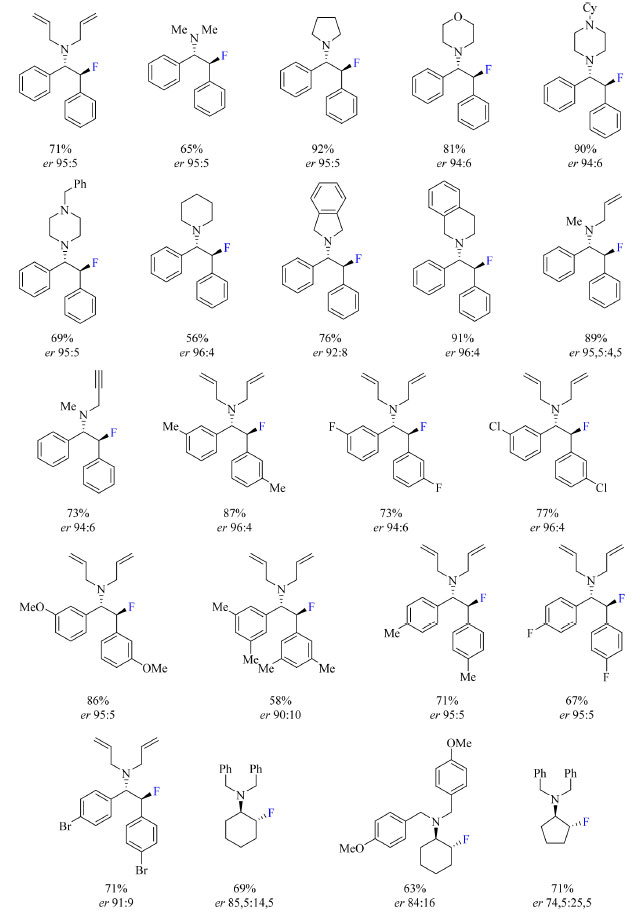 Esquema 20. Escopo do protocolo de síntese enantiosseletiva de β-fluoroaminas a partir de catalisador de ureia com rendimento dos produtos isolados (fonte: adaptado de Gouverneur e colaboradores)6
O mecanismo da reação é constituído da tricoordenação do íon fluoreto com o catalisador formando um complexo que logo em seguida é pareado com o substrato, resultando na fluoração do mesmo e restauração do catalisador, como demostrado no Esquema 21.
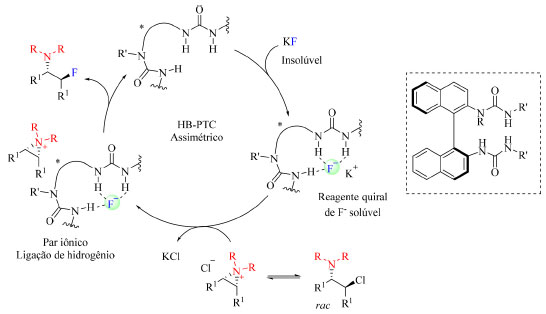 Esquema 21. Mecanismo proposto para síntese de β-fluoroaminas a partir de KF e catalisador com núcleo de ureia (fonte: adaptado de Gouverneur e colaboradores)>68
Apesar da economia associada ao uso de reagentes de fluoração de metais alcalinos, os melhores resultados em questão de enantiosseletividade foram obtidos utilizando catalisadores que empregam em sua síntese reagentes bastante custosos, o que significa que não há grandes melhorias no plano econômico quando comparado com outros trabalhos. Além disso, seria interessante incluir no escopo mais substituintes em R1 que incluam grupos nitro e alquil, por exemplo. Por fim, no Esquema 22 são apresentados outros métodos realizados sob condições nucleofílicas que incluem substituição de um grupo ligante diferente de hidrogênio pelo átomo de flúor (a),69 alquilfluoração de olefinas (b)70 e inserção de flúor em sistemas aromáticos por reação eletroquímica (c).71
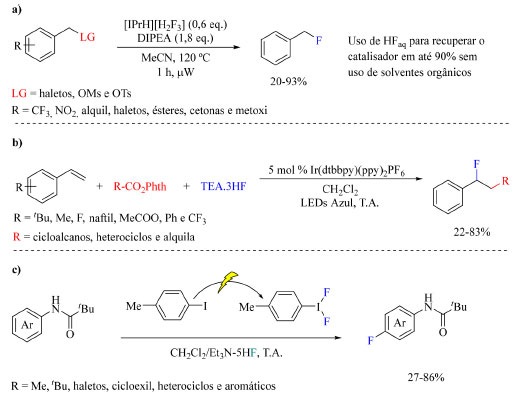 Esquema 22. Métodos alternativos de fluoração sob condições nucleofílicas: (a) substituição de grupo ligante por átomo de flúor,669 (b) alquilfluoração fotoredox de olefinas700 e (c) inserção de F com reação eletroquímica71
Quando comparados com os métodos de fluoração sob condições eletrofílicas as condições nucleofílicas podem apresentar maior economia atômica, menor custo e serem mais amigáveis do ponto de vista ambiental, sendo então mais vantajosas em escala industrial. Contudo, a maioria das fontes de fluoreto tem baixa solubilidade em solventes orgânicos e as reações envolvendo esses reagentes são sensíveis a umidade, uma vez que a água diminui a nucleofilicidade do íon fluoreto e pode ainda levar a formação de produtos de hidrólise.
MÉTODOS DE INSERÇÃO DE ÁTOMOS DE FLÚOR SOB CONDIÇÕES NEUTRAS1, 2, 72 Os métodos de inserção de flúor sob condições neutras compreendem os mecanismos do tipo radicalar, nos quais há a geração do radical F· que reage com um radical de uma molécula orgânica geralmente gerado in situ. Reagentes como F2, hipofluoritos e XeF2 podem ser utilizados na fluoração radicalar, em especial o F2 que tem propensão a clivar homoliticamente sua ligação em razão da sua baixa energia de dissociação de cerca de 37 kcal mol−1. O empecilho ao uso desses reagentes, como já comentado, é sua alta toxidade e difícil manipulação em laboratório. Uma alternativa encontrada para contornar os problemas com os reagentes mencionados acima foi empregar reagentes de fluoração eletrofílica com força de ligação reduzida, tais como os reagentes contendo ligação N-F: NFSI, Selectfluor, os sais de N-fluoropiridínio e N-fluoro-N-arilsulfonamidas, que podem ser utilizados de forma eficiente sob condições brandas. Antes de se aprofundar nos métodos de fluoração é necessário esclarecer conceitos básicos sobre radicais e reações radicalares. Por exemplo, espécies radicalares podem ser formadas através de homólise de ligações σ fracas (a) e transferência de elétron de uma molécula com spin pareado (b), ou através de outros radicais por meio de processos de substituição (c), adição (d) e eliminação (e), como exemplificado no Esquema 23.
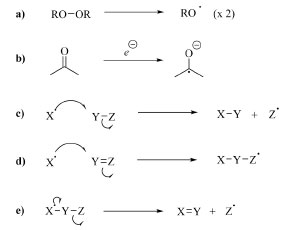 Esquema 23. Métodos de formação de espécies radicalares: (a) homólise, (b) redução, (c) substituição, (d) adição e (e) eliminação (fonte: adaptado de Clayden e colaboradores)72
Radicais podem ser estabilizados por grupos retiradores de elétrons por meio da sobreposição do orbital p do radical (single occupied molecular orbital, SOMO) com o orbital π* vazio do grupo substituinte, gerando um novo orbital SOMO de menor energia (Esquema 24) e de caráter eletrofílico (Esquema 25). Radicais podem ser estabilizados por grupos doadores de elétrons por meio da interação entre o SOMO do radical e o par de elétrons não ligante do grupo doador de elétrons que, apesar de gerar um orbital SOMO de maior energia e de caráter nucleofílico (Esquema 25), gera também um orbital de menor energia para acomodar os elétrons do grupo substituinte (Esquema 26). A terceira e última via de estabilização é por meio da conjugação e deslocalização do radical com os elétrons de ligações σ C-H.
