
 |
 |
 |
 |
 |
Artigo
| Phytochemical study from the aerial parts of Pavonia malacophylla (Malvaceae) and evaluation of its antimicrobial potential |
|
Janderson Barbosa Leite de AlbuquerqueI; Otemberg Souza ChavesI; Wallace Amorim Machado de QueirozI; I. Pós-Graduação em Produtos Naturais e Sintéticos Bioativos, Centro de Ciências da Saúde, Universidade Federal da Paraíba, 58051-900 João Pessoa - PB, Brasil Received: 01/30/2025 *e-mail: mfvanderlei@ltf.ufpb.br Pavonia malacophylla (Link & Otto) Garcke (Malvaceae sensu lato), commonly known as "malva-rosa" or "malva-veludo", is traditionally used to treat flu, cough, and heart pain. The crude ethanolic extract (CEE) of P. malacophylla was obtained through maceration with ethanol, concentrated using a rotary evaporator, and subjected to vacuum liquid chromatography with silica gel 60 as the stationary phase. Hexane, ethyl acetate, and methanol were used as mobile phases in increasing polarity gradients, yielding nine fractions. Chromatographic fractionation led to the isolation and identification of 18 compounds, including one alcohol, three steroids, three triterpenes, four pheophytins, six flavonoids and one phenolic acid. Among these, compounds 1 (decanol), 4a (α-amyrin), 4b (β-amyrin), 11 (5,8-dihydroxy-7,4'-dimethoxyflavone) and 16 (methyl-4-hydroxycinnamate), were identified for the first time in the genus Pavonia. Microbiological assays of the CEE, fractions, and isolated compounds were conducted against Escherichia coli,Candida albicans, and Staphylococcus aureus, demonstrating antimicrobial activity at concentrations ranging from 250 to 400 µg mL-1. INTRODUCTION The development and spread of microbial infections have generated worldwide concern.1 The indiscriminate use and increase in consumption of antimicrobial agents, together with inadequate prescriptions, has contributed to the process of resistance to these drugs, whether through spontaneous mutations or the attribution of new genes that will be inherited by offspring.2,3 Furthermore, the number of infections caused by multidrug-resistant microorganisms has amplified the spectrum of intractable diseases in the current context.4 From the moment penicillin was developed in the mid-1940s, the field of science turned to the search for antimicrobial derivatives from natural products.5 Medicinal plants perform an essential role as the main therapeutic source used in folk medicine. Previous ethnic-medical-botanical studies form the basis for the discovery of new antimicrobials from these herbs.6 Microbiological tests have demonstrated the high antimicrobial power of extracts, fractions, and isolated compounds, such as pheophytins, alkaloids, terpenoids and flavonoids, confirming the potential activity of these natural products.7-11 The Malvaceae sensu lato family comprises around 245 genera and 4465 species distributed throughout tropical and subtropical regions of the world.12 Among the various genera belonging to this family, the genus Pavonia Cav. concentrates approximately 271 species distributed throughout the New and Old World, including Brazil.13 Previous studies11,14 report the prevalence of several classes of secondary metabolites in this genus, such as fatty acids, phenolics, steroids, terpenoids, flavonoids, pheophytins and hydrocarbons. Pavonia malacophylla (Link & Otto) Garcke is popularly known as "malva-rosa" or "malva-veludo", distributed throughout Brazil. Its leaves and roots have been used to treat coughs and flu in the form of syrup, in addition to the use of its raw leaves to treat heart pain.15,16 A preliminary phytochemical screening detected the presence of triterpenes, steroids, flavonoids, coumarins, tannins and alkaloids in the crude ethanolic extract from its aerial parts.17 Considering these aspects and the antimicrobial potential of species of the genus Pavonia linked to the shortage of phytochemical data on the species P. malacophylla, the present study aims to isolate and identify substances from their aerial parts and evaluate their antimicrobial activity.
EXPERIMENTAL General and chemical procedures Glass chromatographic columns were used for the isolation and purification of chemical constituents, packed with silica gel 60 (ASTM, 230-400 mesh, Macherey Nagel®), Sephadex LH-20 (Sigma-Aldrich®) or Amberlite XAD-2 (Sulpeco®). Analytical thin layer chromatography (TLC) was carried out using plates impregnated with silica gel (Whatman®) to analyze the chromatographic profile of the compounds obtained in all processes and analyzed by ultraviolet radiation (UV) at wavelengths of 254 and 366 nm and revealed with diphenylboryloxyethylamine (NP) or p-anisaldehyde reagents, or by impregnating the plates in glass vats saturated with iodine vapors. The identification or structural elucidation of the isolated compounds was carried out using spectroscopic methods (1H and 13C nuclear magnetic resonance (NMR)) and 2D techniques (HSQC (heteronuclear single quantum correlation) and HMBC (heteronuclear multiple bond correlation)) using the BRUKER-ASCEND spectrometer operating at 400 MHz (1H) and 100 MHz (13C) and VARIAN operating at 500 MHz (1H) and 125 MHz (13C) and 200 MHz (1H) and 50 MHz (13C), using deuterated solvents (CDCl3, DMSO-d6 and CD3OD). Chemical shifts (d) were expressed in parts per million (ppm) and coupling constants (J) in Hz. Botanical material The plant material (aerial parts) of Pavonia malacophylla were collected in the municipality of Santa Rita - Paraíba (7º7'42.473"S; 34º59'04.626"W) in June 2011. Botanical identification was carried out by Prof. Dr. Maria of Fátima Agra (CBioTec/UFPB) and an exsiccate was deposited in the Herbarium Prof. Lauro Pires Xavier (Agra 7038). This research was registered in the National System of Genetic Resource Management and Associated Traditional Knowledge (SisGen - A568B8A). Extraction and isolation of compounds The aerial parts of Pavonia malacophylla were dehydrated in an oven with circulating air at an average temperature of 40 ºC for 96 h. Then, the sample was crushed with the aid of a mechanical mill, providing 1050 g of powder. All material was macerated with 95% ethanol for 72 h. The extractive solution obtained was filtered and concentrated in a rotary evaporator under reduced pressure at 40 ºC, providing 403.0 g of crude ethanolic extract (CEE). A sample of CEE (200.0 g) was subjected to vacuum liquid chromatography (VLC) using a Buchner funnel coupled to a kitassato (1 L), and silica gel 60 as stationary phase and hexane (Hex), ethyl acetate (EtOAc) and methanol (MeOH) as mobile phases, alone or in binary mixtures in increasing order of polarity (Hex; Hex:EtOAc (9:1; 7:3; 1:1; 3:7); EtOAc; EtOAc:MeOH (9:1; 7:3; 1:1) obtaining nine fractions that were concentrated in a rotary evaporator and then analyzed by TLC. The Hex:EtOAc (9:1) fraction (6.0 g) was fractionated in column chromatography (CC) using silica gel 60 and Hex, EtOAc and MeOH following the previously described methodology, obtaining nine subfractions. The Hex:EtOAc (9:1) subfraction (930.0 mg) was chromatographed in CC following the same methodology, resulting in 44 subfractions that were analyzed by TLC. It was found that subfractions 14-20 and 26-31 were pure, being coded as compounds 1 (35.0 mg) and 2 (28.0 mg), respectively. The Hex:EtOAc (7:3) fraction (3.5 g) was chromatographed in CC following the previous methodology, obtaining 107 fractions (10 mL each) that were analyzed by TLC. Fractions 47-53 and 6469 were isolated, the compounds coded as 3 (13.0 mg) and 4 and (22.0 mg), respectively. The Hex:EtOAc (3:7) fraction (3.0 g) was subjected to the previous methodology, obtaining 68 fractions that were analyzed by TLC. The fractions 11 and 13 were considered pure and were coded as compounds 5 (15.0 mg) and 6 (12.0 mg), respectively. The combination of fractions 14-24 (220.0 mg) submitted to a CC using Sephadex LH-20 as stationary phase and MeOH and MeOH:CHCl3 (1:1) as mobile phases led to the isolation of compounds 7 (10.0 mg) and 8 (10.0 mg). The 100% EtOAc fraction (5.0 g) was subjected to CC, using Sephadex LH-20 as the stationary phase and MeOH and MeOH:CHCl3 (1:1) as mobile phases. From this process, 42 fractions were obtained, which were analyzed by TLC. Subfraction 26-31 was found to be pure and was coded as compound 9 (25.0 mg). An aliquot of the EtOAc:MeOH (9:1) fraction (5.0 g) was chromatographed using the methodology described previously. From this procedure, 56 fractions were obtained and analyzed by TLC. The subfraction 44-51 (85.0 mg) was subjected to another chromatography, adopting the same methodology. From this, 20 fractions were obtained and analyzed by TLC. The subfractions 15-17 (11.0 mg) and 18-20 (5.0 mg) appeared as pure yellow powders, being then coded as compounds 10 (11.0 mg) and 11 (5.0 mg), respectively. The subfraction 19-43 (714.0 mg) was subjected to the same methodology as subfraction 44-51, resulting in 19 fractions. From this process, the subfraction 6-8 (33.0 mg) was obtained, which appeared pure when analyzed by TLC, in various solvent systems, being coded as compound 12 (33.0 mg). The EtOAc:MeOH (1:1) fraction (7.0 g) was chromatographed in a CC on Amberlite XAD-2 using H2O, MeOH, Hex, acetone and EtOAc as mobile phases. From this procedure, the 100% MeOH subfraction (2.8 g) was chromatographed on Sephadex LH-20 using MeOH and MeOH:CHCl3 (1:1) as mobile phases. Successive chromatographic columns were performed using Sephadex LH-20 as the stationary phase and MeOH and MeOH:CHCl3 (1:1) as mobile phases, resulting in fractions that were analyzed by TLC and then coded as compounds 13 (18.0 mg) and 14 (10.0 mg). Another portion of the CEE (160.0 g) was dissolved in an ethanol:water solution (7:3) with a final volume of 1 L and homogenized with the aid of a mechanical stirrer for 2 h. Subsequently, the hydroalcoholic solution was subjected to liquidliquid chromatography, using Hex, CHCl3 and EtOAc (4 × 500 mL, each solvent), in increasing order of polarity, accompanied by TLC analysis, provided 34.0 g of hexane phase (HP), 37.0 g of CHCl3 phase (CP), 13.0 g of EtOAc phase (AEP), in addition to the hydroalcoholic phase (HAP) (72.0 g). An aliquot of CP (20.0 g) was subjected to CC using silica gel 60 as the stationary phase and Hex, EtOAc, MeOH and H2O as mobile phases alone or in binary mixtures in an increasing degree of polarity, obtaining 67 fractions, which were concentrated in a rotary evaporator and combined through analysis in TLC. Fractions 1516 and 29-30 were pure and were coded as compounds 15 (10.0 mg) and 16 (12.0 mg). Antimicrobial activity assay Test microorganisms To evaluate antimicrobial activity, five strains of microorganisms were selected (Staphylococcus aureus ATCC 15656, Streptococcus mutans ATCC 25175, Pseudomonasaeruginosa ATCC 27853, Escherichiacoli ATCC 25922 and Candidaalbicans ATCC 1106), obtained from the microbiological collection of the Oral Biology Laboratory, from the Health Sciences Center of the Federal University of Paraíba. Determination of minimum inhibitory concentration (MIC) Antifungal and antibacterial activities were determined using dilution assays in 96-well microplates in triplicate. For the cultivation of bacteria and fungi, sterilized test tubes were used, where 0.6 mL of the bacterial/fungal inoculum was added with 7 mL of brain heart infusion (BHI) broth (for bacteria) and 7 mL of Sabouraud broth (for fungi). Then, the tubes were shaken and homogenized in the vortex, then taken to the oven at 35 ± 2 ºC for 24 h for S. aureus,S. mutans,E. coli and P. aeruginosa, and 48 h for C. albicans. The cultivation of S. mutans was carried out using microaerophilia. Stock solutions of extracts, fractions and compounds were prepared separately. These were weighed and mixed with a solution of 10% DMSO-d6 and Tween 80 (5%) in a 1:1 ratio. The solutions were homogenized in the vortex, sonicated, and taken to an oven at 35 ± 2 ºC.18 Secondary solutions SM1, SM2 and SM3 of the samples were prepared separately for three blocks of dilution series. For SM1, 1.5 mL of BHI broth was added with 1.5 mL of SM extracts/fractions/substances, described previously, both in the same proportion (1:1) (v/v), obtaining a final volume of 3 mL and an extract at a concentration of 500 µg mL-1. For SM2, 2.5 mL of BHI broth were added with 0.5 mL of SM extract/substance, in the proportion (1:5) (v/v), obtaining a final volume of 3 mL and an extract at the concentration of 167 µg mL-1. For SM3, 4.5 mL of BHI broth were added with 0.5 mL of extract/substance SM, in the proportion (1:9) (v/v), obtaining a final volume of 5 mL and an extract at the concentration of 100 µg mL-1. The preparation of the bacterial/fungal inoculum remained in an oven for 24 h of cultivation at 37 ºC and 3 mL of the inoculum was removed from each tube and placed in Falcon tubes. Then, the tubes were centrifuged for 10 to 15 min, removing the supernatant and adding 240 µL of the inoculum with 4.8 mL of saline solution to the tube. Once the mixture was homogenized, it was placed in the oven for 1 h, vortexing every 15 min. After this rest, in an oven, the tubes were centrifuged again for 10 to 15 min and the supernatant was discarded and 2.4 mL of saline solution was added, homogenizing in the vortex. The reading was then carried out on the spectrophotometer (OPTIMA) with an absorbance of 0.135 at a wavelength of 640 nm, corresponding to 1.6 × 108 CFU (colony forming unit) mL-1, equivalent to 0.5 on the McFarland scale. After the incubation period, 35 µL of resazurin (100 µg mL-1), prepared in an aqueous solution (1 mg, bring to volume 10 mL) was added to each hole, mixing the contents in the wells of the plate. Chlorhexidine was the antimicrobial used as a positive control.
