
 |
 |
 |
 |
 |
Artigo
| A new alkaloid isolated from Tabernaemontana hystrix Steud. (Apocynaceae) and the antiproliferative potential of the root bark against human cervical cancer (HeLa) |
|
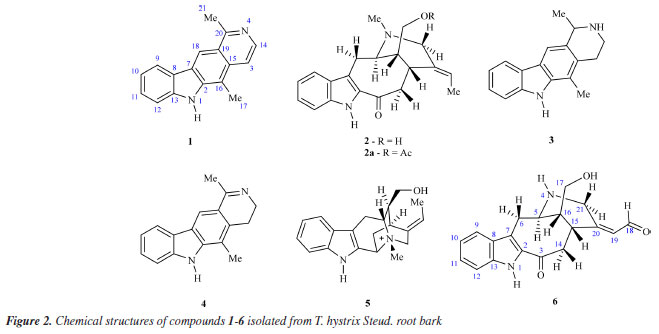
Thalya S. R. NogueiraI,* I. Laboratório de Ciências Químicas, Setor de Química dos Produtos Naturais, Centro de Ciências e Tecnologias, Universidade Estadual do Norte Fluminense Darcy Ribeiro, 28013-602 Campos dos Goytacazes - RJ, Brasil Received: 01/27/2025 *e-mail: thalyasrnogueira@gmail.com Cervical cancer is the fourth most common cancer in the world and the third most common in Brazilian women. The search for compounds with anticancer potential is important, since cancer is a public health problem, and some drugs may lead to side effects. In addition, cases of multidrug resistance may occur. Tabernaemontana species are considered a promising source of bioactive compounds. The antiproliferative potential of the crude extract and fractions of Tabernaemontana hystrix Steud. (Apocynaceae) root bark were evaluated through MTT assays using HeLa cells and NIH-3T3 cells. It was observed that EtOAc and BuOH fractions were selective against tumor cells, with IC50 (halfmaximal inhibitory concentration) values of 3.787 and 8.489 μg mL-1, respectively. The phytochemical investigation of both fractions was carried out, leading to the isolation of six monoterpene indole alkaloids (MIAs): olivacine (1), affinine (2), the mixture of alkaloids 1, janetine (3) and 3,14-dihydro-olivacine (4), and macusine B (5) from the EtOAc fraction. However, the BuOH fraction also provided compounds 1 and 3, and the new MIA, named as Nb-demethyl-18-formyl-affinine (6). Compounds 3 and 4 are being reported for the first time in the species. Further research is needed to evaluate the antiproliferative potential of these compounds. INTRODUCTION Cervical cancer, characterized by disordered replication of the epithelial lining of this organ, is caused by persistent infection by oncogenic types of human papillomavirus (HPV). Although cervical cancer is a preventable disease, it is considered one of the most common cancers. The data point out that this is the fourth most diagnosed type of cancer in women worldwide and the most significant cause of death in young women.1 In Brazil, cervical cancer is the third most common cancer among women, with an estimated 17,010 new cases for each year of the three year period 2023-2025. It is also the fourth leading cause of death among women (6,938 deaths in 2022).2 Treatment depends on the stage of the disease, the size of the tumor, and personal aspects (e.g., age, fertility), but may include surgery, radiotherapy, and chemotherapy.3-5 Pharmacological treatment with antitumor drugs is widely used and is also considered a challenge in cancer treatment. Chemotherapy drugs already available in the clinic can lead to adverse effects, such as nausea and vomiting, loss of appetite and weight, fatigue, leukopenia, and toxic effects, such as nephrotoxicity, hepatotoxicity, cardiotoxicity, and hearing loss, among others. In addition, cancer cells are often resistant to these drugs. Therefore, new forms of treatment are needed that demonstrate safety, effectiveness, and low cost.6-8 In this context, compounds produced by the secondary metabolism of living organisms could be used to treat both the symptoms and the disease. In fact, secondary metabolites are considered an important source of new bioactive compounds or inspiration for drug development, due to their molecular diversity, biological activities, and low side effects.9,10 The Tabernaemontana L. (Apocynaceae) genus includes more than 100 species, distributed in tropical and subtropical regions.11 Species of this genus are known for the bioproduction of monoterpene indole alkaloids (MIAs), a class of compounds considered promising for the discovery of new drugs, since MIAs might be responsible for a wide range of pharmacological activities, including anticancer, antimalarial, and antiarrhythmic.12-14 Tabernaemontana hystrix Steud. (Apocynaceae) is endemic to Brazil, where it can be found in the Brazilian Cerrado and Atlantic Forest and is popularly known as "gancheira" or "janaguba".15 This species is traditionally used by indigenous communities (Te'yikue village at Caarapo and Panambi-Douradina village) from Mato Grosso do Sul, where the leaves and alcohol are employed for mosquito and snake bites, and bark decoction is used for earache.16 Previous studies17,18 reported MIAs structures possessing novel carbon skeletons isolated from T. hystrix. The current article reports the antiproliferative potential of T. hystrix root bark against HeLa and NIH-3T3 cells, and the structural elucidation of a new alkaloid named as Nb-demethyl-18-formyl-affinine (6) together with five known alkaloids, olivacine (1), affinine (2), the mixture of alkaloids 1, janetine (3) and 3,14-dihydro-olivacine (4), and macusine B (5).