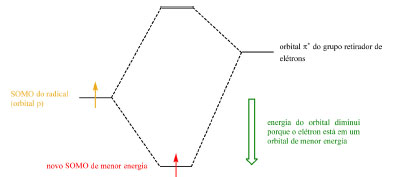 Esquema 24. Estabilização de radical por grupo retirador de elétrons (fonte: adaptado de Clayden e colaboradores)72
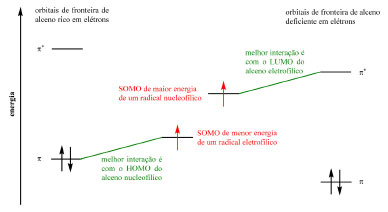 Esquema 25. Representação do caráter eletrofílico e nucleofílico de radicais (fonte: adaptado de Clayden e colaboradores)72
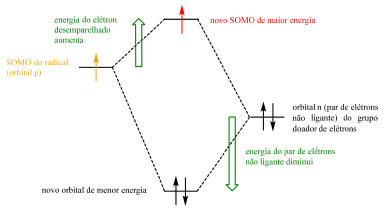 Esquema 26. Estabilização de radical por grupo doador de elétrons (fonte: adaptado de Clayden e colaboradores)72
As reações radicalares são caracterizadas por três etapas: iniciação, como apresentado no Esquema 23, propagação, na qual o radical reage com outra(s) espécie(s) formando outros radicais, e por fim há a etapa de terminação na qual é formada uma espécie não radicalar. Alguns exemplos de reações radicalares serão abordados a seguir. Exemplos modernos de aplicação de fluoração sob condições neutras Em 2022, Pieber e colaboradores73 publicaram um estudo sobre a fluoração de derivados de ácido fenilacético na presença de Selectfluor e 4-(dimetilamino)piridina (DMAP) e surpreendentemente detectaram a formação de dois produtos diferentes quando empregada água ou acetonitrila como solventes da reação (Esquema 27). A formação de ligações C(sp3)-F em sistemas benzílicos na presença de Selectfluor é uma das mais estudadas reações que ocorrem por meio de mecanismo radicalar e seu estudo visa modificar candidatos a medicamentos e prevenir oxidação benzílica indesejada.
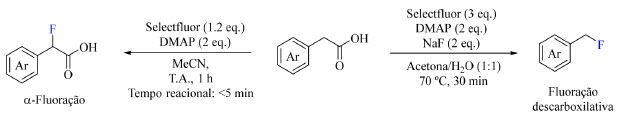 Esquema 27. Condições otimizadas da fluoração de derivados de ácido fenilacético destacando a α-fluoração (à esquerda) e a fluoração descarboxilativa (à direita) (fonte: adaptado de Pieber e colaboradores)73
Os autores pretendiam formar um complexo de transferência de carga (CT) entre o Selectfluor e a piridina, de modo a gerar uma espécie dicatiônica radicalar (TEDA2+·), que é um reagente de transferência de átomo de hidrogênio e potente oxidante que pode gerar o radical no substrato e, posteriormente, auxiliar na fluoração. Ao admitir ácido (4-fluorofenil)acético como substrato foi observada a formação do produto de fluoração descarboxilativa em acetona:água e na presença de NaF com aquecimento, enquanto o produto de fluoração direta foi o majoritário em acetonitrila a temperatura ambiente (Esquema 27). O escopo do trabalho abrange substratos com substituintes doadores e retiradores de densidade eletrônica, tais como: haletos, fenila, alquila, metoxila, CF3 e nitro (Esquema 28). Curiosamente ao substituir o ácido carboxílico do substrato por outros grupos funcionais como cetonas, ésteres e amidas os autores não observaram a formação de produto.
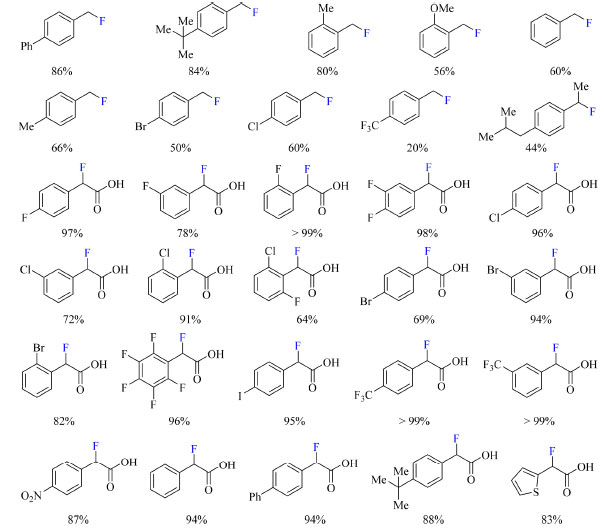 Esquema 28. Escopo do protocolo de fluoração de derivados de ácido fenilacético via α-fluoração ou via fluoração descarboxilativa com rendimento obtido por RMN de 1H (fonte: adaptado de Pieber e colaboradores)73
O produto descarboxilado é obtido através do processo de transferência de elétron (single-electron transfer, SET), enquanto o produto de fluoração direta é obtido pelo processo de transferência de átomo de hidrogênio (HAT, hydrogen atom transfer, do inglês) (Esquema 29). A designação de qual processo ocorre se dá por meio do pKa do substrato e do DMAP nas condições de reação: os substratos estudados apresentam baixa acidez em solventes apróticos polares como acetonitrila e não são desprotonados nesse meio, por consequência não há o íon carboxilato, o que reduz a probabilidade de ocorrer SET.
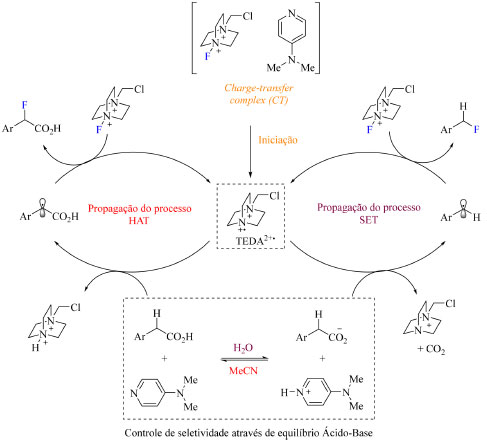 Esquema 29. Mecanismo proposto para a fluoração de derivados de ácido fenilacético indicando os diferentes mecanismos possíveis e a etapa de equilíbrio modulada pelo solvente (fonte: adaptado de Pieber e colaboradores)73
Há uma diferença significativa entre os rendimentos calculados por RMN de 1H e o dos produtos isolados, por exemplo de 95% para 35%, e a explicação dos autores é de que há dificuldades na separação por coluna cromatográfica. Cho e colaboradores74 abordam a trifluorometilação de hidroquinonas por foto-irradiação de derivados de benzoquinonas com CF3SO2Na. Hidroquinonas são conhecidas por suas propriedades antivirais e antifúngicas e com a trifluorometilação dessa classe de moléculas é esperado obter melhoria nas suas atividades biológicas (Esquema 30).
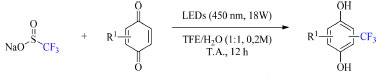 Esquema 30. Condições otimizadas da trifluorometilação de derivados de benzoquinonas (fonte: adaptado de Cho e colaboradores)74
O mecanismo da reação compreende a geração in situ de hidroquinona a partir do substrato de benzoquinona em solvente prótico com foto-irradiação, seguida da formação de um complexo CT entre o substrato (espécie deficiente de elétrons) e a hidroquinona (espécie rica em elétrons), o qual é estabilizado por ligações de hidrogênio e interações π-π. Posteriormente, é adicionado o reagente de trifluorometilação, CF3SO2Na levando a formação de um complexo CT ternário intermediário e, logo em seguida, do produto de acordo como está demostrado no Esquema 31.