RESULTS AND DISCUSSION Identification of isolated compounds Using 1D and 2D NMR spectroscopic methods, it was possible to identify and structurally elucidate 18 compounds from the aerial parts of P. malacophylla, including one aliphatic alcohol (1), three steroids (2) and two in mixture (3a and 3b), three triterpenes, being two in mixture (4a and 4b) and the compound 5, four chlorophyll derivatives (6-9), six flavonoids (10-15) and one phenolic acid (16) (Figure 1).
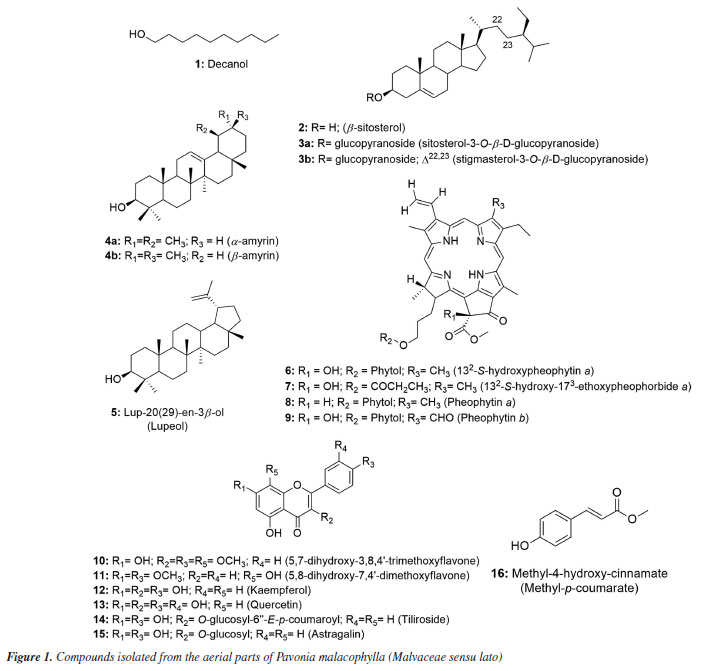
5,7-Dihydroxy-3,8,4'-trimethoxyflavone (10) Melting point of 174-175 ºC; 1H NMR (500 MHz, CDCl3) d 12.43 (s, 1H, 5-OH), 6.42 (s, 1H, s, H-6), 8.11 (d, 2H, J 10.0 Hz, H-2'/6'), 7.05 (d, 2H, J 9.0 Hz, H-3'/5'), 3.86 (s, 3H, 3-OCH3), 3.99 (s, 3H, 8-OCH3), 3.90 (s, 3H, 4'-OCH3); 13C NMR (125 MHz, CDCl3) d 155.8 (C-2), 138.9 (C-3), 179.1 (C-4), 155.1 (C-5), 98.6 (C-6), 157.6 (C-7), 126.8 (C-8), 148.1 (C-9), 105.8 (C-10), 122.9 (C-1'), 130.2 (C-2'/6'), 114.4 (C-3'/5'), 161.9 (C-4'), 60.3 (3-OCH3), 62.1 (8-OCH3), 55.6 (4'-OCH3). The 1H and 13C NMR data were in accordance with published data.11 5,8-Dihydroxy-7,4'-dimethoxyflavone (11) Melting point of 269-270 ºC; 1H NMR (500 MHz, CDCl3) d 12.43 (s, 1H, 5-OH), 6.86 (s, 1H, s, H-3), 6.55 (s, 1H, H-6), 8.11 (d, 2H, J 8.4 Hz, H-2'/6'), 7.12 (d, 2H, J 8.4 Hz, H-3'/5'), 3.90 (s, 3H, 7-OCH3), 3.85 (s, 3H, 4'-OCH3); 13C NMR (125 MHz, CDCl3) d 163.5 (C-2), 103.0 (C-3), 182.4 (C-4), 153.1 (C-5), 95.7 (C-6), 154.3 (C-7), 126.2 (C-8), 144.4 (C-9), 103.9 (C-10), 123.0 (C-1'), 128.5 (C-2'/6'), 114.5 (C-3'/5'), 162.4 (C-4'), 56.5 (7-OCH3), 56.3 (4'-OCH3), 55.6 (4'-OCH3). 1H and 13C NMR data were in agreement with published data.19 3,5,7,4'-Tetrahydroxyflavone (12) Melting point of 277-278 ºC; 1H NMR (500 MHz, CD3OD) d 6.18 (d, J 2.0 Hz, 1H, H-6), 6.39 (d, J 2.0 Hz, 1H, H-8), 8.08 (d, 2H, J 8.9 Hz, H-2'/6'), 6.90 (d, 2H, J 8.9 Hz, H-3'/5'); 13C NMR (125 MHz, CD3OD) d 148.0 (C-2), 137.1 (C-3), 177.3 (C-4), 162.5 (C-5), 99.3 (C-6), 165.6 (C-7), 94.4 (C-8), 158.2 (C-9), 103.7 (C-10), 123.7 (C-1'), 130.6 (C-2'/6'), 116.3 (C-3'/5'), 160.5 (C-4'). 1H and 13C NMR data were in agreement with published data.20 3,5,7,3',4'-Pentahydroxyflavone (13) Melting point of 314-315 ºC; 1H NMR (500 MHz, CD3OD) d 6.17 (d, J 2.0 Hz, 1H, H-6), 6.37 (d, J 2.0 Hz, 1H, H-8), 7.72 (d, 1H, J 2.0 Hz, H-2'), 6.87 (d, 1H, J 8.5 Hz, H-5'), 7.62 (dd, J 8.5 and 2.0 Hz, 1H, H-6'); 13C NMR (125 MHz, CD3OD) d 148.0 (C-2), 137.2 (C-3), 177.3 (C-4), 162.5 (C-5), 99.2 (C-6), 165.6 (C-7), 94.4 (C-8), 158.2 (C-9), 104.5 (C-10), 124.1 (C-1'), 116.2 (C-2'), 146.2 (C-3'), 148.7 (C-4'), 116.0 (C-5'), 121.6 (C-6'). 1H and 13C NMR data were in agreement with published data.8 Kaempferol 3-O-β-D-(6"-E-p-coumaroyl) glucoside (tiliroside) (14) Melting point of 254-255 ºC; 1H NMR (200 MHz, CD3OD) d 6.11 (d, J 2.0 Hz, 1H, H-6), 6.27 (d, J 2.0 Hz, 1H, H-8), 7.96 (d, 1H, J 9.0 Hz, H-2'/6'), 6.79 (d, 1H, J 9.0 Hz, H-3'/5'), 5.23 (d, J 7.6 Hz, 1H, H-1"), 3.15-3.47 (m, H-2"/3"/4"/5"), 4.06 (dd, J 11.8 and 2.2 Hz, H-6a"), 4.19 (dd, J 11.6 and 2.2 Hz, H-6II"), 7.25 (d, J 8.6 Hz, H-2'''/6'''), 6.77 (d, J 8.6 Hz, H-3'''/5'''), 6.05 (d, J 16.0 Hz, 1H, H-α), 7.38 (d, J 16.0 Hz, 1H, H-β); 13C NMR (50 MHz, CD3OD) d 159.2 (C-2), 135.1 (C-3), 179.3 (C-4), 162.8 (C-5), 99.9 (C-6), 165.8 (C-7), 94.8 (C-8), 158.2 (C-9), 105.5 (C-10), 122.6 (C-1'), 132.2 (C-2'/6'), 115.9 (C-3'/5'), 161.4 (C-4'), 104.0 (C-1"), 75.7 (C-2"), 77.9 (C-3"), 71.6 (C-4"), 75.7 (C-5"), 64.3 (C-6"), 127.0 (C-1'''), 131.1 (C-2'''/6'''), 116.7 (C-3'''/5'''), 161.1 (C-4'''), 114.6 (C-α), 146.5 (C-β), 168.8 (COO). 1H and 13C NMR data were in agreement with published data.8 Kaempferol 3-O-β-D-glucopyranoside (astragalin) (15) Melting point of 162-163 ºC; 1H NMR (500 MHz, CD3OD) d 6.12 (d, J 2.0 Hz, 1H, H-6), 6.29 (d, J 2.0 Hz, 1H, H-8), 8.03 (d, 2H, J 9.0 Hz, H-2'/6'), 6.87 (d, 2H, J 9.0 Hz, H-3'/5'), 5.14 (d, J 7.5 Hz, 1H, H-1"), 3.17-3.21 (m, H-2"), 3.41-3.44 (m, H-3"/4"/5"), 3.54 (dd, J 10.4 and 3.9 Hz, H-6a"), 3.68 (dd, J 11.9 and 2.4 Hz, H-6II"); 13C NMR (125 MHz, CD3OD) d 158.5 (C-2), 135.3 (C-3), 178.9 (C4), 162.7 (C-5), 101.3 (C-6), 170.3 (C-7), 95.8 (C-8), 158.8 (C-9), 104.4 (C-10), 122.8 (C-1'), 132.2 (C-2'/6'), 116.1 (C-3'/5'), 161.6 (C-4'), 104.6 (C-1"), 75.7 (C-2"), 78.0 (C-3"), 71.3 (C-4"), 78.3 (C-5"), 62.6 (C-6"). 1H and 13C NMR data were in agreement with published data.21 Methyl-4-hydroxycinnamate (16) Melting point of 135-136 ºC; 1H NMR (500 MHz, CDCl3) d 7.42 (d, J 8.7 Hz, 2H, H-2/6), 6.84 (d, J 8.7 Hz, 2H, H-3/5), 7.63 (d, J 16.0 Hz, 1H, H-7), 6.29 (d, J 16.0 Hz, 1H, H-8), 3.79 (s, 3H, OCH3); 13C NMR (125 MHz, CDCl3) d 127.2 (C-1), 130.1 (C-2/6), 116.1 (C-3/5), 158.2 (C-4), 144.9 (C-7), 115.2 (C-8), 168.1 (C-9), 51.8 (OCH3). 