
 |
 |
 |
 |
 |
Artigo
| 1-Indanone derivatives: a novel one-pot approach to imides via Beckmann-like rearrangement and theoretical insights into their interactions with AChE enzyme |
|
Thiago R. F. BastosI; Telma Leda G. de LemosI; Raimundo Braz FilhoII,III; Aluísio Marques da FonsecaIV I. Departamento de Química Orgânica e Inorgânica, Universidade Federal do Ceará, Campus do Pici, 60021-940 Fortaleza - CE, Brasil Received: 02/12/2025 *e-mail: fmonte@dqoi.ufc.br The reaction of (E)-2,3-dihydro-1H-inden-1-one oxime (3) with carboxylic acid anhydrides and fatty acids aiming at the formation of the respective esters unexpectedly produced imides [5a-5c and 5d-5k] (81-95% yields). This work proposes the formation of these imides from the oxime (3) through a mechanism using an oxaziridine as an intermediate, followed by a Beckmann-like rearrangement. However, in the presence of nucleophilic catalysts [dimethylaminopyridine (DMAP) and dicyclohexylcarbodiimide (DCC)] instead of electrophilic ones. Products were characterized by spectral analysis [NMR (1H / 13C) and MS (EIMS / HRMS)]. In the in silico study, the competition of some oxime derivatives was remarkable compared to galantamine (GNT) and its bioactive interactions with the main amino acid residues that are part of the site of interest of cholinesterase activity. The calculation of the physicochemical properties through molecular modeling also allowed us to understand the interaction between a ligand and its receptor, indicating the derivatives 5i, 5g, and 5h as promising acetylcholinesterase inhibitors. INTRODUCTION Indane or 2,3-dihydro-1H-indene (1) is a mixed hydrocarbon from the petroleum industry with appropriate basic structure for obtaining derivatives. Rigidity and planarity are important characteristics of the indane skeleton,1,2 present in several molecules with biological activity, such as indinavir®, an HIV2,3 (human immunodeficiency virus) protease inhibitor.3,4 This hydrocarbon can be modified in several ways, including the insertion of hydroxyl, carbonyl and carbon-carbon double bond functional groups into the pentacyclic ring. Among some of the possible derivatives (Figure 1), 1-indanone (2) is used as a base intermediate in the synthesis of several therapeutic agents such as eranz® or donepezil hydrochloride, an inhibitor of the enzyme acetylcholinesterase (AChE), used in the treatment of Alzheimer's disease.5,6
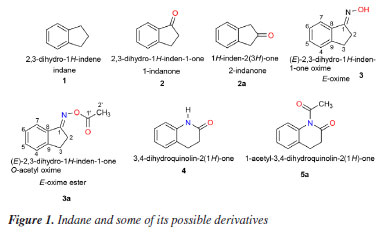
There are reports7 of acetylcholinesterase inhibition with the use of amides and imides, where some new anilide and imide derivatives of 4-aminopyridine (4AP) have been synthesized and evaluated for anamnesis, cognition-enhancing, and anticholinesterase activity by means of their respective in vitro and in vivo models. We are interested in the preparation of esters, such as 3a (Figure 1), from the 1-indanone oxime (3) in view of their possible biological activities. Oximes esters can be used as agrochemical and medicinal agents8-11 and as precursors for the synthesis of bioactive heterocyclic compounds.12 Thus, the cyclic ketone 1-indanone (2) allowed the easy preparation of its oxime which, in turn, would lead to obtaining the desired esters. The oximes allow the production of amides that are important building blocks in organic synthesis, since they are widely used in the production of plastics, rubbers, papers, colored paints and water and sewage treatment.13-15 In addition, amides play an important role as feedstock for the production of analgesic-antipyretic drugs.16,17 The Beckmann rearrangement (BR) is a powerful method in organic synthesis for the preparation of amides or lactams from ketones and is often employed in chemical industry especially for the preparation of e-caprolactam. The conventional BR of ketoximes occurs in the presence of strong Bronsted or Lewis acids such as concentrated sulfuric acid, PCl5 in diethyl ether, hydrogen chloride in acetic anhydride, resulting in large amount of by-products causing environmental hazards and serious corrosion problems.18-20 The BR of oximes is more an important case where the constitution of a reaction product is used to assign configuration to its precursor. In this rearrangement the migrating group retains its configuration. However, even when the migrating group is achiral, there is an important stereochemical aspect to the rearrangement: the group that migrates is the trans group to the OH moiety of the oxime. This fact allows one to infer the stereochemistry of the oxime from the nature of the amide formed from it.21 In this paper, we report efficient syntheses for cyclic imides from an oxime (3) involving Beckmann-type rearrangement in one-pot reactions. The use of bioinformatics in developing new drugs has significantly reduced costs and the time required. Understanding the mechanisms of molecular interaction between the receptor and the ligand and rationalizing how a potential drug can act in this process is one of the most significant challenges of molecular biology and a crucial aspect for success in planning new medicines within the rational drug design approach.22 In this context, research with oximes and their derivatives, such as imides, presents itself as a promising tool in the search for new therapeutic agents through computer simulations. It also enables the investigation of the mechanism of anticholinesterase action.
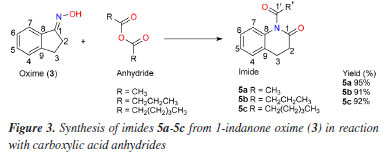
EXPERIMENTAL General experimental procedures The reagents and solvents used were purchased from Sigma-Aldrich® (Saint Louis, USA), Vetec® (Caxias do Sul, Brazil), Synth® (Diadema, Brazil) and Fluka® (Munich, Germany). Low resolution mass spectra (EIMS) were obtained on a Shimadzu GC-2010 Plus instrument coupled to a GCMS-QP2010SE mass spectrometer using capillary column (30 m × 0.25 µm) at flow rate of 1.46 mL min-1 using helium as the carrier gas; high resolution mass spectra (HRMS) were recorded on a Bruker micrOTOF II instrument - method NaFormate_ESI_pos_100-900_Lab-Mass.m. The 1H and 13C NMR (nuclear magnetic resonance) spectra were recorded on a Bruker DPX-300 spectrometer, 300 and 75.5 MHz, respectively, using CDCl3 as solvent; chemical shifts reported in ppm (d scale) with residual CHCl3 (d 7.27) as internal standard for 1H NMR and the central peak of the triplet (d 77.23) of CDCl3 for 13C NMR; spin-spin coupling constant (J) in hertz (Hz). Columns chromatography were performed using silica gel 60 (70-230 mesh, Vetec®) eluted with hexane, hexane:CH2Cl2 9:1, hexane:CH2Cl2 8:2 and CH2Cl2. The chemical reactions were monitored by analytical thin layer chromatography (TLC), performed on Merck silica gel 60 F254 plates, visualized by ultraviolet (UV) irradiation (254 nm) and by spraying with vanilin/perchloric acid/EtOH solution followed by heating. Preparation of (E)-2,3-dihydro-1H-inden-1-one oxime (3) Ketoxime (3) was synthesized from the 1-indanone, following the procedure described in literature.20 In typical preparation, a flask equipment equipped with reflux condenser was charged with 3.9 mL of EtOH, 2.8 mmol (194.46 mg) of hydroxylamine hydrochloride, 4.0 mmol (328.12 mg) of sodium acetate and 1.0 mmol (132.16 mg) of 2-indanone. The reaction system was heated at 70 ºC for 24 h and, after cooling, the white precipitate was collected by filtration, washed with water and dried to give oxime (3) (93% yield). General procedure of imides using acid anhydrides Preparation of imides 5a-5c: reactions of 3 with acid anhydrides The imides 5a-5c (Figure 3) were obtained by the reaction of the corresponding acid anhydrides a-c with the oxime (3). Thus, to a solution of 3 (100 mg, 0.68 mmol) and dimethylaminopyridine (DMAP, 23 mg, 0.2 mmol) in CH2Cl2 (8 mL) were added, in individual experiments, the acid anhydrides a, b and c (2 mmol of each), and the systems were stirred at room temperature for 2 h. Subsequently, the reaction mixtures were extracted with a 5% NaHCO3 solution (3 × 10 mL). The organic phases (dried with Na2SO4) were then evaporated in vacuo and the respective products subjected to silica gel column chromatography using CH2Cl2 as eluent to afford 5a (121 mg, 95%), 5b (133 mg, 91%) and 5c (152 mg, 92%).