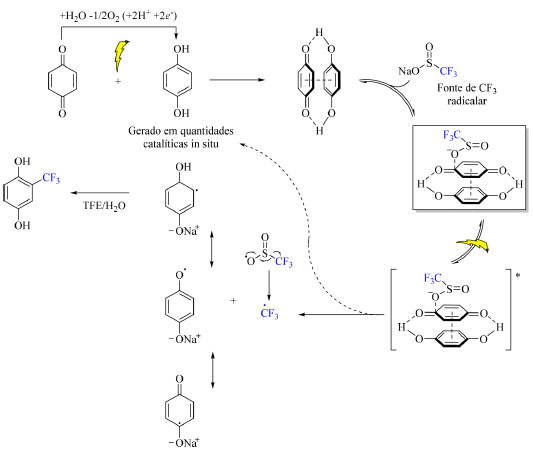 Esquema 31. Mecanismo proposto para trifluorometilação da benzoquinona sob foto-irradiação (fonte: adaptado de Cho e colaboradores)74
Foram realizados testes com diversos solventes, como acetonitrila, trifluoroetanol (TFE), metanol, dimetilformamida, dimetilsulfóxido, benzeno e água, e a mistura 1:1 TFE:H2O exibiu os melhores resultados. A ausência de luz visível ou irradiação abaixo da região do ultravioleta não levou a formação do produto e o uso de aditivos como fotocatalisadores a base de Ir e Ru levaram a diminuição do rendimento causada pela competição entre processos fotoredox. As condições otimizadas se encontram no Esquema 30. Substratos com grupos doadores de elétrons, grupos retiradores de elétrons, e haletos são compatíveis com o protocolo (Esquema 32). Seria interessante investigar o que aconteceria se houvesse um grupo amina no substrato, pois poderia haver competição entre interações do tipo ligação de hidrogênio.
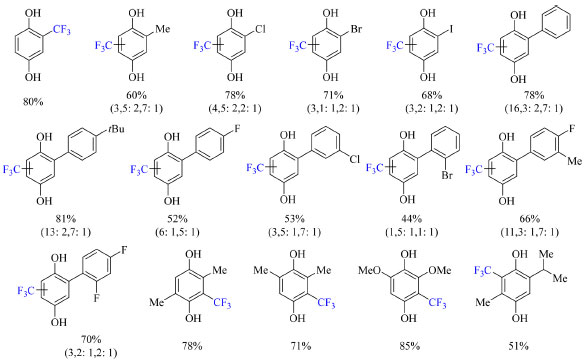 Esquema 32. Escopo do método de trifluorometilação de derivados da benzoquinona via foto-irradiação. Nos casos pertinentes a razão dos regioisômeros formados nas posições 3, 5 e 6 do anel da hidroquinona se encontra seguido do rendimento, o qual é dos produtos isolados ou obtido por RMN de 19F devido a voltatilidade dos produtos (fonte: adaptado de Cho e colaboradores)74
O trabalho desenvolvido por Lectka e colaboradores75 na fluoração de cetais emprega tanto o mecanismo HAT, observado no exemplo de fluoração de derivados do ácido fenilacético,73 quanto o uso da fotoquímica, como apresentado no último exemplo discutido.74 Esse novo método visa a síntese de novos açúcares e esteroides fluorados, o que é desafiador devido a proposta dos autores de realizar a fluoração dos substratos de forma regiosseletiva. O melhor rendimento da reação foi alcançado empregando a luz de uma lâmpada fluorescente compacta (CFL, do inglês compact fluorescent light) na presença de xantona e do reagente de fluoração Selectfluor, tal como é apresentado no Esquema 33.
 Esquema 33. Condições otimizadas da fluoração de moléculas policíclicas complexas contendo grupo cetal (fonte: adaptado de Lectka e colaboradores)75
A otimização da reação também abrangeu o uso de NFSI como reagente de fluoração, luz no comprimento de onda de 300 nm, aumento da temperatura da reação para 80 °C e a substituição da xantona por fluorenona, benzofenona e 5-dibenzosuberenona. Os autores também relatam que o tempo de reação é fundamental para o rendimento do produto de interesse, uma vez que longas durações promoveram a decomposição do produto em alguns casos. O escopo da reação compreende moléculas policíclicas complexas, tais como derivados de glicose, galactose, frutose, manose e vitamina C, e glicoesteroides (Esquema 34). O mecanismo da reação proposto pelos autores (Esquema 35) é apoiado por cálculos de DFT (density functional theory) da estrutura eletrônica das espécies envolvidas, os quais incluem a caracterização do estado de transição e a discussão da regiosseletividade. O mecanismo se inicia pela formação de um radical dicatiônico do Selectfluor via fotoirradiação por CFL, em seguida é abstraído um átomo de hidrogênio geminal à um dos átomos de oxigênio do substrato, levando à formação de um radical estável, que por sua vez é fluorado pelo Selectfluor, formando novamente um radical dicatiônico do Selectfluor constituindo uma reação em cadeia.
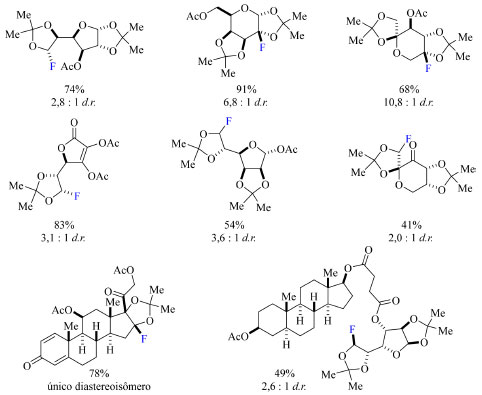 Esquema 34. Escopo do método de fluoração de moléculas policíclicas complexas contendo grupo cetal, com os rendimentos obtidos por RMN de 19F (fonte: adaptado de Lectka e colaboradores)75
Para encerrar a discussão sobre fluorações sob condições neutras no Esquema 36 estão representados alguns métodos adicionais relatados recentemente, especificamente os protocolos de Yu e colaboradores76 (a), Lectka e colaboradores77 (b), Zhu e colaboradores78 (c), Xiao e colaboradores79 (d), e Hong e colaboradores80 (e).
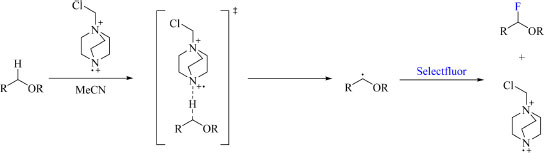 Esquema 35. Mecanismo proposto da fluoração de moléculas policíclicas complexas contendo grupo cetal via reação fotoquímica (fonte: adaptado de Lectka e colaboradores)75
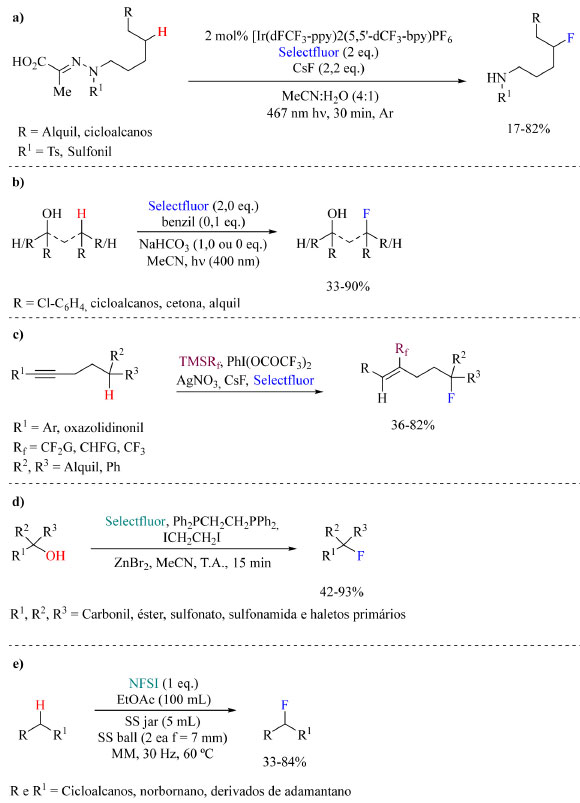 Esquema 36. Métodos alternativos de fluoração sob condições neutras: (a) fluoração δ-C-H,76 (b) fluoração dirigida por hidroxila,77 (c) fluoração dirigida por alcino,78 (d) fluoração dehidroxilativa de álcoois terciários79 e (e) fluoração mecanoquímica80
Uma grande parte dos estudos de fluoração de compostos orgânicos via mecanismo radicalar envolve o uso de Selectfluor como reagente de fluoração. Como comentado previamente, reagentes de fluoração eletrofílicos como o Selectfluor não são econômicos do ponto de vista atômico quando comparados com os reagentes nucleofílicos, no entanto, seu uso em mecanismos radicalares abre a uma nova gama de possíveis fluorações, como as fluorações remotas e fluoração de ligações C(sp3)-H não ativadas, e por consequência disso as condições neutras são as que estão sendo mais exploradas atualmente.