1H and 13C NMR data were in agreement with published data.22 Compound 1 was isolated as a viscous oil and identified as the aliphatic alcohol decanol, a compound produced by plants with ecological relevance that acts as a pesticide in the control of arthropod pests such as Cimex lectularius and as a plant growth regulator in agriculture.23,24 Decanol showed good bactericidal activity due to its ability to damage the cell envelope of Mycobacteria species. Furthermore, the compound reduced biofilm formation by M. smegmatis at concentrations of 0.1 and 0.2 mM, lower than its MIC which was 0.4 mM.25 This compound was previously reported in Helicteres velutina and Helicteres eichleri, species of Malvaceae sensu lato26,27 and for the first time in the genus Pavonia. Compound 2 showed melting point of 141-142 ºC and was identified as β-sitosterol and compounds 3a and 3b were identified as a mixture of two glycosylated phytosteroids: sitosterol- 3OβDglucopyranoside and stigmasterol-3-O-β-D-glucopyranoside, respectively. These compounds are widely found as components of plant cell walls and membranes. Some activities are described in the literature28-32 for the compounds, such as anti-inflammatory, analgesic, anthelmintic, antimutagenic, hypocholesterolemic, antidiabetic, antioxidant and antimicrobial. These compounds are widely found among species of the Malvaceae sensu lato family,27,33 but isolated for the first time in the species P. malacophylla. 1H NMR analysis of compounds 4a and 4b showed absorptions for two olefinic hydrogens at dH 5.16 (t, J 3.0 Hz) and dH 5.10 (t, J 4.0 Hz), indicating the presence of double bonds, suggesting that the compounds would be two triterpenes with unsaturation between C-12 and C-13. The hypothesis that it would be a mixture of triterpenes was corroborated by the characteristic signs of non-hydrogenated olefinic carbons at dC 145.1 (C-13) and monohydrogenated carbons at dC 121.6 (C-12) of triterpenes from the oleanane series, as well as the signs of non-hydrogenated olefinic carbons at dC 139.5 (C-13) and monohydrogenated at dC 124.3 (C-12) of ursane series triterpenes. Furthermore, it was shown that C-13 of the ursane series (dC 139.5) was more protected than C-13 of the oleanane series (dC 145.1), a fact due to the γ-gauche effect of methyl C-29 in the C-13 of the ursane series. Thus, compounds 4a and 4b were identified as a mixture of triterpenes from the ursane (α-amyrin) and oleanane (β-amyrin) series, respectively, isolated for the first time in the genus Pavonia.34,35 A literature36 reports for these triterpenes various activities such as anti-inflammatory, gastroprotective, anti-allergic, and antinociceptive. The analysis of 1H and 13C NMR spectral data compared with literature data allowed us to verify that compound 5 would be lup-20(29)-en-3β-ol, known as lupeol,37 which showed a melting point of 215-216 ºC. This compound is widely distributed in the plant kingdom, including in species of Malvaceae sensu lato, such as Herissantia tiubae,Helicteres eichleri,Waltheria viscosissima and Pavonia distinguenda.7,26,33,38 According to studies,39-43 this compound has great potential acting as an anti-inflammatory, anticancer, antidiabetic, cardioprotective, skin protective, antiprotozoal, antimicrobial, nephroprotective, antiangiogenic and hypocholesterolemic agent. The 1H and 13C NMR spectral data of compounds 6, 7, 8 and 9 indicated that they are chlorophyll derivatives. Comparisons with literature data led to identifying the compounds as 132-S-hydroxypheophytin a (6),44 132-S-hydroxy-173-ethoxypheophorbide a (7),45 pheophytin a (8),10 and pheophytin b (9),44 previously reported in species of Malvaceae sensu lato: Wissadula periplocifolia,Sida rhombifolia,Sida galheirensis,Sidastrummicranthum,Helicteresvelutina,Pavonia glazioviana.9,11,27,45,46 The analysis of one and two-dimensional 1H and 13C NMR spectral data, together with comparisons with literature data,8,11,19 allowed us to identify compound 10 as 5,7-dihydroxy-3,8,4'-trimethoxyflavone and compound 11 as being 5,8-dihydroxy-7,4'-dimethoxyflavone (7,4'-di-O-methylisoscutelarein), flavonoids previously isolated in species belonging to the Malvaceae sensu lato family. The antimicrobial activity of flavonoids has been suggested according to their chemical structure, especially the number and positions of methoxyl and hydroxyl groups. The effect of O-methylation on flavonoids has been reported in the literature with reduced reactivity of hydroxyl groups and increased lipophilicity of these compounds, thus promoting an improvement in antimicrobial activity.47,48 Compounds 12, 13, 14 and 15 were isolated as yellowish powders. Their 1H and 13C NMR spectra showed signals in the region of aromatic hydrogens compatible with flavonoids, making it possible to identify them as 3,5,7,4'-tetrahydroxyflavone (kaempferol) (12), 3,5,7,3',4'-pentahydroxyflavone (quercetin) (13), kaempferol 3-O-β-D-(6"-E-p-coumaroyl) glycoside (tiliroside) (14) and kaempferol 3-O-β-D-glucopyranoside (astragalin) (15). The astragalin was treated against fungal cells producing good antimicrobial and significant antibiofilm activity. Astragalin affected the integrity of the fungal cell membrane and had no cytotoxic effect on human gingival fibroblast cells. It also worked to reduce the expression of genes that encode efflux pumps (CDR1), and when combined with an antifungal drug, it increased its concentration inside cells, making it a therapeutic option for candidiasis.49 Compound 16 was isolated as a white