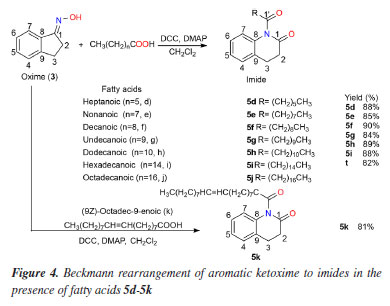
General procedure of imides using fatty acid Preparation of imides 5d-5k: reactions with fatty acids The imides 5d-5k, (Figure 4) were obtained by the reaction of the corresponding fatty acids d-k with the oxime (3). Thus, to a solution of 3 (0.68 mmol, 100 mg), dicyclohexylcarbodiimide (DCC, 268 mg, 1.3 mmol) and DMAP (1.3 mmol, 158 mg) in CH2Cl2 (3 mL) were added, in individual experiments, the fatty acids d (184 μL, 1.3 mmol), e (226 μL, 1.3 mmol), f (224 mg, 1.3 mmol), g (242 mg, 1.3 mmol), h (260 mg, 1.3 mmol), i (333 mg, 1.3 mmol), j (370 mg, 1.3 mmol) and k (410 μL, 1.3 mmol), and the systems were stirred at room temperature for 2 h. Subsequently, CH2Cl2 (10 mL) was added to the mixture, it was filtered and extracted with a 5% NaHCO3 solution (3 × 20 mL). The organic phases (dried with Na2SO4) were then evaporated and the respective products subjected to silica gel chromatographic columns using mixtures of hexane + EtOAc in increasing polarity. The pure products (5d-5k) were obtained in average yields of about 81 to 90%: 5d (154 mg, 88%), 5e (165 mg, 85%), 5f (183 mg, 90%), 5g (179 mg, 84%), 5h (198 mg, 89%), 5i (229 mg, 88%), 5j (229 mg, 82%) and 5k (225 mg, 81%).
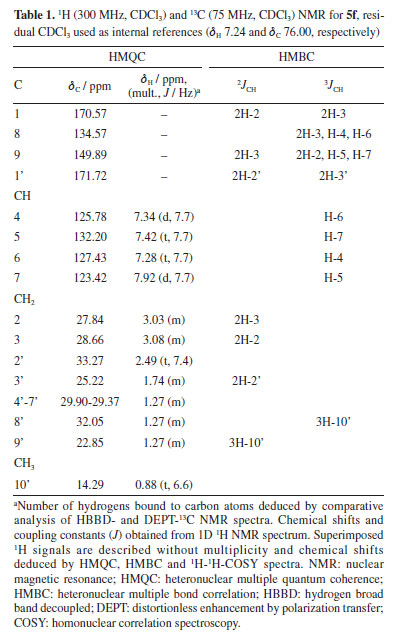
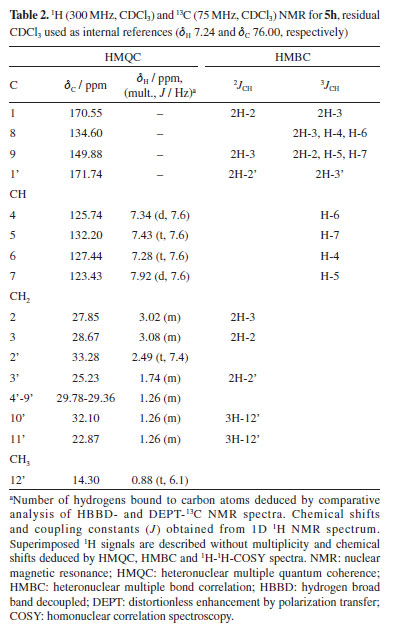
(E)-2,3-Dihydroinden-1-one oxime (3) White solid; mp 147-150 ºC; 1H NMR (300 MHz, CDCl3) d 7.65 (1H, d, J 7.6 Hz, H-7), 7.28 (2H, m, H-4, H-6), 7.19 (1H, m, H-5), 3.09 (2H, m, 2H-2), 2.93 (2H, m, 2H-3); 13C NMR (300 MHz, CDCl3) d 164.26 (C-1), 148.65 (C-8), 136.15 (C-9), 130.61 (CH-6), 127.21 (CH-4), 125.82 (CH-5), 121.82 (CH-7), 28.73 (CH2-2), 26.19 (CH2-3). 1-Acetyl-3,4-dihydroquinolin-2(1H)-one (5a) Yellow solid; mp 200-202 ºC; 1H NMR (300 MHz, CDCl3) d 7.90 (1H, d, J 7.6 Hz, H-7), 7.43 (1H, t, J 7.2 Hz, H-5), 7.34 (1H, br d, H-4), 7.30 (1H, tl, H-6), 3.04 (2H, m, 2H-3), 3.00 (2H, m, 2H-2), 2.23 (3H, s, 3H-2'); 13C NMR (300 MHz, CDCl3) d 170.50 (C-1), 169.28 (C-1'), 149.95 (C-9), 134.49 (C-8), 132.25 (CH-5), 127.44 (CH-6), 125.82 (CH-4), 123.36 (CH-7), 28.67 (CH2-3), 27.84 (CH2-2), 19.90 (CH2-2'); EIMS m/z (rel. int.) [M]+ 189 (48), 147 (100), 119 (30), 91 (22), 43 (37); HRMS m/z [M]+ 189.0789 (calcd. for C11H11NO2, 189.2118), [M + Na]+ 212.0681, [C9H10NO]+ 148.0759, [C9H8N]+ 130.0657. 1-Butyryl-3,4-dihydroquinolin-2(1H)-one (5b) Yellow liquid; 1H NMR (300 MHz, CDCl3) d 7.92 (1H, d, J 7.7 Hz, H-7), 7.43 (1H, t, J 7.6 Hz, H-5), 7.35 (1H, br d, H-4), 7.30 (1H, tl, H-6), 3.08 (2H, m, 2H-3), 3.03 (2H, m, 2H-2), 2.48 (2H, t, J 7.4 Hz, 2H-2'), 1.77 (2H, m, 2H-3'), 1.03 (3H, t, J 6.5 Hz, 3H-4'); 13C NMR (300 MHz, CDCl3) d 171.59 (C-1'), 170.62 (C-1), 149.91 (C-9), 134.56 (C-8), 132.22 (CH-5), 127.45 (CH-6), 125.80 (CH-4), 123.44 (CH-7), 35.13 (CH2-2'), 28.67 (CH2-3), 27.86 (CH2-2), 18.72 (CH2-3'), 13.94 (CH3-4'); EIMS m/z (rel. int.) [M]+ 217 (11), 147 (100), 130 (40), 103 (16), 71 (40), 43 (44); HRMS m/z [M]+ 217.1102 (calcd. for C13H15NO2, 217.2995), [2M + Na]+ 457.2104, [M + Na]+ 240.0988, [C9H10NO]+ 148.0756, [C9H8N]+ 130.0655. 1-Hexanoyl-3,4-dihydroquinolin-2(1H)-one(5c) Yellow liquid; 1H NMR (300 MHz, CDCl3) d 7.92 (1H, d, J 7.7 Hz, H-7), 7.43 (1H, t, J 7.3 Hz, H-5), 7.34 (1H, br d, H-4), 7.30 (1H, tl, H-6), 3.08 (2H, m, 2H-3), 3.02 (2H, m, 2H-2), 2.49 (2H, t, J 7.5 Hz, 2H-2'), 1.74 (2H, m, 2H-3'), 1.37 (4H, m, 4',5'), 0.91 (3H, t, J 6.7 Hz, 3H-6'); 13C NMR (300 MHz, CDCl3) d 171.79 (C-1'), 170.60 (C-1), 149.90 (C-9), 134.57 (C-8), 132.22 (CH-5), 127.45 (CH-6), 125.79 (CH-4), 123.45 (CH-7), 33.23 (CH2-2'), 31.51 (CH2-4'), 28.68 (CH2-3), 27.85 (CH2-2), 24.90 (CH2-3'), 22.51 (CH2-5'), 14.14 (CH3-6'); EIMS m/z (rel. int.) [M]+ 245 (14), 147 (85), 132 (100), 99 (30), 71 (14), 43 (43); HRMS m/z [M]+ 245.1415 (calcd. for C15H19NO2, 245.3261), [C9H10NO]+ 240.0549, [C9H10NO]+ 148.0761, [C9H8N]+ 130.0659. 1-Heptanoyl-3,4-dihydroquinolin-2(1H)-one(5d) Yellow liquid; 1H NMR (300 MHz, CDCl3) d 7.92 (1H, d, J 7.7 Hz, H-7), 7.43 (1H, t, J 7.3 Hz, H-5), 7.34 (1H, br d, H-4), 7.28 (1H, tl, H-6), 3.08 (2H, m, 2H-3), 3.02 (2H, m, 2H-2), 2.49 (2H, t, J 7.4 Hz, 2H-2'), 1.72 (2H, m, 2H-3'), 1.21-1.42 (6H, m, 4'-6'), 0.88 (3H, t, J 6.4 Hz, 3H-7'); 13C NMR (300 MHz, CDCl3) d 171.74 (C-1'), 170.58 (C-1), 149.89 (C-9), 134.59 (C-8), 132.21 (CH-5), 127.44 (CH-6), 125.78 (CH-4), 123.45 (CH-7), 33.26 (CH2-2'), 31.64 (CH2-5'), 29.04 (CH2-4'), 28.67 (CH2-3), 27.84 (CH2-2), 25.17 (CH2-3'), 22.68 (CH2-6'), 14.21 (CH3-7'); EIMS m/z (rel. int.) [M]+ 259 (1), 147 (100), 130 (54), 113 (49), 85 (25), 43 (62.5); HRMS m/z [M]+ 259.1572 (calcd. for C16H21NO2, 259.3506), [C11H9NNaO2]+ 210.1269, [C9H10NO]+ 148.0754, [C9H8N]+ 130.0653. 