EXPERIMENTAL General experimental procedures Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker ASCEND 500 spectrometer (500 MHz for 1H and 125 MHz for 13C), using CDCl3 as solvent. High-resolution mass spectrometry (HRESIMS) data were acquired using an micrOTOFQ II BrukerDaltonics mass spectrometer, with the use of the positive ion mode of analysis. Isolation and purification of the chemical compounds were conducted using several chromatographic methods, including column chromatography (CC), thin-layer chromatography (TLC), vacuum liquid chromatography (VLC), and high-performance liquid chromatography (HPLC). Medium pressure analyses were carried out using a medium pressure liquid chromatography (MPLC) system (Büchi, Flawil) equipped with a C-620 control unit, C-605 module pump, C-640 ultraviolet (UV) detector, and C-660 fraction collector. Analytical and semi-preparative chromatographic analyses were performed using an HPLC system (Shimadzu) equipped with a CBM-20A control unit, LC-20AD binary solvent pump, manual injector, and SPD-M20A diode array detector. A semi-preparative reversed phase (RP) C18 column (Kromasil 100A; 10 mm × 250 mm × 5 µm) was used. Column chromatography was performed on silica gel (0.063-0.200 mm, Merck). All solvents used for extraction and isolation were purchased from Synth with analytical grade. Plant material Root bark was collected at the Sítio Xodó in Varre-Sai, Rio de Janeiro, Brazil (20º54'26.7"S, 41º53'05.4"W), by Antônio Sérgio Nascimento Moreira. The voucher specimen (HUENF 14592) can be found at the herbarium of the State University of the North Fluminense Darcy Ribeiro. The current work was registered in the National System of Management of Genetic Heritage and Associated Traditional Knowledge (SisGen) under No. AC8E4F3. Extraction and isolation procedures Dried and powdered root bark (1.36 kg) from T. hystrix was extracted with MeOH three times at room temperature (each for 5 days), furnishing, after solvent evaporation, 80.3 g of root bark crude extracts. The crude extract was suspended in H2O and partitioned successively with dichloromethane (CH2Cl2), ethyl acetate (EtOAc), and butanol (n-BuOH). Once the EtOAc, and BuOH root bark extracts showed better results than CH2Cl2 in the antiproliferative potential assay, these extracts were selected to be chromatographed to isolation compounds. The EtOAc extract (6.45 g) was subjected to silica gel CC eluted with CH2Cl2:MeOH (100:1 to 1:1) yielding 8 fractions (THRA 1-THRA 8). Fraction THRA 7 (2.22 g) was applied to a neutral alumina CC and eluted with CH2Cl2:MeOH (100:0 to 1:1) to afford 10 subfractions (THRA 7.1-THRA 7.10). THRA 7.1 (46.6 mg) furnished a yellow precipitate, which was washed with acetone to give compound 1 (20.4 mg). THRA 7.3 (391.6 mg), shown to be purified, was identified as compound 2. THRA 7.10 (121.4 mg) was fractionated by a silica gel CC column with CH2Cl2:MeOH (100:0 to 1:1), yielding nine fractions (THRA 7.10.1-THRA 7.10.9). THRA 7.10.9 (19.7 mg) was purified by a silica gel CC column and eluted with CH2Cl2:MeOH (100:0 to 1:1) yielding the mixture (5.4 mg) of compounds 1, 3 and 4. THRA 8 (1.20 g) was fractioned by neutral alumina CC using CH2Cl2:MeOH (100:0 to 1:1) to afford 10 subfractions (THRA 8.1-THRA 8.10). THRA 8.7 (131.7 mg), shown to be purified, was identified as compound 5. Compound 1 (104.6 mg) was obtained as a precipitate from the BuOH fraction after washing with acetone. The BuOH extract (10.0 g) was applied to a neutral alumina CC and eluted with a CH2Cl2:MeOH (100:0 to 1:1) to afford 11 subfractions. After analytical analysis by high-performance liquid chromatography with diode array detector (HPLC-DAD), these fractions were grouped into 9 fractions (THRB 1-THRB 9). THRB 8 (100 