powder. Its 1H NMR spectrum exhibited signals in the region of aromatic hydrogens, with two doublets at dH 7.42 and dH 6.84 (J 8.7 Hz) with an integral for two hydrogens each and with an ortho coupling constant, suggesting the presence of an AA'BB' aromatic system.50 Two other doublets were evidenced in dH 7.63 and dH 6.29 (J 16.0 Hz) with an integral for one hydrogen each, equivalent to the pair of hydrogens linked to the olefinic carbons and trans-type coupling constants, signals attributed to the hydrogens H-7 and H-8 of the caffeoyl unit, respectively. A signal consistent with methoxylic hydrogens in dH 3.79 with an integral for 3H suggested this group as a substituent in its structure.22 When evaluating the expansions of the 13C NMR spectrum, signals were evidenced for 10 carbon atoms. The signal at dC 158.2 was attributed to C-4, due to its deprotection in relation to other aromatic carbons, due to the electronegative effect of the hydroxyl.51 Absorptions at dC 127.2 and dC 168.1 were consistent with the quaternary carbons C-1 and C-9, while signals at dC 130.1 and dC 116.1 were attributed to the methine carbon sets of the AA'BB' (C-2/C-6 and C-3/C-5), respectively. The chemical shifts at dC 144.9 and dC 115.2 were compatible with those of the C-7 and C-8 trans coupling system. The only methyl carbon signal was evident at dC 51.8, thus confirming the proposal observed in the 1H NMR spectrum. Two-dimensional HSQC and HMBC spectra of compound 16 confirmed the proposed molecule.52 The compilation of spectral data and comparison with literature data10,52 allowed the identification of substance 16 as methyl-4hydroxy-cinnamate (methyl-p-coumarate). This compound was previously isolated in the species Hibiscus sabdariffa53 and Abutilon indicum,54 but reported for the first time in the genus Pavonia. Few reports of activity with this phenolic acid show its potency against cells of the acute myeloid lineage in leukemias,55 inhibition of melanoma formation56 and antifungal activity against Alternaria alternata.57 Biological assay The antimicrobial activities of all tested plant drugs are expressed in Table 1. For the Escherichia coli ATCC 25992 strain, the minimum inhibitory concentrations (MIC) of all fractions and extract were 400 µg mL-1 and for the isolated substances the MIC was 300 and 350 µg mL-1. For Staphylococcus aureus ATCC 15656, only the EtOAc and 173-ethoxypheophorbide a fractions presented an MIC of 400 and 250 µg mL-1, respectively. For Candidaalbicans ATCC 1106, only the EtOAc:MeOH (9:1) fraction showed an MIC of 400 µg mL-1, when compared with the chlorhexidine standard.
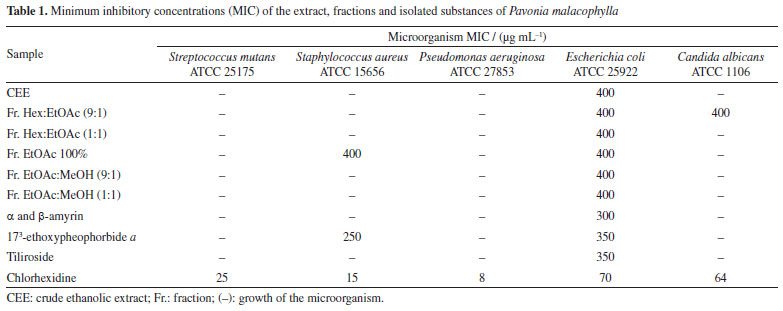
The bioassays performed in this study were carried out using CEE, fractions and compounds isolated against bacterial strains and a yeast strain. The results obtained indicated that all tested samples had an inhibitory effect on the Escherichia coli strain (Table 1). The Hex:EtOAc fraction (9:1) promoted activity against the strain of Candida albicans and Escherichia coli with an MIC of 400 µg mL-1 for both microorganisms, and this action can be justified by the presence of other compounds in the sample, not having specificity between the bacterial and fungal strains tested. The compounds α and β-amyrin (4a/4b), isolated from this fraction, were evaluated as a mixture of triterpenes enhancing antimicrobial activity against the Escherichia coli strain, with an MIC of 300 µg mL-1 when compared to its fraction from Hex:EtOAc (9:1) and its CEE, both with an MIC of 400 µg mL-1. This result was also better when compared to the activity of just the triterpene β-amyrin against the same bacterial strains with an MIC of 10.0 mg mL-1.58 Furthermore, in one study59 the in vitro antibacterial activity against multi-resistant strains of Staphylococcus aureus of a mixture of α-amyrin and β-amyrin was demonstrated through synergistic action with antibiotics. In this same study with in silico tests, these compounds obtained responses due to the presence of the synergistic efflux mechanism, showing greater interaction with the MepA and NorA receptors, which are binding sites for conventional antibiotics, such as ciprofloxacin and norfloxacin, making this association a potential candidate for efflux pump inhibitor. Many reports in the literature60-63 still describe the antitumor, anti-inflammatory, antioxidant, hepatoprotective and antimicrobial potential of terpenoids. The good antimicrobial activity of pentacyclic triterpenes is linked to changes in the structure, morphology and functioning of bacterial cells, with difficulty in biofilm formation, adhesin production and gene expression.64,65 Gerola et al.66 demonstrated the antimicrobial effect of chlorophyll-derived compounds, including pheophorbides. With diverse applicability, pheophytin b has shown potential for the production of biofilms, being a natural biological material used in applications such as electrochemical sensors.67 Another study carried out by de Medeiros et al.68 with pheophytin b demonstrated significant activity against strains of Staphylococcus aureus,Pseudomonas aeruginosa and Escherichia