ESPECTROSCOPIA DE RESSONÂNCIA MAGNÉTICA NUCLEAR - RMN DE 19F Independentemente do método e condição de fluoração empregados nos trabalhos mencionados até o presente momento, todos, sem nenhuma exceção, empregam a técnica de espectroscopia de ressonância magnética nuclear (RMN) para elucidação dos compostos obtidos e até mesmo para cálculo de rendimento das reações sem ser necessária purificação dos compostos previamente a análise. A RMN em solução é imprescindível para a área de síntese orgânica, sendo umas das técnicas mais amplamente empregada atualmente nesta área, devido as suas vantagens como: é uma técnica não destrutível, quantitativa nas condições apropriadas de aquisição e processamento de espectros e de preparo de amostra, com sensibilidade razoável nas concentrações das espécies empregadas na síntese orgânica e de ser uma técnica que abrange uma ampla variedade de núcleos comumente presentes em moléculas utilizadas em reações orgânicas.81 Outro ponto interessante a ser destacado é que não é necessário ser um especialista e possuir conhecimento muito aprofundado dos fundamentos da técnica para obter e interpretar uma análise de RMN. Uma análise quantitativa de RMN permite determinar a concentração das espécies em solução e consequentemente o rendimento da reação.82 Geralmente, espectros quantitativos de 1H são mais explorados pelos químicos sintéticos para alcançar este propósito, no entanto, é possível também empregar os espectros de 19F quantitativos para cálculo de rendimento de brutos reacionais, economizando tempo e recursos, o que inclusive foi realizado por alguns dos trabalhos de fluoração citados.43,68,70,71,74,77,80 O 19F possui spin V2, assim como o 1H, com 100% de abundância natural e razão magnetogírica (γ) de 25,18034 × 107 rad T−1 s−1 muito próxima a razão magnetogírica do 1H que é de 26,75221 × 107 rad T−1 s−1, o que confere frequência de observação 0,94 da frequência de observação do 1H, espectros com excelente dispersão em função da ampla faixa de deslocamento químico (~ 400 ppm) quando comparado com 1H (~ 20 ppm), boa razão sinal/ruído e geralmente em moléculas orgânicas estão presentes em menor quantidade átomos de flúor do que de hidrogênio, o que resulta em espectros de 19F com um menor número de sinais do que um espectro de 1H, o que por sua vez é interessante para amostras que apresentam sobreposição de sinais no espectro de 1H. Combinada às vantagens citadas, a maior faixa de deslocamento químico do espectro de 19F em relação ao de 1H também torna esta opção mais desejável.83,84,85,86 Na Figura 13 é apresentado um espectro de 19F de um amostra contendo uma mistura de 5 espécies fluoradas, exemplificando a ampla faixa de deslocamento químico do 19F e a resolução espectral.
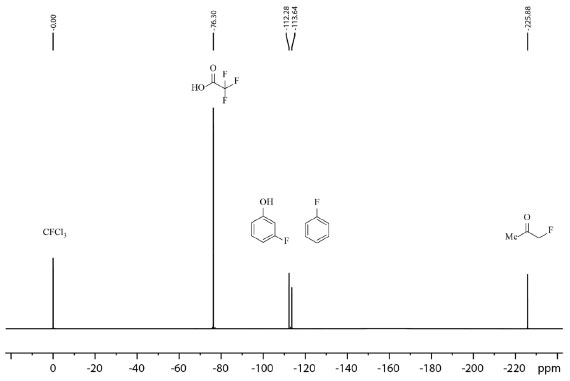 Figura 13. Espectro de 19F de amostra contendo uma mistura de triclorofluorometano, ácido trifluoroacético, 3-fluorofenol, fluorobenzeno e fluoroacetona obtido em equipamento Bruker Avance III a 235 MHz (19F) em CDCl3
Recentemente, as vantagens dos núcleos de 1H e 19F foram combinadas para a análise de amostras complexas e de misturas de compostos fluorados através do desenvolvimento e aplicação dos experimentos pertencentes a família FESTA (fluorine-edited selective TOCSY acquisition),87,88,89 nos quais são obtidos subespectros de 1H que apresentam somente os sinais dos hidrogênios que estão no sistema de spin acoplado a um certo átomo de flúor. Os sinais desses hidrogênios são observados devido a transferência de magnetização entre 19F e 1H acoplados escalarmente e selecionados de forma seletiva e através da transferência de magnetização via TOCSY para a rede de spin 1H-1H. A família FESTA compreende métodos eficientes e de rápida extração de informações estruturais de espécies fluoradas, possibilitando a observação de sinais provenientes de espécies com baixa concentração na mistura, obtenção dos deslocamentos químicos de todos os 1H e dos valores de constante de acoplamento homonuclear (JHH) e heteronuclear (JFH), assim como o sinal, positivo ou negativo, dos acoplamentos heteronucleares JFH. Um exemplo notável é o trabalho de Dal Poggetto e colaboradores87 no desenvolvimento e aplicação do experimento denominado HD-HAPPY-FESTA (homonuclear decoupled heteronuclear antiphase permuted modulated echo yielding fluorine-edited selective TOCSY acquisition) na separação espectral dos quatro isômeros da fluoração e posterior redução da 4-terc-butil-cicloexanona (Esquema 37) e medida do valor e sinal de acoplamentos JFH sem nenhuma etapa de purificação das misturas reacionais. Valores de acoplamento JFH variando entre 51,4 a −2,3 Hz foram medidos através deste experimento da família FESTA, sendo, possível medir valores de acoplamento da ordem de 0,8 Hz, muito próximos a largura de linha.