1-Nonanoyl-3,4-dihydroquinolin-2(1H)-one(5e) Yellow liquid; 1H NMR (300 MHz) d 7.91 (1H, d, J 7.7 Hz, H-7), 7.42 (1H, t, J 7.1 Hz, H-5), 7.34 (1H, br d, H-4), 7.28 (1H, tl, H-6), 3.07 (2H, m, 2H-3), 3.02 (2H, m, 2H-2), 2.48 (2H, t, J 7.5 Hz, 2H-2'), 1.73 (2H, m, 2H-3'), 1.28 (10H, m, 4'-8'), 0.88 (3H, tl, J 7.5 Hz, 3H-9'); 13C NMR (300 MHz, CDCl3) d 171.73 (C-1'), 170.54 (C-1), 149.87 (C-9), 134.58 (C-8), 132.19 (CH-5), 127.4 (CH-6), 125.78 (CH-4) 123.41 (CH-7), 33.25 (CH2-2'), 31.98 (CH2-7'), 29.40-29.30 (CH2-4'-CH2-6'), 28.66 (CH-3), 27.83 (CH2-2), 25.21 (CH2-3'), 22.81 (CH2-8'), 14.26 (C-9'); EIMS m/z (rel. int.) [M]+ 287 (0.1), 147 (100), 130 (70), 115 (30), 103 (74), 89 (23), 77 (60), 63 (29), 51 (43), 39 (25); HRMS m/z [M]+ 287.1885 (calcd. for C18H25NO2, 287.4047), [C9H10NO]+ 148.0760, [C9H8N]+ 130.0658. 1-Decanoyl-3,4-dihydroquinolin-2(1H)-one(5f) Brown liquid; 1H NMR (300 MHz, CDCl3) d 7.92 (1H, d, J 7.7 Hz, H-7), 7.42 (1H, t, 7.4Hz, H-5), 7.34 (1H, br d, H-4), 7.29 (1H, br t, H-6), 3.07 (2H, m, 2H-3), 3.02 (2H, m, 2H-2), 2.48 (2H, t, J 7.5 Hz, 2H-2'), 1.73 (2H, m, 2H-3'), 1.28 (12H, m, 4'-9'), 0.88 (3H, t, J 6.9 Hz, 3H-10'); 13C NMR (300 MHz, CDCl3) d 171.79 (C-1'), 170.59 (C-1), 149.90 (C-9), 134.57 (C-8), 132.21 (C-5), 127.45 (C-6), 125.79 (C-4), 123.45 (C-7), 33.27 (C-2'), 32.06 (C-8'), 29.90-29.37 (C-4'-C-7'), 28.67 (C-3), 27.85 (C-2), 25.22 (C-3'), 22.86 (C-9'), 14.30 (C-10'); EIMS m/z (rel. int.) [M]+ 301 (2), 147 (100), 130 (52), 103 (17), 85 (13), 71 (30), 57 (26), 43 (30); HRMS m/z [M]+ 301.2041 (calcd. for C19H27NO2, 301.4317), [C11H9NNaO2]+ 210.1272, [C9H10NO]+ 148.0756, [C9H8N]+ 130.0653. 1-Undecanoyl-3,4-dihydroquinolin-2(1H)-one(5g) Brown liquid; 1H NMR (300 MHz, CDCl3) d 7.92 (1H, d, J 7.7 Hz, H-7), 7.43 (1H, t, J 7.5 Hz, H-5), 7.34 (1H, br d, H-4), 7.28 (1H, br t, H-6), 3.08 (2H, m, 2H-3), 3.02 (2H, m, 2H-2), 2.49 (2H, t, J 7.5 Hz, 2H-2'), 1.74 (2H, m, 2H-3'), 1.27 (14H, m, 4'-10'), 0.88 (3H, t, J 6.2 Hz, 3H-11'); 13C NMR (300 MHz, CDCl3) d 171.75 (C-1'), 170.56 (C-1), 149.88 (C-9), 134.60 (C-8), 132.20 (CH-5), 127.44 (CH-6), 125.49 (CH-4), 123.44 (CH-7), 33.28 (CH2-2'), 32.08 (CH2-9'), 29.72-29.37 (CH2-4'-CH2-8'), 28.68 (C-3), 27.85 (C-2), 25.23 (CH2-3'), 22.87 (CH2-10'), 14.29 (CH3-11'); EIMS m/z (rel. int.) [M]+ 315 (0.1), 169 (16), 147 (100), 130 (35), 103 (15), 85 (16.5), 57 (29), 43 (25); HRMS m/z [M]+ 315.2198 (calcd. for C20H29NO2, 315.4587), [C9H10NO]+ 148.0759, [C9H8N]+ 130.0656. 1-Dodecanoyl-3,4-dihydroquinolin-2(1H)-one(5h) Brown liquid; 1H NMR (300 MHz, CDCl3) and 13C NMR (300 MHz, CDCl3) see Table 2; EIMS m/z (rel. int.) C21H31NO2, [M]+ 329 (0.1%), [C12H23O]+ 183 (10.5), [C9H10NO]+ 148 (19), [C9H9NO]+ 147 (100), [C9H8N]+ 130 (45.7), [C7H5N]+ 103 (23), [C6H13]+ 85 (17), [C5H11]+ 71 (22), [C4H9]+ 57 (44), [C3H7]+ 43 (40). 1-Palmitoyl-3,4-dihydroquinolin-2(1H)-one (5i) White solid; mp 301-303 ºC; 1H NMR (300 MHz, CDCl3) d 7.91 (1H, d, J 7.7 Hz, H-7), 7.42 (1H, t, J 7.7 Hz, H-5), 7.34 (br d, H-4), 7.29 (1H, br t, H-6), 3.08 (2H, m, 2H-3), 3.02 (2H, m, 2H-2), 2.48 (2H, t, J 7.4 Hz, 2H-2'), 1.73 (2H, m, 2H-3'), 1.24 (24H, m, 4'-15'), 0.87 (3H, t, J 6.9 Hz, 3H-16'); 13C NMR (300 MHz, CDCl3) d 171.74 (C-1'), 170.55 (C-1), 149.88 (C-9), 134.61 (C-8), 132.20 (CH-5), 127.44 (CH-6), 125.79 (CH-4), 123.44 (CH-7), 33.28 (CH2-2'), 32.12 (CH2-14'), 29.89-29.38 (CH2-4'-CH2-13'), 28.68 (CH2-3), 27.85 (CH2-2), 25.24 (CH2-3'), 22.89 (CH2-15'), 14.31 (CH3-16'); EIMS m/z (rel. int.) C25H39NO2, [M]+ 385 (no peak), 147 (100), 130 (42), 115 (17), 103 (51), 91 (13), 77 (24), 51 (15); HRMS m/z [M]+ 385.3056 (calcd. for C19H27NO2, 385.5939), [C10H17O]+ 153.0080, [M + 1]+ 386.3053, [M + Na]+ 408.2873, [M + K]+ 424.2620, [2M + Na]+ 793.5853. 1-Stearoyl-3,4-dihydroquinolin-2(1H)-one(5j) White solid; mp 332-334 ºC; 1H NMR (300 MHz, CDCl3) d 7.92 (1H, d, d, J 7.7 Hz, H-7), 7.42 (1H, t, J 7.3 Hz, H-5), 7.34 (1H, br d, H-4), 7.30 (1H, br t, H-6), 3.07 (2H, m, 2H-3), 3.02 (2H- m, 2H-2), 2.48 (2H, t, J 7.4 Hz, 2H-2'), 1.73 (2H, m, 2H-3'), 1.25 (28H, m, 4'-17'), 0.87 (3H, t, J 6.8 Hz, 3H-18'); 13C NMR (300 MHz, CDCl3) d 171.73 (C-1'), 170.53 (C-1), 149.87 (C-9), 134.58 (C-8), 132.19 (CH-5), 127.43 (CH-6), 125.78 (CH-4), 123.42 (CH-7), 33.26 (CH2-2'), 32.11 (CH2-16'), 29.88-29.37 (CH2-4'-CH2-15'), 28.66 (CH2-3), 27.83 (CH2-2), 25.22 (CH-3'), 22.87 (CH2-17'), 14.30 (CH3-18'); EIMS m/z (rel. int.) C27H43NO2, [M]+ 413 (no peak), 147 (100), 130 (32), 115 (15.5), 103 (35), 91 (12), 77 (20), 51 (13); HRMS m/z [M]+ 413.3293 (calcd. for C27H43NO2, 413.6479), [C10H17O]+ 153.0075, [M + 1]+ 414.3358, [M + Na]+ 436.3177, [2M + Na]+ 849.6468. 