mg) was chromatographed on MPLC using a C18 column (glass column 15/460-044032 manually packed with C18 as stationary phase) and the following gradient elution: solvent A = H2O and formic acid (0.1% v/v); solvent B = MeOH; elution profile = 0.0-70.0 min (5-100% B); 70.0130.0 min (100% B); flow rate of 15.0 mL min-1; 10 fractions were collected (THRB 8.1-THRB 8.10). Fraction correspondent to 6 (THRB 8.6, 18.0 mg) was identified as compound 3. The fraction 9 (THRB 8.9, 4.8 mg) was chromatographed on HPLC-DAD using a C18 column (Kromasil 100A; 10 mm × 250 mm × 5 µm) and the following gradient elution: solvent A = H2O and formic acid (0.1% v/v); solvent B = MeOH; elution profile = 0.0-25.0 min (35-90% B); 26.010.0 min (100% B); and flow rate of 3.0 mL min-1; allowing the isolation of compound 6 (retention time (tR) = 16.265 min, 3.4 mg). Compound characterization Olivacine (1) HRESIMS [M + H]+ m/z 247.1240 (100.0%; C17H15N2+, calcd. m/z 247.1235; Dm/z = 2.0), product ions = 232.0915 (90.0%), 205.0766 (20.0%), 191.0770 (15.0%), 167.0770 (5.0%); 1H NMR (500 MHz, CD3OD) d 8.87 (1H, s, H-18), 8.26 (1H, ddd, J 7.8, 1.1, 0.8 Hz, H-9), 8.10 (1H, d, J 6.6 Hz, H-3), 8.01 (1H, dl, J 6.6 Hz, H-14), 7.55 (1H, td, J 8.1, 1.1 Hz, H-11), 7.52 (1H, ddd, J 8.0, 1.3, 0.7 Hz, H-12), 7.30 (1H, ddd, J 8.1, 6.7, 1.4 Hz, H-10), 3.11 (3H-21, s), 2.80 (3H-17, s); 13C NMR (125 MHz, CD3OD) d 144.32 (C-2), 133.1 (C-3), 128.4 (C-7), 123.8 (C-8), 122.5 (C-9), 121.3 (C10), 129.7 (C-11), 112.2 (C-12), 144.34 (C-13), 118.1 (C-14), 134.6 (C-15), 113.3 (C-16), 12.4 (C-17), 117.5 (C-18), 122.4 (C-19), 159.1 (C-20), 20.4 (C-21). Affinine (2) HRESIMS [M + H]+ m/z 325.1914 (85.0%, C20H25N2O2+, calcd. m/z 325.1911, Dm/z = 0.9), product ions = 307.1709 (42.7%), 265.1706 (100.0%), 152.1082 (48.0%); 1H NMR (500 MHz, CD3OD) d 7.72 (1H, dl, J 8.1 Hz, H-9), 7.43 (1H, d, J 8.3 Hz, H-12), 7.31 (1H, ddd, J 8.3, 8.1, 0.9 Hz, H-11), 7.11 (1H, ddd, J 8.3, 8.1, 0.9 Hz, H-10), 5.54 (1H, ql, J 6.7 Hz, H-19), 3.58 (1H, m, H-21b), 3.483.42 (2H, m, 2H17), 3.40-3.34 (2H, m, 2H-6), 3.32-3.36 (1H, m, H-14b), 3.103.16 (1H, m, H-5), 3.04 (1H, ddd, J 1.5, 7.3, 11.4 Hz, H-15), 2.90 (1H, dl, J 10.8 Hz, H-21a), 2.51 (1H, dd, J 7.4, 13.0 Hz, H-14a), 2.48 (3H, s, Me-N-4), 1.94 (1H, t, J 6.5 Hz, H-16); 13C NMR (125 MHz, CD3OD) d 191.5 (C-3), 137.2 (C-13), 136.0 (C-2), 134.7 (C-20), 128.2 (C-8), 126.0 (C-11), 121.3 (C-19), 120.4 (C-7), 120.2 (C-9), 119.7 (C-10), 112.0 (C-12), 64.1 (C-17), 55.6 (C-5), 52.0 (C-21), 42.8 (C-14), 41.0 (Me-N-4), 40.0 (C-16), 29.8 (C-15), 19.4 (C-6), 10.8 (C-18). Janetine (3) HRESIMS [M + H]+ m/z 251.1535 (8.5%, C17H19N2+, calcd. m/z 251.1543; Dm/z = -3.2), product ions = m/z 249.1387 (22.2%), 234.1298 (100.0%), 219.1052 (44.0%), 208.1131 (39.0%); 1H NMR (500 MHz, CD3OD) d 8.04 (1H, d, J 7.8 Hz, H-9), 7.87 (1H, s, H-18), 7.49 (1H, d, J 8.2 Hz, H-12), 7.38 (1H, ddd, J 8.0, 7.2, 1.0 Hz, H-11), 7.16 (1H, ddd, J 8.0, 7.2, 1.0 Hz, H-10), 5.58-5.67 (1H, m, H-20), 3.44-3.69 (1H, m, H-3), 3.06-3.25 (1H, m, H-14), 2.48 (3H, s, 3H17), 1.80 (3H, d, J 6.5 Hz, 3H-21); 13C NMR (125 MHz, CD3OD) d 142.3 (C-13), 140.8 (C-2), 127.7 (C-19), 127.1 (C-11), 125.6 (C-15), 124.5 (C-8), 123.3 (C-7), 121.2 (C-9), 120.1 (C-10), 119.2 (C-16), 116.3 (C-18), 112.1 (C-12), 53.5 (C-20), 40.8 (C-3), 24.7 (C-14), 20.5 (C-21), 13.3 (C-17). 