coli at concentrations ranging from 25 to 50 µg mL-1. From this, it was decided to test the compound 173-ethoxypheophorbid a (7), since pheophytins have this antimicrobial power and share the same main skeleton, the porphyrin nucleus. The EtOAc 100% fraction and the compound 173-ethoxypheophorbid a (7) tested showed activity of 400 and 250 µg mL-1, respectively, against the Staphylococcus aureus strain, and both samples showed activity against Escherichia coli with MIC of 400 and 350 µg mL-1, respectively1. A study carried out by Gomes et al.46 further showed that pheophytin a also has a significant antimicrobial effect against the bacterial and fungal strains tested, where the minimum inhibitory concentrations ranged from 38 to 150 µg mL-1. The tested fraction EtOAc:MeOH (9:1) presented an MIC of 400 µg mL-1 against Candidaalbicans. From this fraction, a glycosylated flavone, tiliroside (14), was isolated, which showed good antimicrobial activity against a strain of Escherichia coli, with an MIC of 350 µg mL-1. Tiliroside has an amphipathic characteristic, due to the presence of a p-coumaroyl group in its chemical structure, ensuring greater lipophilicity.69 This physicochemical characteristic determines its antimicrobial power, as it can bind to protein structures, such as efflux pumps produced by resistant bacteria, preventing other substances from diffusing to the outside of the microbial cell and not exerting their bactericidal/bacteriostatic activity.70 Hence to support this evidence, in our study E. coli was the only strain susceptible to all tested fractions and isolated substances < 400 µg mL-1 (Table 1). These results suggest that Gram-negative bacteria with the outer membrane containing lipopolysaccharide are potentially more susceptible than Gram-positive microorganisms. Tiliroside isolated from the aerial parts of Herissantia tiubae demonstrated in vitro modulating activity of bacterial resistance against strains of Staphylococcus aureus (MIC = 256 µg mL-1), modulating the activity of the tested antibiotics, which when administered concomitantly, reduced the MIC of norfloxacin and ciprofloxacin (16 times), lomefloxacin (four times) and ofloxacin (twice), and reduction to acriflavine (128 times), suggesting a possible inhibitor of the efflux pump in bacteria.69 Following recent classifications for antimicrobial activity, the extract, fractions and isolated compounds of Pavonia malacophylla reached levels of moderate activity (100-625 µg mL-1) and with potentially useful activities.71,72 In this way, the antimicrobial activity presented by the extract, fractions and compounds isolated from Pavonia malacophylla can determine its main role in the environment, which is protection against pathogenic agents, thus reinforcing the need to intensify and deepen the mechanisms of action of these promising substances with antimicrobial potential.
CONCLUSIONS The phytochemical studies with aerial parts of Pavonia malacophylla resulted in isolation and identification of 18 compounds, including one alcohol, three steroids, three triterpenes, four chlorophyll derivatives, six flavonoids and one phenolic acid. The crude ethanolic extract, fractions, and four isolated compounds (α and β-amyrin, 173-ethoxypheophorbide a and tiliroside) were analyzed against the Escherichia coli,Candida albicans, and Staphylococcus aureus strains, showing activity in concentrations ranging from 250 to 400 µg mL-1, thus providing an antimicrobial alternative in controlling infections. These results contributing to the chemotaxonomic and ethnopharmacological knowledge of the Malvaceae sensu lato family.
ACKNOWLEDGMENTS The authors would like to thank the National Council for Scientific and Technological Development, Brazil (CNPq) for the financial support, and the Coordination for the Improvement of Higher Education Personnel - Brazil (CAPES) and the Multiuser Characterization and Analysis Laboratory (LMCA-UFPB) for obtaining spectra, and to the Oral Biology Laboratory (CCS-UFPB) for assistance with microbiological tests.
AUTHOR CONTRIBUTIONS JBLA (author), OCS, WAMQ, DAF, BFAR, MFA and MFVS carried out the phytochemical study and identification of the compounds. MFVS supervised the work. FCS performed the microbiological tests.
REFERENCES 1. Keihanian, F.; Saeidinia, A.; Abbasi, K.; Keihanian, F.; Infect. Drug Resist. 2018, 11, 1723. [Crossref] 2. Reygaert, W. C.; AIMS Microbiol. 2018, 4, 482. [Crossref] 3. Cox, G.; Wright, G. D.; Int. J. Med. Microbiol. 2013, 303, 287. [Crossref] 4. Blair, J. M. A.; Webber, M. A.; Baylay, A. J.; Ogbolu, D. O.; Piddock, L. J. V.; Nat. Rev. Microbiol. 2015, 13, 42. [Crossref] 5. Brito-Filho, S. G.; Maciel, J. K. S.; Teles, Y. C. F.; Fernandes, M. M. M. S.; Chaves, O. S.; Ferreira, M. D. L.; Fernandes, P. D.; Felix, L. P.; Cirino, I. C. S.; Siqueira-Júnior, J. P.; Braz-Filho, R.; de Souza, M. F. V.; Rev. Bras. Farmacogn. 2017, 27, 453. [Crossref] 6. Mohammed, F.; Shifa, S. P.; J. Pharmacogn. Phytochem. 