1-Oleoyl-3,4-dihydroquinolin-2(1H)-one (5k) Brown liquid; 1H NMR (300 MHz, CDCl3) d 7.92 (1H, d, J 7.7 Hz, H-7), 7.42 (1H, t, J 7.0 Hz, H-6), 7.34 (1H, br d, H-4), 7.30 (1H, br t, H-5), 5.34 (2H, t, J 5.4 Hz, H-9, H-10), 3.07 (2H, m, 2H-3), 3.02 (2H, m, 2H-2), 2.48 (2H, t, J 7.6 Hz, 2H-2'), 2.02 (4H, m, 2H-8', 2H-11'), 1.73 (2H, m, 2H-3'), 1.32-1.26 (20H, m, 8'-17'), 0.87 (3H, t, J 6.8 Hz, 3H-18'); 13C NMR (300 MHz, CDCl3) d 171.71 (C-1'), 170.54 (C-1), 149.86 (C-9), 134.56 (C-8), 132.19 (CH-5), 130.19 (=CH-9'), 129.91 (=CH-10'), 127.43 (CH-6), 125.77 (CH-4), 123.42 (CH-7), 33.23 (CH2-2'), 32.08 (CH2-16'), 29.95-29.28 (CH2-4'-CH2-7' / CH-12'-CH-15'), 28.66 (CH2-3), 27.83 (CH2-2), 27.40 (C-8'),a 27.35 (C-11')a, 25.18 (CH2-3'), 22.86 (CH2-17'), 14.87 (CH3-18'); EIMS m/z (rel. int.) C27H41NO2, [M]+ 411 (no peak), 147 (100), 130 (32), 115 (15.5), 103 (35), 91 (12), 77 (20), 51 (13); HRMS m/z [M]+ 411.3137 (calcd. for C27H41NO2, 411.6320), [C9H8N]+ 130.0654, [C9H10NO]+ 148.0752, [C27H38NO2)]+ 408.2897. aInterchangeable assignments. Preparation of binders The molecules 1, 2, 3, 3a, 4, and 5a-5k were used for the simulation and had their 3D structures created by Chem3D software.23 The design and structural optimization of each compound was performed using the software Avogadro®,24 configured to use the Merk molecular force field (MMFF94),25 with cycles of 500 interactions and the steepest algorithm, having the convergence limit of 10e-7 and later converted to the pdbqt format. Preparation of proteins The X-ray crystal structures of protein of AChE (PDB ID: 4EY6) was obtained from the Protein Data Bank repository files, named as the crystalline structure of recombinant human acetylcholinesterase on galantamine complex, classified as hydrolase inhibitor, expression system, and Homo sapiens organism.26 In order for the coupling to occur without interfering with other chemical compounds, the residues present in the PDB file of the AChE that are part of the protein structure, such as ions, molecular water, and the native binder, were removed, loads, and lost atoms. Polar hydrogens were added through the AutoDock suite, version 4.2, and the AutoDock Tools auxiliary tool, version 1.5.4.27 Molecular docking Molecular docking was performed by using the AutoGrid-AutoDock Vina28 on MGL Tools platform, version 1.5.7.27 The default parameters of the automatic settings were used to set the genetic algorithm parameters. For docking, the active site of AChE (4EY6) with a radius of 6 Å (binding area around inhibitor) was used. The input ligands with full protonation were used in .mol2 format. The cholinergic hypothesis29 was used for the study on Alzheimer's disease, the enzyme acetylcholinesterase. For the docking of the main protease AChE (4EY6), the following parameters were used: number of grid points in xyz (40 40 40), spacing (0.642), grid center in xyz (-13.50 -43.04 27.64). Other parameters were set to default. To validate the simulations, the redocking technique was performed on the co-crystallized ligand, galantamine®. The results of predicting protein-ligand interactions by docking studies were visualized using Pymol.30 The 2D-schematic representations of the protein-ligand interactions were generated using Discovery Studio.31 To obtain a larger data set, for all simulations (docking and redocking), 50 independent simulations were performed, being possible to obtain 20 poses per simulation. The exhaustiveness criterion equal to 64 was used to improve the partial refinement of the individual coupling calculations.32 The protein structure was kept rigid, while all binding and twists of the ligands were adjusted to rotate.33 Docking validation For statistical validation of the simulations, redocking procedures were performed and the RMSD (root mean square deviation) values were evaluated, having as parameter of choice of the best pose, values less than 2 Å.34 To analyze the stability of the complex (proteins/ligand) the affinity energy was used as parameter, which has ideality parameter values below -6.0 kcal mol-1.35 To assess the strength of the H-bond, the values of the distances between the donor and recipient atoms were used, being classified as strong, the interactions that were between 2.5 and 3.1 Å; as average those that were between 3.1 and 3.55 Å, and weak those with a distance greater than 3.55 Å.36 To validate the simulations, the redocking technique was performed on the co-crystallized ligand galantamine®, used in antiacetylcholinergic test.37 In addition to reference ligand, the analogues compounds were subjected to the same criteria and simulation conditions.