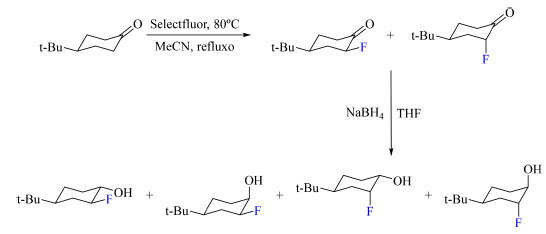 Esquema 37. Reação de α-fluoração da 4-terc-butil-cicloexanona com SelectFluor como agente de fluoração seguida da reação de redução gerando quatro isômeros de álcoois fluorados (fonte: adaptado de Dal Poggetto e colaboradores)87
Aplicações de RMN de 19F quantitativos requerem atenção especial uma vez que em função da ampla janela espectral (400 ppm) pulsos de radiofrequência com banda de excitação homogênea são difícies de serem obtidos. Em situações onde a janela espectral não é excitade de forma homogênea, distorções nas intensidades dos sinais mais afastados do centro da janela espectral ocorrem, comprometendo a acurácia da integração dos sinais e consequentemente da análise quantitativa. Este é um problema que pode ser observado até mesmo em espectros de 1H, no entanto, os efeitos são mais proiminentes para núcleos com janelas espectrais maiores, tais como 19F e 13C.90 O experimento CHORUS (chirped, ordered pulses for ultra-broadband spectroscopy) é composto de uma sequência de pulsos capaz de excitar com amplitude e fase constantes grandes janelas espectrais (250 kHz) usando uma amplitude máxima de pulsos de radiofrequência de 15 kHz, amplitude essa inferior que a de um típico pulso duro de 90° empregado por exemplo na sequência de RMN de 1H quantitativo. Morris e colaboradores90 demostraram a repetibilidade e robustez do método através de séries de medidas sob condições controladas, garantindo 99,8% de excitação da janela espectral para o núcleo de 19F, portanto, contornando o problema mencionado e garantindo a acurácia na integração dos sinais do espectro de 19F e sua análise quantitativa. O emprego da RMN para os químicos orgânicos não se restringe somente a obtenção de rendimento e experimentos para elucidação estrutural de produtos de reação, a técnica também pode ser empregada para monitorar reações e realizar estudos da cinética e análises do mecanismo da reação por meio da quantificação da concentração dos reagentes e produtos ao longo do tempo e da identificação dos intermediários.81 Os métodos explorados no monitoramento de reação por RMN incluem métodos ex situ e in situ. O método ex situ é compreendido pela condução da reação em bancada nas condições otimizadas com adição de um padrão interno, o qual irá viabilizar que a concentração das espécies em solução seja determinada através dos espectros quantitativos de RMN, uma alíquota é retirada antes de iniciar a reação e outras são retiradas em quenched ao longo do tempo. Por fim, as alíquotas são transferidas para o tubo de RMN e diluídas com solvente deuterado, e então são obtidos os espectros.81 Para o monitoramento in situ a reação é conduzida inteiramente dentro de um tubo de RMN, todavia não é possível agitar o tubo enquanto o mesmo estiver inserido na sonda do equipamento. Antes de iniciar a reação é obtido um espectro para que os parâmetros de aquisição, lock e shimming do equipamento sejam ajustados, em seguida é adicionado o último componente da reação que dará início a mesma e são obtidos espectros em tempo real em intervalos pre-estabelecidos. O tubo de RMN pode permanecer no equipamento durante todo o período da reação (monitoramento in situ contínio), pode ser retirado e inserido de tempos em tempos (in situ interrompido) ou se a reação for controlada por um estímulo externo, como calor e luz, pode-se empregar o monitoramento in situ por ativação periódica.81 A escolha do método é dependente da natureza da reação, do tempo e da disponibilidade do equipamento, em alguns casos podem ser empregadas técnicas especializadas em fluxo,81,91 como a de quenched-flow e RMN on-line, no entanto, são necessários outros tipos de instrumentação juntamente com o equipamento de RMN, o que traz uma limitação. Uma revisão recente81 foi publicada na forma de tutorial para monitoramento de reação por RMN, explicando em detalhes os métodos de monitoramento, a matemática por trás do estudo de cinética, as condições requeridas para ser realizado um experimento quantitativo, preparo de amostra, uso de padrões internos, tipos de tubos de RMN, detalhes da instrumentação e dos passos para se adquirir e processar um espectro, entre outros pontos que completam o que está sendo abordado neste presente artigo de revisão. Os autores também abordaram 15 estudos de casos que ilustram a aplicação das técnicas e experimentos e que demostram a eficiência e poder da RMN. Alguns dos maiores destaques no monitoramento de reação e processos por RMN de 19F residem nas áreas biológicas, como por exemplo no estudo da atividade de derivados fluorados do cofator da enzima sulfotransferase, responsável por processos biológicos envolvendo troca do grupo SO3.92 No estudo da interação entre proteínas (com resíduos de triptofano fluorados) e carboidratos (glicanos),93 na expressão em células humanas de proteínas a partir de aminoácidos fluorados com a intenção de observar a interação entre um alvo intracelular e seus inibidores,94 na identificação e quantificação de aminoácidos de uma mistura complexa (amostra metabolômica) através da reação dos mesmos com uma molécula fluorada,95 e um último exemplo é um estudo que visou monitorar a desativação de partículas semelhantes a vírus fluoradas,96 dentre outros exemplos atuais.97,98,99,100,101 Finalizando este tema, com o intuito de auxiliar no monitoramento de reações é possível agregar aos espectros em uma dimensão de 19F, como 19F e 19F{1H}, que são os mais conhecidos para esse núcleo, os experimentos em duas dimensões envolvendo 19F, tais como 1H-19F COSY,102 1H-19F HOESY103 e 19F DOSY.104,105 O experimento de COSY (correlation spectrocopy) tem como objetivo observar correlações entre os núcleos de 1H e 19F, o experimento de HOESY (heteronuclear overhauser effect spectroscopy) de detectar proximidade através do espaço (NOE heteronuclear), e o experimento de DOSY (diffusion ordered spectroscopy) de separar na dimensão de difusão compostos fluorados presentes em misturas de acordo com seu coeficiente de difusão na amostra. Na Figura 14 estão apresentados os mapas de contorno dos experimentos de 1H-19F COSY e 1H-19F HOESY do fármaco voriconazol, o qual é usado no combate a micoses nas unhas, a fim de visualização da aplicabilidade dos experimentos. Através do experimento COSY é possível observar correlação entre o átomo de flúor do anel da pirimidina e os hidrogênios deste mesmo anel e entre esse átomo de flúor e os hidrogênios ligados aos carbonos com hibridização sp3. Também é observada correlação entre o flúor na posição orto do benzeno com os dois hidrogênios aromáticos mais próximos e com os hidrogênios ligados aos carbonos com hibridização sp3. O átomo de flúor na posição para correlaciona somente com os três hidrogênios do benzeno. O experimento de HOESY esclarece quais são os hidrogênios próximos espacialmente a cada átomo de flúor: para o flúor da pirimidina somente é observado NOE com seu hidrogênio vizinho do anel e com o H (CH) ligado ao carbono sp3 mais próximo, para o flúor na posição orto do benzeno é observado NOE com o hidrogênio aromático vizinho, com H (CH) e H (CH2) ligados a carbonos sp3, e já para o flúor na posição para somente é observado NOE com os seus dois hidrogênios aromáticos vizinhos.
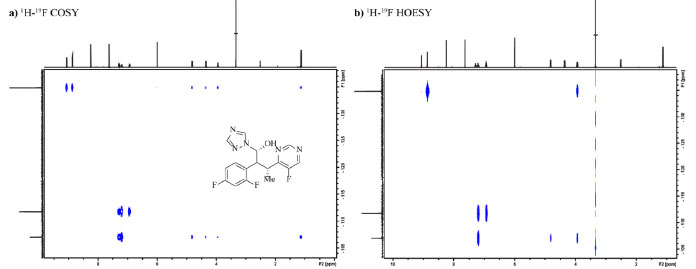 Figura 14. Mapas de contorno dos experimentos 1H-19F COSY (a) e 1H-19F HOESY (b) do fármaco voriconazol em DMSO-d6 obtidos em equipamento Bruker Avance III a 600 MHz (1H)
As versões homonucleares (1H) desses experimentos são rotineiros em síntese orgânica, no entanto, ainda se faz necessário difundir suas versões heteronucleares, uma vez que são experimentos que proporcionam muitas informações estruturais e que são ferramentas poderosas no monitoramento e elucidação estrutural de compostos orgânicos.