2019, 8, 1887. [Link] accessed in April 2025 7. Silva, D. A.; Falcão-Silva, V. S.; Gomes, A. Y. S.; da Costa, D. A.; Lemos, V. S.; Agra, M. F.; Braz-Filho, R.; Siqueira-Junior, J. P.; de Souza, M. F. V.; Pharm. Biol. 2009, 47, 279. [Crossref] 8. Gomes, R. A.; Ramirez, R. R. A.; Maciel, J. K. S.; Agra, M. F.; de Souza, M. F. V.; Falcão-Silva, V. S.; Siqueira-Junior, J. P.; Quim. Nova 2011, 34, 1385. [Crossref] 9. Teles, Y. C. F.; Gomes, R. A.; Oliveira, M. S.; de Lucena, K. L.; do Nascimento, J. S.; Agra, M. F.; Igoli, J. O.; Gray, A. I.; de Souza, M. F. V.; Quim. Nova 2014, 37, 1491. [Crossref] 10. Chaves, O. S.; Teles, Y. C. F.; Monteiro, M. M. O.; Mendes Junior, L. G.; Agra, M. F.; Braga, V. A.; Silva, T. M. S.; de Souza, M. F. V.; Molecules 2017, 22, 94. [Crossref] 11. Oliveira, M. S.; Chaves, O. S.; Teles, Y. C. F.; Fernandes, D. A.; MacaúbasSilva, C.; Queiroz, W. A. M.; Lima, J. B.; Mazzotti, M. R. R. M.; Lima, E. O.; Fernandes, G. L.; Conceição, A. S.; de Souza, M. F. V.; Quim. Nova 2025, 48, e20250011. [Crossref] 12. The Plant List, version 1.1, Malvaceae, http://www.theplantlist.org/1.1/browse/A/Malvaceae/, accessed in April 2025. 13. Grings, M.; Boldrini, I. I.; Revista Brasileira de Biociências 2013, 11, 352. [Link] accessed in April 2025 14. de Albuquerque, J. B. L.; da Silva, C. M.; Fernandes, D. A.; de Souza, P. I. V.; de Souza, M. F. V.; Quim. Nova 2022, 45, 977. [Crossref] 15. Furtado, L. G.; Souza, R. C.; van den Berg, M. E.; Boletim do Museu Paraense Emílio Goeldi, Nova Série: Antropologia 1978, 70, 01. [Link] accessed in April 2025 16. Ribeiro, D. A.; de Oliveira, L. G. S.; de Macêdo, D. G.; de Menezes, I. R. A.; da Costa, J. G. M.; da Silva, M. A. P.; Lacerda, S. R.; Souza, M. M. A.; J. Ethnopharmacol. 2014, 155, 1522. [Crossref] 17. Lima, B. A. T.; Teles, Y. C. F.; Monteiro, G. F.; Silva, C. M.; Chaves, O. S.; Souza, M. F. V.; 3º Simpósio Nordestino de Química; Campina Grande, Brasil, 2017. [Link] accessed in April 2025 18. Sampaio, F. C.; Pereira, M. S. V.; Dias, C. S.; Costa, V. C. O.; Conde, N. C. O.; Buzalaf, M. A. R.; J. Ethnopharmacol. 2009, 124, 289. [Crossref] 19. Teles, Y. C. F.; Chaves, O. S.; Agra, M. F.; Batista, L. M.; de Queiroz, A. C.; de Araújo, M. V.; Alexandre-Moreira, M. S.; Braz-Filho, R.; de Souza, M. F. V.; Rev. Bras. Farmacogn. 2015, 25, 363. [Crossref] 20. Maciel, J. K. S.; Chaves, O. S.; Brito Filho, S. G.; Teles, Y. C. F.; Fernandes, M. G.; Assis, T. S.; De Andrade, A. P.; Felix, L. P.; Silva, T. M. S.; Ramos, N. S. M.; Silva, G. R.; De Souza, M. F. V.; Molecules 2016, 21, 11. [Crossref] 21. Dantas, C. A. G.: Estudo Fitoquímico e Investigação da Atividade Imunomoduladora de Metabólitos Secundários de Erythroxylum simonis Plowman (Erythroxylaceae); Dissertação de Mestrado, Universidade Federal da Paraíba, João Pessoa, Brasil, 2017. [Link] accessed in April 2025 22. Sahidin, I.; Bahrun, A.; Taufik, M.; Yodha, A. W. M.; Sabandar, C. W.; Imran, I.; Kadidae, L. O.; Darmawan, A.; Widodo, H.; Diantini, A.; J. Nat. Pharm. Prod. 2019, 15, e64788. [Crossref] 23. Perry, S. C.; US pat.US20140303262 2014. 24. Xie, F.; China pat.CN107691525 2018. 25. Mukherjee, K.; Tribedi, P.; Mukhopadhyay, B.; Sil, A. K.; FEMS Microbiol. Lett. 2013, 338, 177. [Crossref] 26. de Assis, E. B.: Estudo Fitoquímico e Abordagem sobre a Atividade Larvicida de Helicteres eichleri K. Schum (Sterculiaceae) Frente a Aedes aegypti L. (Diptera: Culicidae); Dissertação de Mestrado, Universidade Federal da Paraíba, João Pessoa, Brasil, 2019. [Link] accessed in April 2025 27. Fernandes, D. A.; Souza, M. S. R.; Teles, Y. C. F.; Oliveira, L. H. G.; Lima, J. B.; Conceição, A. S.; Nunes, F. C.; Silva, T. M. S.; de Souza, M. F. V.; Molecules 2018, 23, 2784. [Crossref] 28. Villaseñor, I. M.; Angelada, J.; Canlas, A. P.; Echegoyen, D.; Phytother. Res. 2002, 16, 417. [Crossref] 29. Loizou, S.; Lekakis, I.; Chrousos, G. P.; Moutsatsou, P.; Mol. Nutr. Food Res. 2010, 54, 551. [Crossref] 30. Gupta, R.; Sharma, A. K.; Dobhal, M. P.; Sharma, M. C.; Gupta, R. S.; J. Diabetes 2011, 3, 29. [Crossref] 31. Baskar, A. A.; Al Numair, K. S.; Paulraj, M. G.; Alsaif, M. A.; Muamar, M. A.; Ignacimuthu, S.; J. Med. Food 2012, 15, 335. [Crossref] 32. Subramaniam, S.; Keerthiraja, M.; Sivasubramanian, A.; Rev. Bras. Farmacogn. 2014, 24, 44. [Crossref] 33. Ferreira, M. D. L.; Fernandes, D. A.; Nunes, F. C.; Teles, Y. C. F.; Rolim, Y. M.; da Silva, C. M.; de Albuquerque, J. B. L.; Agra, M. F.; de Souza, M. F. V.; Rev. Bras. Farmacogn. 2019, 29, 582. [Crossref] 34. Bandeira, P. N.; Lemos, T. L. G.; Costa, S. M. O.; dos Santos, H. S.; Rev. Bras. Farmacogn. 2007, 17, 204. [Crossref] 35. Pereira Júnior, R. J.: Estudo Fitoquímico de Stachytarpheta sessilis Moldenk; Dissertação de Mestrado, Universidade Federal do Ceará, Fortaleza, Brasil, 2008. [Link] accessed in April 2025 36. Barros, F. W. A.; Bandeira, P. N.; Lima, D. J. B.; Meira, A. S.; de Farias, S. S.; Albuquerque, M. R. J. R.; dos Santos, H. S.; Lemos, T. L. G.; de Morais, M. O.; Costa-Lotufo, L. V.; Pessoa, C. O.; Bioorg. Med. Chem. 2011, 19, 1268. [Crossref] 37. Alhassam, A. M.; Ahmed, Q. U.; Latip, J.; Shah, S. A. A.; Khan, A. A. Y. F.; Sarian, M. N.; Wahab, R. A.; Taher, M.; Abdullahi, M. I.; Khatib, A.; S. Afr. J. Bot. 2018, 116, 16. [Crossref] 38. Garcia, C. M.: Estudo Fitoquímico e Atividade Biológica de Pavonia distinguenda A. St.