3,14-Dihydro-olivacine (4) GC-MS [M]+ m/z 248 (100.0%, C17H16N2, calcd. m/z 248), product ions: m/z 247 (95.0%), 233 (50%); 1H NMR (500 MHz, CD3OD) d 8.87 (1H, sl, H-9), 8.40 (1H, s, H-21), 7.45 (1H, dt, J 8.2, 1.0 Hz, H-12), 7.35 (1H, ddd, J 8.2, 7.2, 1.0 Hz, H-11), 7.13 (1H, ddd, J 8.0, 7.1, 0.8 Hz, H-10), 3.69 (2H, t, J 7.5 Hz, 2H-3), 3.00 (2H, t, J 7.5 Hz, 2H-3), 2.53 (3H, s, 3H-17), 2.48 (3H, s, 3H-18); 13C NMR (125 MHz, CD3OD) d 144.3 (C-2), 143.8 (C-19), 142.1 (C-13), 134.0 (C-15), 128.7 (C-20), 126.6 (C-11), 124.5 (C-8), 122.8 (C-7), 120.8 (C-9), 120.0 (C-21), 119.8 (C-10), 118.0 (C-16), 111.8 (C-12), 45.5 (C-3), 24.01 (C-14), 13.0 (C-18), 12.8 (C-17). Macusine B (5) HRESIMS [M + H]+ m/z 309.1972 (85.5%, C20H25N2O+, calcd. m/z 309.1967, Dm/z = 1.6), product ions = m/z 291.1872 (100.0%), 261.1380 (2.5%), 160.1126 (8.3%), 148.1122 (7.1%); 1H NMR (500 MHz, CD3OD) d 7.51 (1H, dt, J 7.8, 0.8 Hz, H-9), 7.40 (1H, dt, J 8.2, 0.8 Hz, H-12), 7.19 (1H, ddd, J 8.2, 7.1, 1.1 Hz, H-11), 7.08 (1H, ddd, J 8.2, 7.1, 1.1 Hz, H-10), 5.65 (1H, q, J 6.8 Hz, H-19), 4.94 (1H, d, J 10.2 Hz, H-3), 4.45 (1H, dquint, J 16.0, 2.5 Hz, H-21b), 4.23 (1H, dt, J 16.0, 1.6 Hz, H-21a), 3.58-3.53 (1H, m, H-5), 3.58-3.53 (2H, m, 2H-17), 3.32-3.34 (H, m, H-6b), 3.11-3.13 (1H, m, H-6a), 3.11-3.13 (1H, m, H-15), 3.10 (3H, s, Me-N-4), 2.53 (1H, ddd, J 13.4, 10.2, 1.8 Hz, H-14b), 2.14-2.17 (1H, m, H-14a), 2.162.19 (1H, m, H-16), 1.71 (3H, ddd, J 6.8, 2.5, 1.1 Hz); 13C NMR (125 MHz, CD3OD) d 38.8 (C-13), 132.5 (C-2), 129.0 (C-20), 127.5 (C-8), 123.8 (C-11), 122.1 (C-19), 121.0 (C-10), 119.3 (C-9), 122.7 (C-12), 102.0 (C-7), 66.8 (C-5), 65.8 (C-21), 63.8 (C-17), 62.4 (C-3), 48.1 (Me-N-4), 45.1 (C-16), 33.4 (C-14), 27.4 (C-15), 25.3 (C-6), 13.0 (C-18). Nb-Demethyl-18-formyl-affinine (6) HRESIMS [M + H]+ m/z 325.1536 (100.0%, C19H21N2O3+, calcd. m/z 325.1547 Dm/z = -3.4), product ions = m/z 307.1414 (15.4%), 283.1414 (9.5%), 265.1313 (3.2%), 152.0719 (18.1%); 1H NMR (500 MHz, CD3OD) d 8.85 (1H, sl, H-18), 7.86 (1H, d, J 8.2 Hz, H-9), 7.44 (1H, d, J 8.3 Hz, H-12), 7.32-7.34 (1H, m, H-19), 7.32 (1H, ddd, J 8.3, 8.2, 0.7 Hz, H-11), 7.15 (1H, ddd, J 8.2, 8.3, 0.7 Hz, H-10), 4.13-4.21 (1H, m, H-6a), 3.67-3.72 (1H, m, H-5), 3.60 (1H, dd, J 11.1, 5.0 Hz, H-17b), 3.52 (1H, dd, J 11.1, 6.0 Hz, H-17a), 3.41-3.47 (1H, m, H-16), 3.34 (1H, dd, J 7.7, 5.0 Hz, H-14a), 3.13 (1H, dd, J 10.9, 6.0 Hz, H-21a), 3.05 (1H, dd, J 10.9, 8.1 Hz, H-21b), 2.98 (1H, sl, H-15), 2.84-2.91 (1H, m, H-6b), 2.75-2.79 (1H, m, H-14b); 13C NMR (125 MHz, CD3OD) d 192.2 (C-3), 188.4 (C-18), 156.2 (C-19), 138.5 (C-20), 138.4 (C-13), 136.8 (C-2), 128.1 (C-8), 127.4 (C-11), 121.6 (C-9), 121.5 (C-7), 121.3 (C-10), 113.4 (C-12), 64.4 (C-17), 63.6 (C-21), 56.3 (C-5), 48.0 (C-14), 42.6 (C16), 29.8 (C-6), 25.6 (C-15). Biological assays Cell viability assay and selectivity On 96-well plates, cells were seeded in triplicate with 3 × 104 cells per well (HeLa or NIH-3T3). After 24 h, the samples were added, and the cells were incubated for 48 h. Subsequently, treatments were withdrawn and the cells were incubated with 0.5 mg mL-1 of MTT reagent (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide) (Sigma-Aldrich) for 4 h. The formazan crystals were dissolved in DMSO and the absorbance was measured at 570 nm using an Epoch microplate spectrophotometer (BioTek Instruments). Carboplatin (Libbs Farmacêutica) was used as a positive control. Non-linear regression was used to calculate the concentration capable of inhibiting cell viability by 50% (IC50) using GraphPad Prism (GraphPad Software, version 8.0.2). The selectivity index of a compound was evaluated by the ratio between the IC50 of the compound in non-tumor cells and the IC50 of the compound in tumor cells, in this case NIH-3T3 and HeLa cells, respectively. Statistical analysis The statistical program GraphPad Prism 8 was used to perform statistical analyses. Results are displayed as the average ± standard error. In cell viability and cell migration experiments, a one-way parametric analysis of variance (ANOVA) test was performed, followed by the Tukey test to evaluate which samples differed statistically from the negative control group. p-Values less than 0.05 were considered significant.