PERSPECTIVAS FUTURAS: O QUE PODE VIR A SEGUIR? Os avanços no desenvolvimento de reagentes de fluoração, tanto de caráter eletrofílico quanto nucleofílico, proporcionaram como consequência o desenvolvimento de inúmeros métodos de fluoração, tais como os apresentados nessa revisão, no entanto, com a finalidade de sanar as recentes demandas relacionadas a sustentabilidade e a Química Verde, é esperado que ainda surjam no mercado mais reagentes de fluoração que viabilizem reações de fluoração mais brandas e com maior economia atômica, e que requerem e gerem espécies com menor nível de toxicidade. O reagente de fluoração eletrofílico Selectfluor vem sendo umas das principais opções dos químicos na fluoração de moléculas orgânicas devido a sua ampla gama de aplicações e facilidade de manipulação, entretanto, seu custo elevado impossibilita sua expansão para os mais diversos laboratórios do Brasil e do mundo. Combinada com sua baixa economia atômica, as desvantagens do Selectfluor devem incentivar o impulso no desenvolvimento de outros reagentes de fluoração, principalmente do grupo de reagentes que apresentam a ligação N-F. Com o surgimento de novos reagentes de fluoração que atendam a nova demanda e que proporcionem o desenvolvimento de novos métodos de fluoração, uma nova porta é aberta para a inclusão desta classe de reações nas disciplinas experimentais de cursos de graduação, de modo a abordar um dos tópicos que estão em alta e que apresentam caráter multidisciplinar, uma vez que a química de organofluorados é pertinente também nas áreas de farmácia, medicina e outras ciências da área da saúde. As estratégias de late-stage fluorination, fluoração enantiosseletiva, quimiosseletiva e regiosseletiva, e a química de 18F também têm previsão de entrarem em grande destaque devido aos seus impactos tanto em pesquisas básicas quanto em pesquisas aplicadas principalmente a área de saúde envolvendo os exames de PET (positron emission tomography). A técnica de espectroscopia de RMN voltada para o átomo de 19F é uma ferramenta rápida e eficaz na identificação e caracterização de compostos fluorados, como apresentado em detalhes nesta revisão. Os mesmos experimentos de 1H empregados no monitoramento de reações e na identificação e caracterização de intermediários podem ser aplicados ao 19F, ou seja, experimentos de 19F para os fins mencionados já estão bem estabelecidos. O desafio na área é de expandir o conhecimento sobre a técnica e de implementar nos equipamentos de ressonância magnética os experimentos de 19F, que, embora não seja difícil, necessita de um especialista da área de RMN. Após a implementação, devidos ajustes das sequências de pulsos e treinamento dos usuários, assim como já é realizado para os experimentos de rotina envolvendo 1H, os experimentos de 19F podem ser realizados de forma também rotineira, e a interpretação dos resultados obtidos é muitas vezes mais simples que para os experimentos envolvendo 1H.
CONCLUSÕES Os compostos organofluorados vêm emergindo de forma rápida em diversas áreas, tais como farmacêutica e agroquímica, e sua utilidade nessas áreas tem origem no impacto que a introdução que o átomo de flúor ocasiona nas propriedades químicas e biológicas de moléculas. Atualmente há três métodos de inserção desse átomo: fluoração eletrofílica, nucleofílica e radicalar (sob condições neutras). Cada condição apresenta suas vantagens, no entanto, as condições neutras apresentam mais estudos no âmbito de sustentabilidade e as nucleofílicas em economia atômica, e consequentemente são reportados mais trabalhos utilizando essas duas condições. Os reagentes de fluoração contendo ligação N-F são de fácil manuseio, eficientes, seletivos, estáveis e não são sensíveis a umidade ou higroscópicos. Eles podem ser empregados tanto sob condições eletrofílicas quanto neutras e por conta dos levamentos apresentados, acreditamos serem a melhor escolha para fluorar uma molécula orgânica. Em relação a condição de fluoração, acreditamos que a neutra seja a melhor, uma vez que há mais estudos mostrando como reações dentro dessa categoria podem ser conduzidas de formas mais brandas. Vale ressaltar que apesar do grande desenvolvimento da química de organofluorados ainda há diversos desafios envolvendo melhoria em enantiosseletividade, quimiosseletividade, rendimento e sustentabilidade, e em especial no desenvolvimento de estratégias condizentes com a Química Verde que possam ser aplicadas industrialmente para a síntese de compostos químicos fluorados. Uma ferramenta poderosa para auxiliar a superar esses desafios é a técnica de espectroscopia de ressonância magnética nuclear envolvendo o átomo de 19F, a qual propociona uma economia de tempo e de recursos pois não é necessária uma amostra pura para a obtenção da análise de RMN, é sensível, não destrutiva e apresenta mais vantagens para misturas complexas quando comparado com o espectro de 1H. A técnica também permite o monitoramento de reações orgânicas, estudo da cinética e análise do mecanismo, principalmente quando explorados os experimentos 2D envolvendo o átomo de 19F.
AGRADECIMENTOS Os autores agracedem a Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP) pelo apoio financeiro à C. C. (Processo 2021/05095-6) e à C. F. T. (Processo 2020/10246-0). Os autores também agradecem ao Professor Dr. Marco Antônio Barbosa Ferreira (UFSCar) pelas importantes correções do manuscrito.