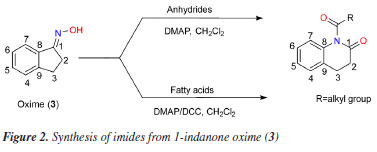
RESULTS AND DISCUSSION Synthesis and characterization of the compounds Prior to the preparation of desired esters (3a, for example; Figures 1 and 2), our attention was focused on the obtention of 2,3-dihydro-1H-indene-1-one oxime (3), easily prepared from 1-indanone (2) by reaction with hydroxylamine hydrochloride in the presence of sodium acetate. Thus, following the literature procedure,38-40 the transformation of 2 into 3 was performed at excellent yield (93%) suggesting an easy oximation stage. Then, to obtain esters, 3 was treated with carboxylic acid anhydrides in the presence of DMAP as a catalyst and then, with aliphatic carboxylic acids in the presence of DMAP with DCC as a coupling agent, according to the Steglich esterification method. Initially, the reaction with the oxime (3) (Figure 2) was investigated using acetic anhydride as an acyl group donor reagent, hoping to obtain the corresponding oxime ester 3a. The reaction, according to the literature procedure,41 occurred in the presence of DMAP as catalyst and CH2Cl2 as a solvent. Analysis of the 1H NMR spectrum showed that the product of this reaction had an acetyl group, indicated by the signal at δH 2.23 (s, 3H), which is in agreement with the 13C NMR spectrum through the two signals at δC 170.50 (C=O) and 19.90 (CH3). The fragments peaks at m/z 147 [M - C2H2O]+ (100%) and m/z 43 [CH3CO]+ (30%) in the EIMS, are also in agreement with the presence of the acetyl group. The 1H NMR spectrum also displayed signals to one isolated four-proton (-CH2-CH2-) spin fragment {d 3.00 (m, 2H); 3.04 (m, 2H)} and for four aromatic protons d 7.30 (tl, 1H), 7.34 (dl, 1H), 7.43 (t, J 7.2 Hz, 1H) and 7.90 (d, J 7.6 Hz, 1H). The 13C NMR spectrum, in turn, revealed two signals due to methylene carbons (d 28.67 and 27.84) and six signals for aromatic carbons d 149.95 and 134.49 (C-2), 132.25, 127.44, 125.82 and 123.36 (CH-4), all confirmed by distortionless enhancement by polarization transfer (DEPT) experiment. Thus, all these data would be in agreement with the formation of the desired ester, that is, 3a. However, the 13C NMR spectrum of the product, in addition to the signal at δC 170.50, recorded another well-resolved signal at δC 169.28, that is, consistent with the presence of another carbonyl group. In addition, in this spectrum, the absence of the signal around δC 164.00—corresponding to the oxime carbon (C-1) in the benz-fused cyclopentanone oxime observed in the 13C NMR spectrum of compound 3 (or 3a)—supports the presence of the molecular ion peak at m/z 189 ([M]+, 48%) and a prominent base peak at m/z 147 (100%), which can be attributed to a McLafferty rearrangement. In turn, the high-resolution mass spectrum (HRMS) suggested the molecular formula C11H11NO2 ([M]+ m/z 189.0789) and contained ion peaks at m/z 130.0657 [C9H8N]+, 148.9759 [C9H10NO]+ and 212.0683 [C11H11NNaO2]+. From these data we conclude that the product obtained was not the ester 3a, but 5a, an imide, i.e., 1-acetyl-3,4-dihydroquinolin-2(1H)-one in 95% yield (Figure 3). The chemical shifts at δC 170.50 and 169.28 were attributed to carbons C-1 and C-1', respectively, in 5a. Heteronuclear multiple bond correlation (HMBC) experiment showed the correlation of the methyl protons at δH 2.23 (CH3-1') with carbon at δC 169.28 (C-1'). In particular, in the structure of imide 5a (as in the other examples), the system 3,4-dihydroquinolin-2-(1H)-one is inserted, admitting, at first hand, that the formation of imides (5a, for example) in the present work, required the generation of amide 4 from oxime (3) (Figure 1). In fact, there are already reports in the literature19 of the formation of the amide 3,4-dihydro-2(1H)-quinolinone (Figure 2) through the rearrangement of the 1-indanone oxime via Beckmann rearrangement. Therefore, the production of imides in this work would have occurred through a reaction between 4 and carboxylic acid anhydrides (or carboxylic acids). This is unlikely, due to important theoretical and experimental aspects: (i) the very low nucleophilic power of the nitrogen of the amide and, in the case of carboxylic acids, the low electrophilic power of the carbonyl carbon, do not justify the high yields observed in the formation of imides; (ii) normally, the Beckmann rearrangement requires the presence of strong acids such as sulfuric acid or Lewis acids (SnCl4, SOCl2, PCl5, etc.), that is, it occurs under conditions of electrophilic activation of the hydroxyl group of the oxime. Nevertheless, in this study, the reactions occurred in the presence of DMAP with carboxylic acid anhydrides and DMAP/DCC with fatty acids, that is, in both cases under the action of nucleophilic agents, therefore, in basic conditions. Thus, in this work, we are proposing a hypothesized mechanism for the reaction of 3 with carboxylic acid anhydrides in the presence of DMAP using CH2Cl2 as a solvent to form imide 5a (as well as 5b and 5c) as described in Scheme 1S, presented in the Supplementary Material. First, the DMAP nucleophilic species interacts with the anhydride to generate the acylpyridinium intermediate (6). Then, the activated carbonyl carbon of the acylpyridinium is attacked by the oxime (3) to form an oxaziridine, followed by a Beckmann-type rearrangement to give imide 5a (as well as 5b, 5c). The E stereochemistry of the oxime was inferred from the nature of the imide (5a, final product), in view of the two possible regioisomeric lactams of the oxime (3). This is also in agreement with the product 5a, obtained from oxaziridine as a reaction intermediate, as the expected stereochemistry in a Beckmann rearrangement. We extended this methodology by exploring reactions of oxime (3) with other acid anhydrides (butanoic and hexanoic anhydrides). The results shown in Figure 3 reveal that the reaction worked well, providing the corresponding imides (5b and 5c) also in excellent yields. Complete 1H and 13C NMR data for compounds 5a-5c have been in the Experimental section. The investigation was then directed to the reactions of oxime (3) with fatty acids. Thus, following the same experimental procedure,41 we first investigated the reaction of 3 with decanoic acid (5f) hoping to obtain the corresponding oxime ester (oxime decanoate). In these cases, reactions in addition to DMAP also required the presence of DCC as coupling reagent (see Scheme 2S, in Supplementary Material). The proposed mechanism for the reaction of 3 with fatty acids in the presence of DMAP/DCC is similar to that of reactions with anhydrides. Initially, the protonation of the carbodiimide group of DCC by the carboxylic acid induces the attack of the carboxylate anion, originating the reactive O-acylurea. This, in turn, is attacked by DMAP, a nucleophile stronger than the oxime, on the acyl group, originating the highly reactive intermediate acylpyridinium assisted by the expulsion of dicyclohexylurea (DCU) as an excellent leaving group. Finally, the reaction of the oxime with acylpyridinium leads to the oxaziridine which follows the same paths as in the previous cases (Scheme 1, Supplementary Material), leading to the production of imide 5f (a well as 5d, 5e, 5g-5k). Analysis of the 1H and 13C NMR data, showed that the compound obtained was also an imide, i.e., 1-decanoyl-3,4-dihydroquinolin-2-(1H-one) (5f), 90% yield. 