RESULTS AND DISCUSSION The antiproliferative potential of the crude extract, CH2Cl2, EtOAc, and BuOH fractions from T. hystrix root bark was evaluated against human cervical cancer (HeLa cell line) and non-cancerous cell lines (fibroblast cell line, NIH-3T3) through the MTT assay. According to Indrayanto et al.,19 all samples were considered to present good activity, being more cytotoxic to cancerous cells than non-cancerous. In particular, ethyl acetate fraction was considered the most promising, since half maximal inhibitory concentration (IC50) value of 3.787 µg mL-1 was observed to HeLa cells, which was lower than observed for the positive control, carboplatin (IC50 = 17.84 µg mL-1). According to the National Cancer Institute (NCI) guidelines, extracts with IC50 values below 20 µg mL-1 are considered promising for potential anticancer activity.20 In addition, the selectivity index (SI) found for all samples (values ≥ 2) indicates that the samples affect the cancer cells more than normal cells. This analysis also emphasizes the potential of the ethyl acetate fraction (SI = 11.25) as a prospective anticancer sample, since it showed a SI value ≥ 10.19 The percentage of viable HeLa cells for each sample when treated with different concentrations of treatments are shown in Figure 1, while the IC50 values for HeLa and NIH-3T3 cell lines, and the SI are presented in Table 1.
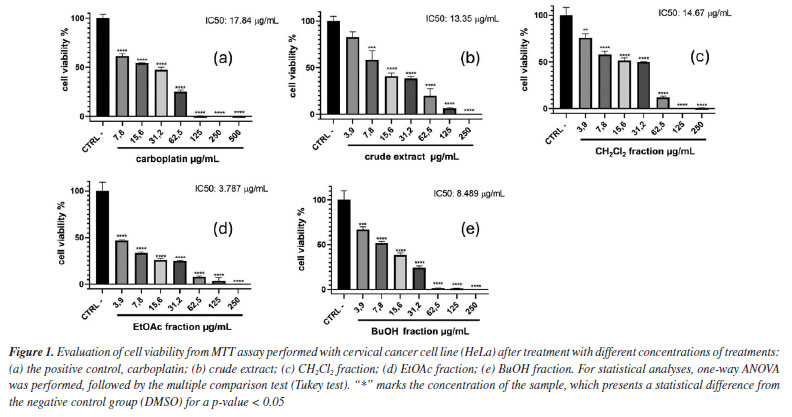
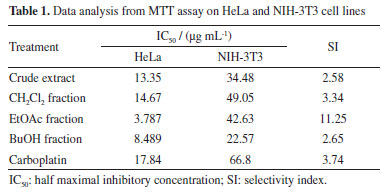
The results of the biological activity of the extract and fractions presented here are consistent with those reported in the literature14,21 for Tabernaemontana species. Due to the results observed in the biological assay, the ethyl acetate fraction from T. hystrix root bark was selected for the isolation of compounds. The butanol fraction was also chosen because of its chemical profile, which indicates the presence of more nitrogenated compounds (alkaloids) than the other fractions on TLC revealed with Dragendorff. Moreover, articles on Tabernaemonta rarely work with polar fractions. They usually work with acid-base extracts. From the ethyl acetate fraction, the compounds obtained were identified as olivacine22 (1), affinine23 (2), the mixture of 1 (2.41%), janetine24 (3, 52.17%) and 3,14-dehydro-olivacine24 (4, 45.41%), and macusine B25 (5), whereas the butanol fraction provided compounds 1, 3, and one previously unknown compound (6). The chemical structures reported here are shown in Figure 2 and have been characterised by 1H and 13C NMR (1D and 2D), and HRESIMS experiments, and compared with data described in the literature.
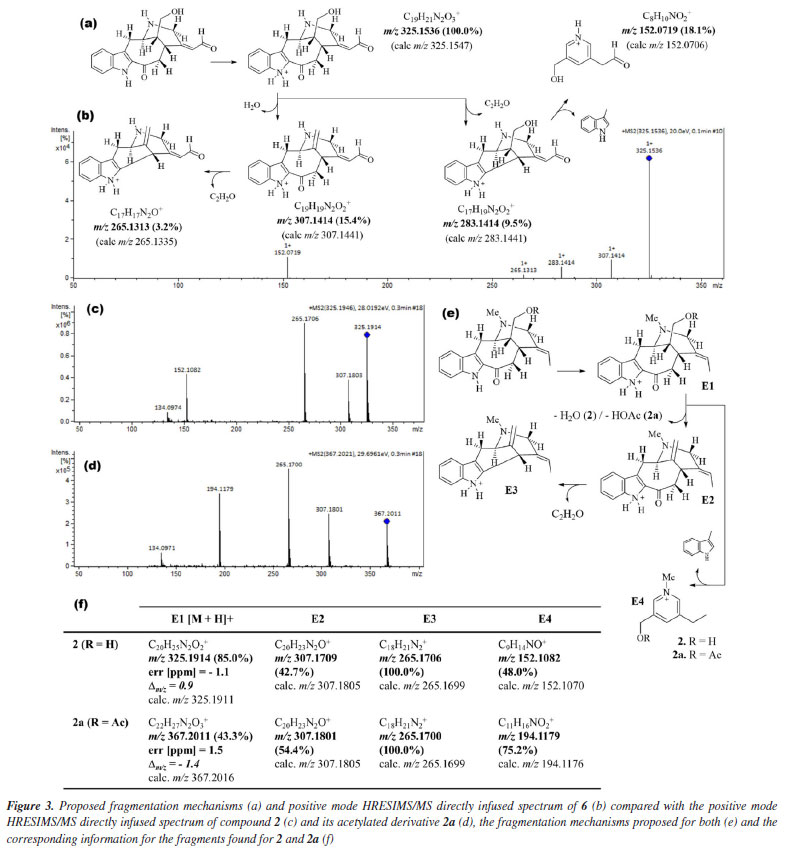
Compound 6 was isolated as a brown ceric-spray. The molecular formula, C19H20N2O3, was found from the protonated molecular ion [M + H]+ at m/z 325.1536 (Dm/z = -3.4), implying a hydrogen deficiency index of 11 (6 attributed to an indole unit, 2 to a bicycle system in the monoterpene moiety, 2 to carbonyl functions of ketone and aldehyde groups, and 1 to an unsaturation in the monoterpene moiety). The directly infused HRESIMS/MS spectrum and the proposed fragmentation mechanisms of 6 are shown in Figures 3a and 3b.