REFERÊNCIAS 1. Liang, S.; Hammond, G. B.; Xu, B.; Chem. - Eur. J. 2017, 23, 17850. [Crossref] 2. Caron, S. S.; Org. Process Res. Dev. 2020, 24, 470. [Crossref] 3. Carvalho, N. F.; Pliego Junior, J. R.; Phys. Chem. Chem. Phys. 2015, 17, 26745. [Crossref] 4. Liang, T.; Neumann, C. N.; Ritter, T.; Angew. Chem., Int. Ed. 2013, 52, 8214. [Crossref] 5. Britton, R.; Gouverneur, V.; Lin, J. H.; Meanwell, M.; Ni, C.; Pupo, G.; Xiao, J. C.; Hu, J.; Nat. Rev. Methods Primers 2021, 1, 47. [Crossref] 6. Ogawa, Y.; Tokunaga, E.; Kobayashi, O.; Hirai, K.; Shibata, N.; iScience 2020, 23, 101467. [Crossref] 7. Jeschke, P. Em Organofluorine Chemistry: Synthesis, Modeling, and Applications; Szabo, K. J.; Selander, N., eds.; Wiley: Weinheim, 2021. 8. Wang, Q.; Song, H.; Wang, Q.; Chin. Chem. Lett. 2022, 33, 626. [Crossref] 9. Gouverneur, V.; Müller, K.; Fluorine in Pharmaceutical and Medicinal Chemistry, 1st ed.; Imperial College Press: London, 2012. 10. Kazmierczak, M.; Bilska-Markowska, M.; Eur. J. Org. Chem. 2021, 41, 5585. [Crossref] 11. Reddy, V. P.; Organofluorine Compounds in Biology and Medicine, 1st ed.; Elsevier: Oxford, 2015. 12. Yerien, D. E.; Bonesi, S.; Postigo, A.; Org. Biomol. Chem. 2016, 14, 8398. [Crossref] 13. Dong, T.; Tsui, G. C.; Chem. Rec. 2021, 21, 4015. [Crossref] 14. Dalvit, C.; Piotto, M.; Vulpetti, A.; J. Fluorine Chem. 2017, 202, 34. [Crossref] 15. Cesarin-Sobrinho, D.; Ferreira Neto, J. C.; Quim. Nova 2001, 24, 604. [Crossref] 16. Bõrgel, J.; Ritter, T.; Chem 2020, 6, 1877. [Crossref] 17. Ajenjo, J.; Destro, G.; Cornelissen, B.; Gouverneur, V.; EJNMMI Radiopharmacy and Chemistry 2021 , 6, 1. [Crossref] 18. Boechat, N.; Pinto, A. C.; Bastos, M. M.; Quim. Nova 2015, 38, 1323. [Crossref] 19. Umemoto, T.; Yang, Y.; Hammond, G. B.; Beilstein J. Org. Chem. 2021, 17, 1752. [Crossref] 20. Harsanyi, A.; Sandford, G.; Green Chem. 2015, 17, 2081. [Crossref] 21. Champagne, P. A.; Desroches, J.; Hamel, J. D.; Vandamme, M.; Paquin, J. F.; Chem. Rev. 2015, 115, 9073. [Crossref] 22. Rozatian, N.; Hodgson, D. R. W.; Chem. Commun. 2021, 57, 683. [Crossref] 23. Umemoto, T.; Kawada, K.; Tomita, K.; Tetrahedron Lett. 1986, 27, 4465. [Crossref] 24. Umemoto, T.; Harasawa, K.; Tomizawa, G.; Kawada, K.; Tomita, K.; Bull. Chem. Soc. Jpn. 1991, 64, 1081. [Crossref] 25. Differding, E.; Ofner, H.; Synlett 1991, 3, 187. [Crossref] 26. Banks, R. E.; Mohialdin-Khaffaf, S. N.; Lal, G. S.; Sharif, I.; Syvret, R. G.; Chem. Commun. 1992, 8, 595. [Crossref] 27. Stavber, S.; Zupan, M.; Poss, A. J.; Shia, G. A.; Tetrahedron Lett. 1995, 36, 6769. [Crossref] 28. Yasui, H.; Yamamoto, T.; Ishimaru, T.; Fukuzumi, T.; Tokunaga, E.; Akikazu, K.; Shiro, M.; Shibata, N.; J. Fluorine Chem. 2011, 132, 222. [Crossref] 29. Zhu, C. L.; Maeno, M.; Zhang, F. G.; Shigehiro, T.; Kagawa, T.; Kawada, K.; Shibata, N.; Ma, J. A.; Cahard, D.; Eur. J. Org. Chem. 2013, 2013, 6501. [Crossref] 30. Wolstenhulme, J. R.; Rosenqvist, J.; Lozano, O.; Ilupeju, J.; Wurz, N.; Engle, K. M.; Pidgeon, G. W.; Moore, P. R.; Sandford, G.; Gouverneur, V.; Angew. Chem., Int. Ed. 2013, 52, 9796. [Crossref] 31. Pereira, R.; Wolstenhulme, J.; Sandford, G.; Claridge, T. D. W.; Gouverneur, V.; Cvengros, J. N.; Chem. Commun. 2016, 52, 1606. [Crossref] 32. Meyer, D.; Jangra, H.; Walther, F.; Zipse, H.; Renaud, P.; Nat. Commun. 2018, 9, 1. [Crossref] 33. Haynes, W. M.; Lide, D. R.; Bruno, T. J.; CRC Handbook of Chemistry and Physics, 95th ed.; CRC Press: Boca Raton, 2014. 34. Rappoport, Z.; CRC Handbook of Tables for Organic Compound Identification, 3rd ed.; CRC Press: Boca Raton, 1967. 35. Gehring, P. J.; Torkelson, T. R.; Oyen, F.; Toxicol. Appl. Pharmacol. 1967, 11, 361. [Crossref] 36. Nyffeler, P. T.; Durón, S. G.; Burkart, M. D.; Vincent, S. P.; Wong, C. H.; Angew. Chem., Int. Ed. 2005, 44, 192. [Crossref] 37. Banks, R. E.; Lawrence, N. J.; Besheesh, M. K.; Popplewell, A. L.; Pritchard, R. G.; Chem. Commun. 1996, 14, 1629. [Crossref] 38. Differding, E.; Rüegg, G. M.; Tetrahedron Lett. 1991, 32, 3815. [Crossref] 39. Umemoto, T.; Fukami, S.; Tomizawa, G.; Harasawa, K.; Kawada, K.; Tomita, K.; J. Am. Chem. Soc. 1990, 112, 8563. [Crossref] 40. Geng, C.; Du, L.; Liu, F.; Zhu, R.; Liu, C.; RSC Adv. 2015, 5, 33385. [Crossref] 41. Wannenmacher, N.; Pfeffer, C.; Frey, W.; Peters, R.; J. Org. Chem. 2022, 87, 670. [Crossref] 42. Park, H.; Verma, P.; Hong, K.; Yu, J. Q.; Nat. Chem. 2018, 10, 755. [Crossref] 43. Yamamoto, K.; Li, J.; Garber, J. A. O.; Rolfes, J. D.; Boursalian, G. B.; Borghs, J. C.; Genicot, C.; Jacq, J.; Van Gastel, M.; Neese, F.; Ritter, T.; Nature 2018, 554, 511. [Crossref] 44. Testa, C.; Roger, J.; Fleurat-Lessard, P.; Hierso, J. C.; Eur. J. Org. Chem. 2019, 2019, 233. [Crossref] 45. Hollingworth, C.; Gouverneur, V.; Chem. Commun. 2012, 48, 2929. [Crossref] 46. See, Y. Y.; Morales-Colón, M. T.; Bland, D. C.; Sanford, M. S.; Acc. Chem. Res. 2020, 53, 2372. [Crossref] 47. Kim, D. W.; Ahn, D. S.; Oh, Y. H.; Lee, S.; Kil, H. S.; Oh, S. J.; Lee, S. J.; Kim, J. S.; Ryu, J. S.; Moon, D. H.; Chi, D. Y.; J. Am. Chem. Soc. 2006, 128, 16394. [Crossref] 48. Pliego Junior, J. R.; J. Mol. Liq. 2017, 237, 157. [Crossref] 49. Pliego Junior, J. R.; Org. Biomol. Chem. 2021, 19, 1900. [Crossref] 50. Jadhav, V. H.; Choi, W.; Lee, S. S.; Lee, S.; Kim, D. W.; Chem. - Eur. J. 2016, 22, 4515. [Crossref] 51. Kang, S. M.; Kim, C. H.; Lee, K. C.; Kim, D. W.; Org. Lett. 2019, 21, 3062. [Crossref] 52. Silva, S. L.; Valle, M. S.; Pliego Junior, J. R.; J. Org. Chem. 2020, 85, 15457. [Crossref] 53. Melo, A. S.; Valle, M. S.; Pliego Junior, J. R.; J. Fluorine Chem. 2023, 269, 110146. [Crossref] 54. Silva, S. L.; Valle, M. S.; Pliego Junior, J. R.; J. Mol. Liq. 2020, 319, 114211. [Crossref] 55. Watson, D. A.; Su, M.; Teverovskiy, G.; Zhang, Y.; García-Fortanet, J.; Kinzel, T.; Buchwald, S. L.; Science 2009, 325, 1661. [Crossref] 56. Mu, X.; Zhang, H.; Chen, P.; Liu, G.; Chem. Sci. 2014, 5, 275. [Crossref] 57. Pliego Junior, J. R.; Mol. Catal. 2022, 529, 112560. [Crossref] 58. Pliego Junior, J. R.; Mol. Catal. 