1H-1H COSY (homonuclear correlation spectroscopy) NMR spectrum in conjunction with 1H-13C HMQC (heteronuclear multiple quantum coherence) and 1H-13C HMBC experiments confirmed the presence of one isolated four-hydrogen (-CH2-CH2-) spin system; furthermore, the HMBC plot showed correlations of the carbonyl carbons δC 170.59 (C-1) and 171.79 (C-1') with the hydrogen atoms δH 3.02 (m), 2H-2 / 3.08 (m), 2H-3 and 2.49 (t, J 7.4 Hz), 2H-2' / 1.74 (m), 2H-3', respectively. The 1H and 13C NMR spectra of 5f have been rigorously assigned (Table 1) with the aid of heteronuclear single-quantum correlation (HSQC) and HMBC NMR experiments - chemical shifts (d) and multiplicities (J, Hz) of 1H and 13C (as derived from DEPT experiment). The HRMS contained ions at m/z 301.2041 (for C19H27NO2 [M]+), 210.1272 [C11H9NNaO2]+, 148.9759 [C9H10NO]+ and 130.0657 [C9H8N]+. Fragment peaks at m/z (%) 155 (20) [C10H19O]+, 147 (100) [C9H9NO]+, 130 (55) [C9H8N]+, 103 (19) [C8H7]+, 85 (14) [C6H13]+, 71 (30) [C5H11]+ and 43 (29) [C3H7]+ in the EIMS {[M]+ 301 (1)} also indicated the presence of aromatic and aliphatic moieties. The peaks at m/z (%) 147 (100) [C9H9NO]+ and 148 (100) [C9H10NO]+ recorded in the EIMS and HRMS spectra, respectively, can be explained by McLafferty rearrangement from the molecular ion. In order to confirm the results presented in Table 1, the reaction of 3 using another fatty acid, 5h (dodecanoic acid), as substrate was carried out under the same experimental conditions. Comparison of the 1H and 13C NMR data of 5h with those of 5f showed that 5h is also an imide, in this case, 1-dodedecanoyl-3,4-dihydroquinolin-2-(1H-one) (5h), in 89% yield. 1H-1H COSY NMR spectrum in conjunction with 1H-13C HMQC and 1H-13C HMBC experiments confirmed the presence of one isolated four-hydrogen (-CH2-CH2-) spin system; furthermore, the HMBC plot showed correlations of the carbonyl carbons δC 170.57 (C-1) and 171.75 (C-1') with the hydrogen atoms δH 3.03 (m), 2H-2 / 3.08 (m), 2H-3 and δH 2.49 (t, J 7.4 Hz), 2H-2' / 1.74 (m), 2H-3', respectively. The 1H and 13C NMR spectra of 5h have been rigorously assigned (Table 2) with the aid of HSQC and HMBC NMR experiments - chemical shifts (d) and coupling constants (J, Hz) of 1H and 13C (as derived from DEPT experiment).
Hassner and Vazken42 observed that after the formation of O-acylurea, another reactive intermediate is formed - a symmetrical anhydride of the carboxylic acid [(RCO)2O]. In this mechanistic proposal (see Scheme 3S, in Supplementary Material), DMAP reacts with this anhydride to form the activated intermediate acylpyridinium which reacts with the oxime as shown in Schemes 1S and 2S. By analogy to the above cases, this methodology was then extending by exploring the oxime (3) reactions with other acid fatty acids [heptanoic (5d), nonanoic (5e), undecanoic (5g), hexadecanoic (5i), octadecanoic (5j) and, 5-en-octadecanoic (5k)]. The results shown in Figure 4 reveal that the reaction worked well by affording the corresponding imides (5d, 5e, 5g and 5i-5k) with high yields. All imides obtained have very similar structures, with differences only in the carbon chain originating from the carboxylic acid that reacted with oxime (3). Thus, in the 1H and 13C NMR spectra, the chemical shifts and assignments for 5b-5e, 5g and 5i-5k (see Experimental section) were compared with the respective data from 5a, 5f and 5h. In silico results Molecular docking results The molecular docking method is employed to identify potential non-covalent inhibitors of acetylcholinesterase (AChE). This computational approach, facilitated by a combination of binding affinity (kcal mol-1) and RMSD using AutoDock Vina, significantly reduces the time required to screen and identify promising compounds from extensive ligand libraries. In the initial phase of the in silico study of oxime derivatives as AChE inhibitors, a preliminary virtual ranking was established. The binding affinity score from AutoDock Vina was selected due to its strong correlation with binding affinity (primary) energy.43 Consequently, all tested derivatives were identified as the most promising ligands in comparison to the reference ligand, galantamine, following refinement through molecular docking. The RMSD was initially used for the statistical validation of the complex formation simulations and to determine the best binding pose. The RMSD is calculated by measuring the average distance between atoms of two ligand poses, with validation criteria being RMSD values close to 2 Å. All simulations (docking and redocking) produced RMSD values of 2 Å or less, with the derivative 5i/AChE complex showing a particularly low RMSD of 0.98 Å (Table 3).
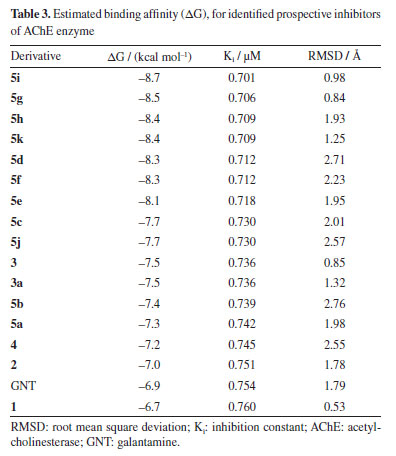
The best poses, selected based on RMSD ranking and binding energy, revealed that the complexes 5i/AChE, 5g/AChE, and 5h/AChE had RMSD values between 0.98 and 1.93 Å, and binding energies of -8.7, -8.5, and -8.4 kcal mol-1, respectively. In the redocking simulations, the GNT/AChE complex exhibited a binding energy of -6.9 kcal mol-1. All other derivatives also demonstrated favorable binding energies and RMSD values below 2 Å, surpassing the reference ligands in these metrics. The structure of AChE is characterized by several key regions that play a crucial role in its catalytic function. These include the catalytic active site (CAS), the acyl binding pocket, the choline binding site (CBS), represented by the Trp86 residue, the peripheral active site (PAS), and a narrow but deep gorge (~ 20 Å) that connects the CAS to the PAS. The CAS, located near the bottom of this gorge, is formed by the catalytic triad residues Ser203, His447, and Glu534,44 which are responsible for the hydrolysis of acetylcholine (Figure 5).