Analysis of the 1H, attached proton test (APT)-13C, and heteronuclear single quantum correlation (HSQC) NMR spectra indicated signals corresponding to 19 C-atoms, including 12 sp2 carbon atoms [six methines and six non hydrogenated: assigned as one indole ring - dC 136.8 (C-2), 121.5 (C-7), 128.1 (C-8), 121.6 (C9), 121.3 (C-10), 127.4 (C-11), 113.4 (C-12), and 138.4 (C-13), two carbonyl groups - dC 192.2 (C-3) and 188.4 (CH-18), and one double bond - dC 156.2 (C-19) and 138.5 (C-20)], 3 sp3 methines {CH-5, CH-15, and CH16 [including one linked to an N-atom at dC 56.3 (C-5)]}, and 4 sp3 methylenes {CH2-6, CH2-14, CH2-17 and CH2-21 [including one linked to an N-atom at dC 63.5 (CH2-21)]}. In the 1H NMR spectrum, it was possible to recognize the presence of four signals characteristic of aromatic hydrogen atoms of a non-substituted indole ring24 at dH 7.86 (1H, d, J 8.2 Hz, H-9), 7.15 (1H, ddd, J 8.3, 8.2, 0.7 Hz, H-10), 7.32 (1H, ddd, J 8.3, 8.2, 0.7 Hz, H-11), and 7.44 (1H, d, J 8.3 Hz, H-12), correlated in the HSQC spectrum with the carbon signals at dC 121.6 (CH-9), 121.3 (CH-10), 127.4 (CH-11), and 113.4 (CH-12), respectively. The 1H-1H-COSY spectrum of 6 was used to establish homonuclear hydrogen atom interactions (geminal and vicinal), and HSQC and HMBC (heteronuclear multiple bond correlation) experiments to recognize heteronuclear correlations through interactions between C- and H-atoms via direct (1JHC) and long-range couplings, involving two (2JHC) and three (3JHC) bonds. All NMR data are presented in Table 2, and Figure 4 summarizes the observed 1H-1H-COSY, HMBC, and 1H-1H-NOESY (nuclear Overhauser enhancement spectroscopy) correlations. These data contribute to confirming the presence of a non-substituted benzene ring in the indole moiety. Furthermore, a similarity between compounds 6 and 2 could be suggested. The principal difference between these two alkaloids lies in the absence of the methyl group linked to the aliphatic N-4 (or Nb), and the presence of carbonyl of aldehyde group (CH-18) in 6, replacing the methyl group (CH3-18) in 2. The 1H and 13C NMR chemical shift assignments for compound 2 are also presented in Table 2.
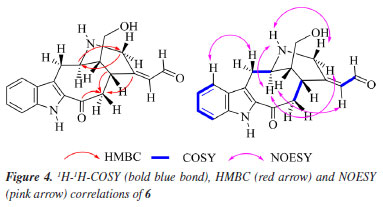
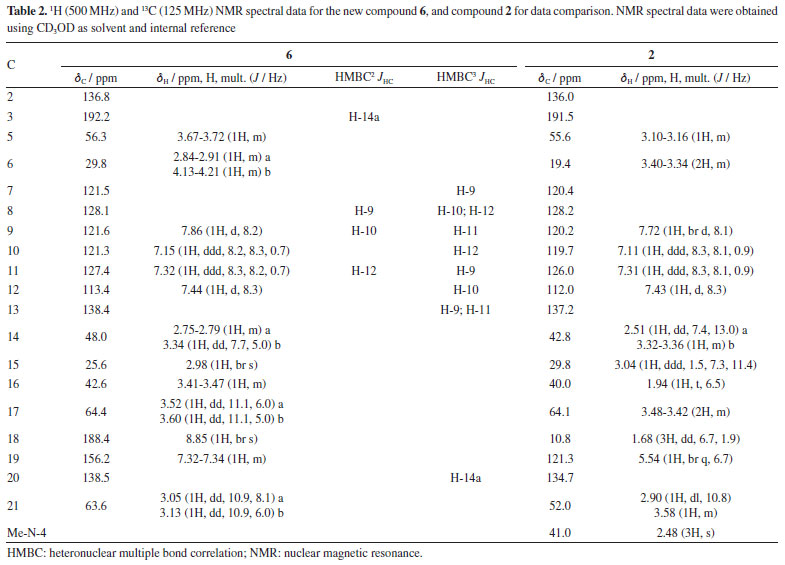
In the 1H NMR spectrum of compound 6, it was possible to observe the absence of the signal characteristic of hydrogen atoms of a methyl group linked to the aliphatic N-4, when compared with the 1H NMR spectrum of compound 2 [signal at dH 2.48 (s, 3H) with 1JHC HSQC correlation at dC 41.0 (Me-N-4)] or literature.17 In addition, when the compound has the Me-N-4 system, it is possible to observe structural effects affecting the chemical shift. In this case, due to the steric interactions, exemplified by the γ-effect, "the introduction of a methyl group into a cyclic molecule leads to new interactions with an adjacent hydrogen atom, resulting in the shielding of the carbon nuclei that are involved".26 In agreement with this, a difference in the signals of CH2-6 (dC 29.8 and dC 19.4, for compounds 6 and 2, respectively) and CH-16 (dC 42.6 and dC 40.0, for compounds 6 and 2, respectively) was observed in the APT-13C NMR spectra between compounds 6 and 2, where both carbon atoms in compound 2 presented shielded values of chemical shift due to the presence of Me-N-4, confirming the proposed structure of compound 6. A difference in the chemical shift of hydrogen can also be observed for H-6 and H-16 in the 1H NMR spectrum. Another difference in the 1H NMR spectrum of compound 6 compared to 2 was the absence of two signals characteristic of a monoterpene indole alkaloid structure,23 signals related to methine and methyl hydrogens, H-19 [dH 5.54 (1H, br q, J 6.7 Hz)] and 3H-18 [dH 1.68 (3H, dd, J 6.7, 1.9 Hz)], respectively, which can be observed in compound 2. On the other hand, based on the COSY, HMBC, and NOESY correlations (see correlations in Figure 4), two unshielded signals were assigned to H-18 [dH 8.85 (1H, br s)] and H-19 [dH 7.32 (1H, m)]. Furthermore, due to the 2JHC heteronuclear correlation in the HSQC spectrum dH 8.85 (H-18) and dC 188.4 (C18), and COSY correlations observed between H-18 and H-19, these signals are related to a