-Hill. et Naudin e Dorstenia brasiliensis Lam.; Tese de Doutorado, Universidade Federal de Santa Maria, Santa Maria, Brasil, 2007. [Link] accessed in April 2025 39. Rauth, S.; Ray, S.; Bhattacharyya, S.; Mehrotra, D. G.; Alam, N.; Mondal, G.; Nath, P.; Roy, A.; Biswas, J.; Murmu, N.; Mol. Cell. Biochem. 2016, 417, 97. [Crossref] 40. Amoussa, A. M. O.; Lagnika, L.; Boujot, M.; Vonthron-Senecheau, C.; Sanni, A.; BMC Complementary Altern. Med. 2016, 16, 284. [Crossref] 41. Devkar, R. A.; Chaudhary, S.; Adepu, S.; Xavier, S. K.; Chandrashekar, K. S.; Setty, M. M.; Pharm. Biol. 2016, 54, 1237. [Crossref] 42. Singh, A.; Mukhtar, H. M.; Kaur, H.; Kaur, L.; Nat. Prod. Res. 2020, 34, 2514. [Crossref] 43. Thirumalaisamy, R.; Amenn, F.; Subramanian, A.; Selvankumar, T.; Alwakeel, S. S.; Govarthanan, M.; Int. J. Pept. Res. Ther. 2020, 26, 2179. [Crossref] 44. de Brito-Filho, S. G.; Fernandes, M. G.; Chaves, O. S.; Chaves, M. C. O.; Araruna, F. B.; Eiras, C.; Leite, J. R. S. A.; Agra, M. F.; BrazFilho, R.; de Souza, M. F. V.; Quim. Nova 2014, 37, 603. [Crossref] 45. Chaves, O. S.; Gomes, R. A.; Tomaz, A. C. A.; Fernandes, M. G.; Mendes-Junior, L. G.; Agra, M. F.; Braga, V. A.; de Souza, M. F. V.; Molecules 2013, 18, 2769. [Crossref] 46. Gomes, R. A.; Teles, Y. C. F.; Pereira, F. O.; Rodrigues, L. A. S.; Lima, E. O.; Agra, M. F.; de Souza, M. F. V.; Braz. J. Pharm. Sci. 2015, 51, 861. [Crossref] 47. Liu, Y.; Fernie, A. R.; Tohge, T.; Plants 2022, 11, 564. [Crossref] 48. Förster, C.; Handrick, V.; Ding, Y.; Nakamura, Y.; Paetz, C.; Schneider, B.; Castro-Falcón, G.; Hughes, C. C.; Luck, K.; Poosapati, S.; Kunert, G.; Huffaker, A.; Gershenzon, J.; Schmelz, E. A.; Köllner, T. G.; Plant Physiol. 2022, 188, 167. [Crossref] 49. Ivanov, M.; Kannan, A.; Stojković, D.; Glamočlija, J.; Grdadolnik, S. G.; Sanglard, D.; Soković, M.; EXCLI Journal 2020, 19, 1436. [Crossref] 50. Moura, A. C. S.; Vilegas, W.; dos Santos, L. C.; Quim. Nova 2011, 34, 1136. [Crossref] 51. Khalil, H. E.; Kamel, M. S.; J. Pharm. Sci. Res. 2015, 7, 509. [Link] accessed in April 2025 52. Pinheiro, A. A. V.: Contribuição para o Conhecimento Fitoquímico de Sida rhombifolia L. (Malvaceae) e Avaliação da Atividade Antimicrobiana do seu Óleo Essencial; Dissertação de Mestrado, Universidade Federal da Paraíba, João Pessoa, Brasil, 2016. [Link] accessed in May 2025 53. Thien, T. V. N.; Do, L. T. M.; Dang, P. H.; Huynh, N. V.; Dang, H. P.; Nguyen, T. T.; Tran, K. T.; Huu, D. M. N.; That, Q. T.; Nat. Prod. Res. 2019, 35, 2218. [Crossref] 54. Khan, R. S.; Senthi, M.; Rao, P. C.; Basha, A.; Alvala, M.; Tummuri, D.; Masubuti, H.; Fujimoto, Y.; Begum, A. S.; Nat. Prod. Res. 2015, 29, 1069. [Crossref] 55. Trachtenberg, A.; Muduli, S.; Sidoryk, K.; Cybulski, M.; Danilenko, M.; Front. Pharmacol. 2019, 10, 507. [Crossref] 56. Kubo, I.; Nihei, K.; Tsujimoto, K.; Bioorg. Med. Chem. 2004, 12, 5349. [Crossref] 57. Yuan, S.; Li, W.; Li, Q.; Wang, L.; Cao, J.; Jiang, W.; J. Agric. Food Chem. 2019, 67, 2801. [Crossref] 58. Musa, N. M.; Sallau, M. S.; Oyewale, A. O.; Ali, T.; Adv. J. Chem., Sect. A 2024, 7, 1. [Crossref] 59. Oliveira, R. C.; Bandeira, P. N.; Lemos, T. L. G.; Santos, H. S.; Scherf, J. F.; Rocha, J. E.; Pereira, R. L. S.; Freitas, T. S.; Freitas, P. R.; Pereira-Junior, F. N.; Marinho, M. M.; Marinho, E. M.; Marinho, E. S.; Nogueira, C. E. S.; Coutinho, H. D. M.; Teixeira, A. M. R.; J. Biomol. Struct. Dyn. 2021, 40, 12785. [Crossref] 60. Somova, L. O.; Nadar, A.; Rammanan, P.; Shode, F. O.; Phytomedicine 2003, 10, 115. [Crossref] 61. Tsai, S. J.; Yin, M.-C.; J. Food Sci. 2008, 73, 174. [Crossref] 62. Sultana, T.; Rashid, M. A.; Ali, M. A.; Mahmood, S. F.; Bangladesh J. Sci. Ind. Res. 2010, 45, 27. [Crossref] 63. Shanmugan, M. K.; Dai, X.; Kumar, A. P.; Tan, B. K.; Sethi, G.; Bishayee, A.; Biochem. Pharmacol. 2013, 85, 1579. [Crossref] 64. Huang, L.; Luo, H.; Li, Q.; Wang, D.; Zhang, J.; Hao, X.; Yang, X.; Eur. J. Med. Chem. 2015, 95, 64. [Crossref] 65. Navina, R.; Lee, Y. G.; Kim, S. M.; Curr. Bioact. Compd. 2017, 13, 177. [Crossref] 66. Gerola, A. P.; Santana, A.; França, P. B.; Tsubone, T. M.; de Oliveira, H. P.; Caetano, W.; Kimura, E.; Hioka, N.; Photochem. Photobiol. 2011, 87, 884. [Crossref] 67. Pauli, G. E. N.; Araruna, F. B.; Eiras, C.; Leite, J. R. S. A.; Chaves, O. S.; Brito Filho, S. G.; de Souza, M. F. V.; Chavero, L. N.; Sartorelli, M. L.; Bechtold, I. H.; Materials Science and Engineering: C 2015, 47, 339. [Crossref] 68. de Medeiros, F. A.; Medeiros, A. A. N.; Tavares, J. F.; Barbosa Filho, J. M.; Lima, E. O.; da Silva, M. S.; Quim. Nova 2012, 35, 1179. [Crossref] 69. Falcão-Silva, V. S.; Silva, D. A.; Souza, M. F. V.; Siqueira-Júnior, J. P.; Phytother. Res. 2009, 23, 1367. [Crossref] 70. Farhadi, F.; Khameneh, B.; Iranshahi, M.; Iranshahy, M.; Phytother. Res. 2019, 33, 13. [Crossref] 71. Freires, I.; Denny, C.; Benso, B.; de Alencar, S.; Rosalen, P.; Molecules 2015, 20, 7329. [Crossref] 72. Van Vuuren, S.; Holl, D.; J. Ethnopharmacol. 2017, 208, 236. [Crossref]
Editor handled this article: Jorge M. David |

On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access