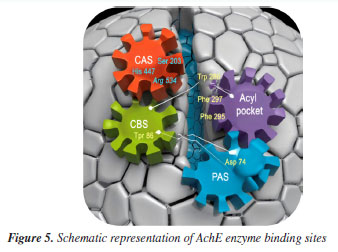
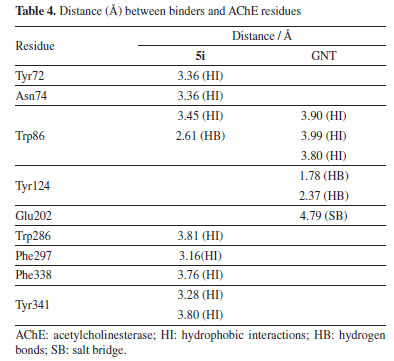
The PAS, situated near the surface of the enzyme, acts as an initial substrate recognition site and facilitates substrate entry. This region is defined by residues such as Asp74 and Trp286, which play a role in the binding and guidance of the substrate toward the active site. Additionally, the PAS has been implicated in the formation of amyloid-β fibrils, further underscoring its relevance beyond the enzymatic activity itself.45,46 The calculated binding fitness of the ligand 5i surpassed that of the AChE inhibitor galantamine (GNT). Both ligands are well accommodated within the active site of AChE, with their functional groups occupying key pockets of the enzyme. Notably, their ring structures engage with the catalytic triad residues and the acyl pocket through hydrogen bonding and hydrophobic interactions, particularly with Trp86. Additionally, the ligands form hydrogen bonds with other relevant residues. Furthermore, ligand 5i exhibits hydrophobic interactions with Asp74, located within the peripheral active site (PAS), which accounts for its higher predicted affinity compared to GNT.47 This ligand also interacts with the acyl pocket at the Trp286 residue, strengthening its binding profile. In the case of GNT, the polar environment is stabilized by two hydrogen bonds with Tyr124. The ligand also engages in π-alkyl interactions with Trp86 at the choline-binding site. In addition to these interactions, a salt bridge was observed with Glu202, situated near the CAS. These interactions are summarized in Table 4 and Figures 6a and 6b.
CONCLUSIONS In conclusion, an efficient conversion (82 to 95% yield) of oxime in cyclic imides, involving the Beckmann rearrangement (BR), has been developed. The key features of this method are to avoid using catalysts that result in undesired by-products and that requires large amounts of strong acids (sulfuric acid, for example), in addition to occur in one-pot reactions. The mechanism for the formation of imides via a oxaziridine intermediate, followed by BR under Lewis base catalysis, represents a mechanistic proposal. The operationally simple protocol with available substrates offers an efficient access to cyclic imides. Calculating the physicochemical properties through molecular modeling allowed us to understand the interaction between a ligand and its receptor. The results showed that most derivatives were energetically influenced, especially the 5i analog. They showed a better classification than the others analyzed and the reference ligand, GNT. It should be noted that several more tests and analyses must be carried out until the final goal is completed to optimize time and cost and present significant advantages.
SUPPLEMENTARY MATERIAL Supplementary information (1H, 13C NMR, MS and mechanistic hypothesis of the formation of the main derivatives) is available at http://quimicanova.sbq.org.br/, as a PDF file, with free access.
DATA AVAILABILITY STATEMENT All data are available in the text and supplementary material.
ACKNOWLEDGMENTS This work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Brasil and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Brasil. We thank Dr. Antonio J. R. da Silva e Ari M. da Silva (UFRRJ) for obtaining mass spectra in high-resolution.
REFERENCES 1. Schmidt, R.; Griesbaum, K.; Behr, A.; Biedenkapp, D.; Voges, H. W.; Garbe, D.; Paetz, C.; Collin, G.; Mayer, D.; Höke, H.; Hydrocarbons, ULLMANN'S Encyclopedia of Industrial Chemistry; Wiley‐VCH Verlag GmbH & Co. KGaA: Weinheim, 2014. [Crossref] 2. Gabriele, B.; Mancuso, R.; Veltri, L.; Chem. - Eur. J. 2016, 22, 1767. [Crossref] 3. Dorsey, B. D.; Levin, R. B.; McDaniel, S. L.; Vacca, J. P.; Guare, J. P.; Anderson, P. S.; Huff, J. R.; Darke, P. L.; Zugay, J. A.; Emini, E. A.; Schleif, W. A.; Quintero, J. C.; Lin, J. H.; Chen, I. W.; Holloway, M. K.; Fitzgerald, P. M. D.; Axel, M. G.; Ostovic, D.; J. Med. Chem. 1994, 37, 3443. [Crossref] 4. Ghosh, A. K.; Bilcer, G.; Schiltz, G.; Synthesis 2001, 15, 2203. [Crossref] 5. Sugimoto, H.; Iimura, Y.; Yamanishi, Y.; Yamatsu, K.; J. Med. Chem. 1995, 38, 4821. [Crossref] 6. Sugimoto, H.; Pure Appl. Chem. 1999, 71, 2031. [Crossref] 7. Sinha, S. K.; Shrivastava, S. K.; Bioorg. Med. Chem. Lett. 2013, 23, 2984. [Crossref] 8. Liu, X. H.; Zhi, L. P.; Song, B. A.; Xu, H. L.; Chem. Res. Chin. Univ. 2008, 24, 454. [Crossref] 9. Bachovchin, D. A.; Wolfe, M. R.; Masuda, K.; Brown, S. J.; Spicer, T. P.; Fernandez-Vega, V.; Chase, P.; Hodder, P. S.; Rosen, H.; Cravatt, B. F.; Bioorg. Med. Chem. Lett. 2010, 20, 661. [Crossref] 10. Gao, Y.; Song, J.; Shang, S.; Wang, D.; Li, J.; BioResources 2012, 7, 4150. [Crossref] 11. Bindu, P. J.; Mahadevan, K. M.; Satyanarayan, N. D.; Naik, T. R. R.; Bioorg. Med. Chem. Lett. 2011, 22, 898. [Crossref] 12. Hwu, J. R.; Tsay, S. C.; Hong, S. C.; Hsu, M. H.; Liu, C. F.; Chou, S. S. P.; Bioconjugate Chem. 2013, 24, 1778. [Crossref] 13. Steyermark, A.; Kirk-Othmer Encyclopedia of Chemical Technology, vol. 11, 3rd ed.; Wiley-Interscience: New York, 1984. [Crossref] 14. Clayden, J.; Angew. Chem., Int. Ed. 2023, 42, 1788. [Crossref] 15. Dunetz, J. R.; Magano, J.; Weisenburger, G. A.; Org. Process Res. Dev. 2016, 20, 140. [Crossref] 16. Ayoub, S. S.; Temperature 2021, 8, 351. [Crossref] 17. Shen, B.; Makley, D. M.; Johnston, J. N.; Nature 2010, 465, 1027. [Crossref] 18. Aricò, F.; Quartarone, G.; Rancan, E.; Ronchin, L.; Tundo, P.; Vavasori, A.; Catal. Commun. 