α,β-unsaturated carbonyl group. The ketone group localized at position C-3 (dC 192.2) was confirmed by 2JHC heteronuclear correlation with H-14a [dH 2.51 (1H, dd, J 7.4, 13.0 Hz)] in the HMBC spectrum. A signal at dC 64.4 observed in the APT-13C NMR spectra, characteristic of a C-atom bonded to a hydroxyl group, correlated with two signals of diastereotopic hydrogens in the 1H NMR spectrum at dH 3.52 (1H, dd, J 11.1, 6.0 Hz, H-17a) and dH 3.60 (1H, dd, J 11.1, 5.0 Hz, H-17b), which were assigned to CH2-17, and indicates the presence of a methylene carbinol carbon. Although there were fewer correlations in the HMBC spectrum, information from the MS/MS analysis of the directly infused compounds 6, 2, and its acetylated derivative 2a supported this proposed structure. As shown in Figure 3, these three molecules exhibited a similar fragmentation pattern, confirming that they belong to the same type of monoterpene indole alkaloid structure, the corynanthean type (vobasan subtype).27 The product ion at m/z 307.1414 (C19H19N2O2+) for compound 6, m/z 307.1709 (C20H23N2O+) and m/z 307.1801 (C20H23N2O+) for compounds 2 and 2a, respectively, indicates the loss of H2O (-18 Da) for compounds 6 and 2, and HOAc (-60 Da) for 2a. Another product ion and suggested molecular formula that could provide key information about the chemical structure of compound 6 is the one at m/z 152.0719 (C8H10NO2+). For compounds 2 and 2a these were observed at m/z 152.1082 (C9H14NO+) and m/z 152.1082 (C11H16NO2+), respectively. The similarities and differences between these two alkaloids (6 and 2) are confirmed by these fragments, which aid in the suggestion of carbinol carbon at CH2-17 and carbonyl of the aldehyde group at CH-18. The relative stereochemistry for the C-16 was proposed according to the literature.23 Compound 2 and its epimer present different values for the chemical shift of C-16. The literature23,28-30 shows a dC ~ 38.5 for C-16 from epi-affinine.Herein, values of dC 40.0 and dC 42.6 were observed for C-16 in compounds 2 and 6, respectively, showing that these compounds have a different stereochemistry compared to the epimer. Compound 6 was named as Nb-demethyl-18-formyl-affinine. Figure 3 shows the HRESIMS/MS spectrum for compounds 6, 2, and 2a (Figures 3b, 3c, 3d, respectively), the proposed fragmentation mechanisms for both (Figures 3a and 3e, respectively), and the corresponding information for the fragments found for 2 and 2a (Figure 3f).
CONCLUSIONS Based on the results of the present study, it can be concluded that the root bark samples of T. hystrix showed promising antiproliferative activity against HeLa cervical cancer cells, being selective for this cell line. In general, the samples were more active than the positive control used (carboplatin), especially the EtOAc fraction selected for phytochemical investigation. Consequently, a phytochemical study of the most active fraction of the T. hystrix root bark was carried out. The EtOAc fraction resulted in the isolation of the following compounds: olivacine (1), affinine (2), the mixture of 1, janetine (3) and 3,14-dihydro-olivacine (4), and macusine B (5). The BuOH fraction (an alkaloid-rich fraction) also provided compounds 1 and 3, and Nb-demethyl-18-formyl-affinine (6). Compounds 3, 4, and 6 are being reported for the first time in the species, with compound 6 being new to the existing literature. As future perspectives, all compounds should be assayed to evaluate their antiproliferative potential against HeLa cervical cancer cells, with promising results being expected, since the fractions showed antiproliferative activity against this cell line. In addition, it will be possible to verify the hypothesis that the activity is related to the presence of compound 1 in both fractions, since this substance has known anticancer activity.31
SUPPLEMENTARY MATERIAL Complementary material for this work (NMR spectra and HRESIMS/MS of all compounds) is available at http://quimicanova.sbq.org.br/, as a PDF file, with free access
ACKNOWLEDGMENTS The authors are grateful to Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), and Coordenação de Aperfeiçoamento de Pessoal de Nivel Superior (CAPES) - finance code 001.