2021, 506, 111540. [Crossref] 59. Okoromoba, O. E.; Han, J.; Hammond, G. B.; Xu, B.; J. Am. Chem. Soc. 2014, 136, 14381. [Crossref] 60. Olah, G. A.; Welch, J. T.; Vankar, Y. D.; Nojima, M.; Kerekes, I.; Olah, J. A.; J. Org. Chem. 1979, 44, 3872. [Crossref] 61. Middleton, W. J.; J. Org. Chem. 1975, 40, 574. [Crossref] 62. Lal, G. S.; Pez, G. P.; Pesaresi, R. J.; Prozonic, F. M.; Cheng, H.; J. Org. Chem. 1999, 64, 7048. [Crossref] 63. Beaulieu, F.; Beauregard, L. P.; Courchesne, G.; Couturier, M.; Laflamme, F.; L'Heureux, A.; Org. Lett. 2009, 11, 5050. [Crossref] 64. Lheureux, A.; Beaulieu, F.; Bennett, C.; Bill, D. R.; Clayton, S.; Laflamme, F.; Mirmehrabi, M.; Tadayon, S.; Tovell, D.; Couturier, M.; J. Org. Chem. 2010, 75, 3401. [Crossref] 65. Umemoto, T.; Singh, R. P.; Xu, Y.; Saito, N.; J. Am. Chem. Soc. 2010, 132, 18199. [Crossref] 66. Tang, P.; Wang, W.; Ritter, T.; J. Am. Chem. Soc. 2011, 133, 11482. [Crossref] 67. Zhang, Z.; Luo, J.; Gao, H.; Org. Lett. 2021, 23, 9332. [Crossref] 68. Pupo, G.; Chiara Vicini, A.; Ascough, D. M. H.; Ibba, F.; Christensen, K. E.; Thompson, A. L.; Brown, J. M.; Paton, R. S.; Gouverneur, V.; J. Am. Chem. Soc. 2019, 141, 2878. [Crossref] 69. Alič, B.; Petrovč, J.; Jelen, J.; Tavcar, G.; Iskra, J.; J. Org. Chem. 2022, 87, 5987. [Crossref] 70. Jang, E.; Kim, I.; Jang, H. S.; Sim, J.; J. Org. Chem. 2022, 87, 2640. [Crossref] 71. Berger, M.; Lenhard, M. S.; Waldvogel, S. R.; Chem. - Eur. J. 2022, 28, e2022010. [Crossref] 72. Clayden, J.; Greeves, N.; Warren, S.; Wothers, P.; Organic Chemistry, 2nd ed.; Oxford University Press: Oxford, 2001. 73. Madani, A.; Anghileri, L.; Heydenreich, M.; Möller, H. M.; Pieber, B.; Org. Lett. 2022, 24, 5376. [Crossref] 74. Lee, D. S.; Soni, V. K.; Heo, S.; Hwang, H. S.; You, Y.; Cho, E. J.; Green Chem. 2022, 24, 6481. [Crossref] 75. Capilato, J. N.; Pitts, C. R.; Rowshanpour, R.; Dudding, T.; Lectka, T.; J. Org. Chem. 2020, 85, 2855. [Crossref] 76. Herron, A. N.; Hsu, C. P.; Yu, J. Q.; Org. Lett. 2022, 24, 3652. [Crossref] 77. Harry, S. A.; Xiang, M. R.; Holt, E.; Zhu, A.; Ghorbani, F.; Patel, D.; Lectka, T.; Chem. Sci. 2022, 13, 7007. [Crossref] 78. Shu, C.; Feng, J.; Zheng, H.; Cheng, C.; Yuan, Z.; Zhang, Z.; Xue, X. S.; Zhu, G.; Org. Lett. 2020, 22, 9398. [Crossref] 79. Zhang, W.; Gu, Y. C.; Lin, J. H.; Xiao, J. C.; Org. Lett. 2020, 22, 6642. [Crossref] 80. Min, S.; Park, B.; Nedsaengtip, J.; Hyeok Hong, S.; Adv. Synth. Catal. 2022, 364, 1975. [Crossref] 81. Ben-Tal, Y.; Boaler, P. J.; Dale, H. J. A.; Dooley, R. E.; Fohn, N. A.; Gao, Y.; García-Domínguez, A.; Grant, K. M.; Hall, A. M. R.; Hayes, H. L. D.; Kucharski, M. M.; Wei, R.; Lloyd-Jones, G. C.; Prog. Nucl. Magn. Reson. Spectrosc. 2022, 129, 28. [Crossref] 82. Sigma-Aldrich; Quantitative NMR Technical Details and TraceCERT® Certified Reference Materials, 2017. [Link] acessado em Novembro 2023 83. Howe, P. W. A.; Prog. Nucl. Magn. Reson. Spectrosc. 2020, 118-119, 1. [Crossref] 84. Xu, Z.; Zhao, Y.; J. Fluorine Chem. 2023, 266, 110089. [Crossref] 85. Dolbier, W. R.; Guide to Fluorine NMR for Organic Chemists, 2nd ed.; John Wiley & Sons: New Jersey, 2016. 86. Branco, F. S. C.; Silva, B. V.; Do Rio, G. F.; Santana, M. J.; Queiroz Júnior, L. H. K.; Pinto, A. C.; Boechat, N.; Lião, L. M.; Quim. Nova 2015, 38, 1237. [Crossref] 87. Dal Poggetto, G.; Soares, J. V.; Tormena, C. F.; Anal. Chem. 2020, 92, 14047. [Crossref] 88. Barbosa, T. M.; Castañar, L.; Moutzouri, P.; Nilsson, M.; Morris, G. A.; Rittner, R.; Tormena, C. F.; Anal. Chem. 2020, 92, 2224. [Crossref] 89. Castanar, L.; Moutzouri, P.; Barbosa, T. M.; Tormena, C. F.; Rittner, R.; Phillips, A. R.; Coombes, S. R.; Nilsson, M.; Morris, G. A.; Anal. Chem. 2018, 90, 5445. [Crossref] 90. Power, J. E.; Foroozandeh, M.; Adams, R. W.; Nilsson, M.; Coombes, S. R.; Phillips, A. R.; Morris, G. A.; Chem. Commun. 2016, 52, 2916. [Crossref] 91. Schotten, C.; Howard, J. L.; Jenkins, R. L.; Codina, A.; Browne, D. L.; Tetrahedron 2018, 74, 5503. [Crossref] 92. Mlynarska-Cieslak, A.; Chrominski, M.; Spiewla, T.; Baranowski, M. R.; Bednarczyk, M.; Jemielity, J.; Kowalska, J.; ACS Chem. Biol. 2022, 17, 661. [Crossref] 93. Lete, M. G.; Franconetti, A.; Bertuzzi, S.; Delgado, S.; Azkargorta, M.; Elortza, F.; Millet, O.; Jiménez-Osés, G.; Arda, A.; Jiménez-Barbero, J.; Chem. - Eur. J. 2023, 29, e202202208. [Crossref] 94. Pham, L. B. T.; Costantino, A.; Barbieri, L.; Calderone, V.; Luchinat, E.; Banci, L.; J. Am. Chem. Soc. 2023, 145, 1389. [Crossref] 95. Montgomery, K.; Elhabashy, A.; Chen, G.; Chen, Q. H.; Krishnan, V. V.; J. Fluorine Chem. 2023, 266, 110084. [Crossref] 96. Leung, R. L. C.; Robinson, M. D. M.; Ajabali, A. A. A.; Karunanithy, G.; Lyons, B.; Raj, R.; Raoufmoghaddam, S.; Mohammed, S.; Claridge, T. D. W.; Baldwin, A. J.; Davis, B. G.; J. Am. Chem. Soc. 2017, 139, 5277. [Crossref] 97. Sugiki, T.; Ito, A.; Hatanaka, Y.; Tsukamoto, M.; Murata, T.; Miyanishi, K.; Kagawa, A.; Fujiwara, T.; Kitagawa, M.; Morita, Y.; Negoro, M.; NMR Biomed. 2023, 36, 1. [Crossref] 98. Li, Q.; Trajkovski, M.; Fan, C.; Chen, J.; Zhou, Y.; Lu, K.; Li, H.; Su, X.; Xi, Z.; Plavec, J.; Zhou, C.; Angew. Chem., Int. Ed. 2022, 61, 1. [Crossref] 99. Krempl, C.; Sprangers, R.; J. Biomol. NMR 2023, 1, 1. [Crossref] 100. Christensen, M. V.; Kongstad, K. T.; Sondergaard, T. E.; Staerk, D.; Nielsen, H. M.; Franzyk, H.; Wimmer, R.; J. Biomol. NMR 2019, 73, 167. [Crossref] 101. Baranowski, M. R.; Warminski, M.; Jemielity, J.; Kowalska, J.; Nucleic Acids Res. 2020, 48, 8209. [Crossref] 102. Macheteau, J. P.; Oulyadi, H.; Van Hemelryck, B.; Bourdonneau, M.; Davoust, D.; J. Fluorine Chem. 2000, 104, 149. [Crossref] 103. Dewis, L.; Crouch, R.; Russell, D.; Butts, C.; Magn. Reson. Chem. 2019, 57, 1143. [Crossref] 104. Dal Poggetto, G.; Favaro, D. C.; Nilsson, M.; Morris, G. A.; Tormena, C. F.; Magn. Reson. Chem. 2014, 52, 172. [Crossref] 105. Barbosa, T. M.; Morris, G. A.; Nilsson, M.; Rittner, R.; Tormena, C. F.; RSC Adv. 2017, 7, 34000. [Crossref] |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access