2014, 49, 47. [Crossref] 19. Umanadh, Y.; Reddy, N. S.; Mukkanti, K.; Omprakash, G.; Asian J. Chem. 2015, 27, 1209. [Crossref] 20. Yamamoto, H.; Komeda, M.; Ozaki, A.; Sumimoto, M.; Hori, K.; Sugimoto, T.; Int. J. Org. Chem. 2015, 05, 147. [Crossref] 21. Ahluwalia, V. K.; Stereochemistry of Organic Compounds; Springer: Switzerland, 2022. [Crossref] 22. de Azevedo Junior, W.; Dias, R.; Curr. Drug Targets 2008, 9, 1031. [Crossref] 23. Ahmadi, M.; Motlagh, M. J.; Rahmani, A. T.; Zolfagharzadeh, M. M.; Shariatpanahi, P.; Chermack, T. J.; Coons, L. M.; Cotter, J.; Eyiah-Donkor, E.; Poti, V.; Derbyshire, J.; Dolan, T. E.; Fuller, T.; Kishita, Y.; McLellan, B. C.; Giurco, D.; Aoki, K.; Yoshizawa, G.; Handoh, I. C.; Kuhmonen, T.; Kuusi, O.; Lauhakangas, O.; Ruttas-Küttim, R.; Mason-D'Croz, D.; Vervoort, J.; Palazzo, A.; Islam, S.; Lord, S.; Helfgott, A.; Havlok, P.; Peou, R.; Sassen, M.; Veeger, M.; van Soesbergen, A.; Arnell, A. P.; Stuch, B.; Arslan, A.; Lipper, L.; Roper, J.; Tarasti, E.; Dearden, P.; Kowalski, B.; Lowe, J.; Roland, R.; Surridge, M.; Thomas, S. S.; Jones, S.; Travesset-Baro, O.; Gallacheir, B. P.; Jover, E.; Rosas-Casals, M.; Veenman, S.; Leroy, P.; Zackery, A.; Shariatpanahi, P.; Zolfagharzadeh, M. M.; Pourezzat, A. A.; Lee, C.; Pschorn, U.; Rössler, E.; Sillescu, H.; Kaufmann, S.; Schaefer, D.; Spiess, H. W.; Palierne, J. F.; Seo, Y.; Paeng, K.; Park, S.; Reignier, J.; Favis, B. D.; Maeda, H.; Ueda, M.; Morinaga, T.; Matsumoto, T.; Ma, Z.; Lacroix-Desmazes, P.; O'Reilly, J. M.; Matusinovic, Z.; Shukla, R.; Manias, E.; Hogshead, C. G.; Wilkie, C. A.; Yin, Z.; Koulic, C.; Pagnoulle, C.; Jérôme, R.; Tanaka, T.; Nakatsuru, R.; Kagari, Y.; Saito, N.; Okubo, M.; Sikka, M.; Pellegrini, N. N.; Schmitt, E. A.; Winey, K. I.; Ray, D. K.; Himanshu, A. K.; Sinha, T. P.; Soto-Figueroa, C.; Rodríguez-Hidalgo, M. D. R.; Martínez-Magadán, J. M.; Utracki, L. A.; Kamal, M. R.; Shi, G. Z. H.; Rodrigue, D.; Gonzalez-Nüñez, R.; Logothetidis, S.; Zallen, R.; Tsagaropoulos, G.; Eisenberg, A.; Boudenne, A.; Ibos, L.; Candau, Y.; Thomas, S. S.; Barlow, J. W.; Paul, D. R.; Ye, Y. S.; Shen, W. C.; Tseng, C. Y.; Rick, J.; Huang, Y. J.; Chang, F. C.; Hwang, B. J.; Braunecker, W. A.; Pintauer, T.; Tsarevsky, N. V.; Kickelbick, G.; Matyjaszewski, K.; Schwahn, D.; Han, C. D.; Freed, K. F.; Dudowicz, J.; Fu, X.; Qutubuddin, S.; Fayt, R.; Jérôme, R.; Teyssié, P. H.; Graebling, D.; Muller, R.; Palierne, J. F.; Chernikova, E.; Terpugova, P.; Bui, C.; Charleux, B.; Fayt, R.; Jerome, R.; Teyssié, P. H.; Cognard, J.; Laborde, J.; Deraeve, C.; Lecoq, L.; Sournia-Saquet, A.; Stigliani, J. L.; Orena, B. S.; Mori, G.; Pratviel, G.; Bernardes-Génisson, V.; Lee, M.; Lodge, T.; Macosko, C.; Heitz, W.; Brügging, W.; Freund, L.; Gailberger, M.; Greiner, A.; Jung, H.; Kampschulte, U.; Nießner, N.; Osan, F.; Schmidt, H. W.; Wicker, M.; Guo, S.; Ait-Kadi, A.; Jaramillo-Soto, G.; García-Morán, P. R.; Enríquez-Medrano, F.; Maldonado-Textle, H.; Albores-Velasco, M. E.; Guerrero-Santos, R.; Vivaldo-Lima, E.; Ishihara, N.; Seimiya, T.; Kuramoto, M.; Uoi, M.; Rohini, R.; Bose, S.; Macromolecules 2005, 24, 1; ChemDraw 15.0 User Guide, https://www.drugfuture.com/UploadFiles/2016-2/75/W130996343785884.pdf, accessed in June 2025 24. Hanwell, M. D.; Curtis, D. E.; Lonie, D. C.; Vandermeersch, T.; Zurek, E.; Hutchison, G. R.; J. Cheminf. 2012, 4, 17. [Crossref] 25. Halgren, T. A.; J. Comput. Chem. 1996, 17, 490. [Crossref] 26. Cheung, J.; Rudolph, M. J.; Burshteyn, F.; Cassidy, M. S.; Gary, E. N.; Love, J.; Franklin, M. C.; Height, J. J.; J. Med. Chem. 2012, 55, 10282. [Crossref] 27. Morris, G. M.; Ruth, H.; Lindstrom, W.; Sanner, M. F.; Belew, R. K.; Goodsell, D. S.; Olson, A. J.; J. Comput. Chem. 2009, 30, 2785. [Crossref] 28. Trott, O.; Olson, A. J.; J. Comput. Chem. 2010, 31, 455. [Crossref] 29. Francis, P. T.; Palmer, A. M.; Snape, M.; Wilcock, G. K.; J. Neurol., Neurosurg. Psychiatry 1999, 66, 137. [Crossref] 30. The PyMOL Molecular Graphics System; Pymol, version 2.3; Schrödinger, LLC, USA, 2020. 31. BIOVIA Discovery Studio Modeling Environment, version 4.5; Dassault Systèmes, San Diego, USA, 2015. 32. Marinho, E. M.; Andrade Neto, J. B.; Silva, J.; da Silva, C. R.; Cavalcanti, B. C.; Marinho, E. S.; Nobre Junior, H. V.; Microb. Pathog. 2020, 148, 104365. [Crossref] 33. Nguyen, D. D.; Xiao, T.; Wang, M.; Wei, G. W.; J. Chem. Inf. Model. 2017, 57, 1715. [Crossref] 34. Yusuf, D.; Davis, A. M.; Kleywegt, G. J.; Schmitt, S.; J. Chem. Inf. Model. 2008, 48, 1411. [Crossref] 35. Shityakov, S.; Förster, C.; Adv. Appl. Bioinf. Chem. 2014, 7, 23. [Crossref] 36. Imberty, A.; Hardman, K. D.; Carver, J. P.; Perez, S.; Glycobiology 1991, 1, 631. [Crossref] 37. Inazu, M.; Yamada, T.; Kubota, N.; Yamanaka, T.; Pharmacol. Res. 2013, 76, 119. [Crossref] 38. Dimmock, J. R.; Sidhu, K. K.; Chen, M.; Li, J.; Quail, J. W.; Allen, T. M.; Kao, G. Y.; The Journal of Pharmaceutical Sciences 1994, 83, 852. [Crossref] 39. Araújo, C. R. M.; Gonsalves, A. A.; Rev. Virtual Quim. 2015, 7, 1469. [Crossref] 40. Irie, H.; Tanaka, S.; Zhang, Y.; Koyama, J.; Taga, T.; Machida, K.; Chem. Pharm. Bull. 1988, 36, 3134. [Crossref] 41. Neises, B.; Steglich, W.; Angew. Chem., Int. Ed. 1978, 17, 522. [Crossref] 42. Hassner, A.; Alexanian, V.; Tetrahedron Lett. 1978, 19, 4475. [Crossref] 43. Verdonk, M. L.; Cole, J. C.; Hartshorn, M. J.; Murray, C. W.; Taylor, R. D.; Proteins: Struct., Funct., Genet. 2003, 52, 609. [Crossref] 44. Colovic, M. B.; Krstic, D. Z.; Lazarevic-Pasti, T. D.; Bondzic, A. M.; Vasic, V. M.; Curr. Neuropharmacol. 2013, 11, 315. [Crossref] 45. Inestrosa, N. C.; Alvarez, A.; Calderón, F.; Mol. Psychiatry 1996, 1, 359. [Link] accessed in May 2025 46. Inestrosa, N.; Urra, S.; Colombres, M.; Curr. Alzheimer Res. 2005, 1, 249. [Crossref] 47. Olazaran, J.; Garcia, G.; Neurologia 2002, 17, 429. [Link] accessed in May 2025
Editor handled this article: Jorge M. David |

On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access