REFERENCES 1. World Health Organization, Cancer Today, https://gco.iarc.fr/en, accessed in April 2025. 2. Instituto Nacional do Câncer (INCA), Câncer do Colo do Útero, https://www.gov.br/inca/pt-br/assuntos/cancer/tipos/colo-do-utero, accessed in April 2025. 3. Yang, S. T.; Wang, P. H.; Liu, H. H.; Chang, W. H.; Chou, F. W.; Lee, W. L.; Taiwanese Journal of Obstetrics and Gynecology 2024, 63, 320. [Crossref] 4. Yang, S. T.; Wang, P. H.; Liu, H. H.; Chang, C. W.; Chang, W. H.; Lee, W. L.; Taiwanese Journal of Obstetrics and Gynecology 2024, 63, 637. [Crossref] 5. Cohen, P. A.; Jhingran, A.; Oaknin, A.; Denny, L.; The Lancet 2019, 393, 169. [Crossref] 6. Meschini, S.; Marra, M.; Calcabrini, A.; Federici, E.; Galeffi, C.; Arancia, G.; Int. J. Oncol. 2003, 23, 1505. [Crossref] 7. Condello, M.; Pellegrini, E.; Multari, G.; Gallo, F. R.; Meschini, S.; Toxicol. in Vitro 2020, 65, 104819. [Crossref] 8. Yuan, R.; Hou, Y.; Sun, W.; Yu, J.; Liu, X.; Niu, Y.; Lu, J. J.; Chen, X.; Ann. N. Y. Acad. Sci. 2017, 1401, 19. [Crossref] 9. Thawabteh, A.; Juma, S.; Bader, M.; Karaman, D.; Scrano, L.; Bufo, S.; Karaman, R.; Toxins 2019, 11, 656. [Crossref] 10. Newman, D. J.; Cragg, G. M.; J. Nat. Prod. 2020, 83, 770. [Crossref] 11. Simões, A. O.; Endress, M. E.; Conti, E.; Taxon 2010, 59, 772. [Crossref] 12. Athipornchai, A.; Asian J. Pharm. Clin. Res. 2018, 11, 45. [Crossref] 13. O'Connor, S. E.; Maresh, J. J.; Nat. Prod. Rep. 2006, 23, 532. [Crossref] 14. Nogueira, T. S. R.; Vieira, M. G. C.; Robaina, R. R. S.; Braz-Filho, R.; Gontijo, D. C.; de Oliveira, A. B.; Vieira, I. J. C.; J. Ethnopharmacol. 2024, 328, 117921. [Crossref] 15. Koch, I.; Rapini, A.; Simões, A. O.; Kinoshita, L. S.; Spina, A. P.; Castello, A. C. D.; Tabernaemontanain Lista de Espécies da Flora do Brasil, 2015. [Link] accessed in April 2025 16. Pavão, S.; Lopes, I. G.; Vilharva, K. N.; Peralta, A.; da Silva Pedro, M.; Benites, E.; Gisloti, L. J.; Espaço Ameríndio 2021, 15, 160. [Crossref] 17. Monnerat, C. S.; de Souza, J. J.; Mathias, L.; Braz-Filho, R.; Vieira, I. J. C.; J. Braz. Chem. Soc. 2005, 16, 1331. [Crossref] 18. de Souza, J. J.; Mathias, L.; Braz-Filho, R.; Vieira, I. J. C.; Helv. Chim. Acta 2010, 93, 422. [Crossref] 19. Indrayanto, G.; Putra, G. S.; Suhud, F. In Profiles of Drug Substances, Excipients and Related Methodology; Al-Majed, A. A., ed.; Academic Press: New York, 2021, p. 273. [Crossref] 20. Boik, J. In Natural Compounds in Cancer Therapy; Farnell, S., ed.; Oregon Medical Press LLC: Princeton, 2001. 21. Gonçalves, B. M. F.; Duarte, N.; Ramalhete, C.; Barbosa, F.; Madureira, A. M.; Ferreira, M. J. U.; Phytochem. Rev. 2024. [Crossref] 22. Santos, A. K. L.; Machado, L. L.; Bizerra, A. M. C.; Monte, F. J. Q.; Santiago, G. M. P.; Braz-Filho, R.; Lemos, T. L. G.; Nat. Prod. Commun. 2012, 7, 1. [Crossref] 23. Braga, R. M.; Leitáo Filho, H. F.; Reist, F. A. M.; Phytochemistry 1984, 23, 175. [Crossref] 24. Azoug, M.; Phytochemistry 1995, 39, 1223. [Crossref] 25. Coune, C. A.; Angenot, L. J. G.; Denoël, J.; Phytochemistry 1980, 19, 2009. [Crossref] 26. Kleinpeter, E.; Seidl, P. R.; J. Phys. Org. Chem. 2004, 17, 680. [Crossref] 27. Danieli, B.; Palmisano, G. In The Alkaloids: Chemistry and Pharmacology, vol. 27; Brossi, A., ed.; Academic Press: New York, 1986, p. 1-130. [Crossref] 28. Araujo, A. R.; Kascheres, C.; Fujiwara, F.; Marsaioli, A. J.; Phytochemistry 1984, 23, 2359. [Crossref] 29. Arambewela, L. S. R.; Khuong-Huu, F.; Phytochemistry 1981, 20, 349. [Crossref] 30. Lavaud, C.; Massiot, G.; Vercauteren, J.; Le Men-olivier, L.; Phytochemistry 1982, 21, 445. [Crossref] 31. Tylińska, B.; Wiatrak, B.; Biology 2021, 10, 564. [Crossref]
Guest Editor handled this article: Antonio E. M. Crotti |

On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access