
 |
 |
 |
 |
 |
Artigo
| LC-MS/MS based dereplication of isoquinoline-derived alkaloids from the trunk bark of Unonopsis rufescens (Baill.) R.E.Fr. (Annonaceae) |
|
Lucas Antônio F. de AlmeidaI,II; Felipe Moura A. da SilvaI,II,* I. Programa de Pós-Graduação em Química, Universidade Federal do Amazonas, 69080-900 Manaus - AM, Brasil Received: 02/02/2025 *e-mail: lbelem@ufam.edu.br; felipemas@ufam.edu.br Mass spectrometry-based approaches constitute the primary strategy for the dereplication of isoquinoline-derived alkaloids, which are widely distributed in various genera of the Annonaceae family. On the other hand, although many species from the Annonaceae family have been studied regarding their alkaloid content, others remain largely unexplored, such as U. rufescens. In order to dereplicate isoquinoline-derived alkaloids from the trunk bark of U. rufescens, a liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) analysis in positive ionization mode was conducted with its alkaloid fraction and the fragmentation data were subsequently interpreted manually. Based on the fragmentation pattern knowledge regarding isoquinoline-derived alkaloids, pallidine (1), along with the benzyltetrahydroisoquinolines coclaurine (2), N-methylcoclaurine (3), reticuline (5), norarmepavine (6), norjuziphine (7), and N-methylnorjuziphine (8); proaporphine stepharine (4); oxoaporphines liriodenine (10) and lysicamine (13); C-7 substituted aporphine norushinsunine (11); and the aporphines asimilobine (9), isopiline (12), nornuciferine (14), anonaine (15), and O-methylisopiline (16) were tentatively identified. In addition, a mixed fragmentation pathway was observed for isopiline (12), and a proposed fragmentation pathway for this compound was presented. Overall, this study expands the knowledge of U. rufescens, demonstrating the rich chemical diversity present in this species. Furthermore, it contributes to the research on the fragmentation patterns of isoquinoline-derived alkaloids. INTRODUCTION Isoquinoline-derived alkaloids are widely distributed in living organisms, particularly within the plant kingdom, and are divided into subclasses such as simple isoquinolines, benzylisoquinolines, berberine, protoberberines, and aporphinoids.1,2 On the other hand, aporphinoids can be divided into several subclasses, such as aporphines, oxoaporphines, C-7 and/or C-4 substituted aporphines, dehydroaporphines, phenanthrenes, and miscellaneous aporphinoids.3 Aporphines, in turn, include noraporphines, aporphines, quaternary aporphines, aporphine N-oxides, and natural N-acylated noraporphines.3 Isoquinoline-derived alkaloids present a growing list of biological activities, including antibacterial, antifungal, antidiabetic, antitumor, antioxidant, anti-inflammatory, antiparasitic, insecticidal, neuroprotective, antiviral, and others.1,2 This has attracted the attention of researchers worldwide. In this context, chemical analyses, especially those based on mass spectrometry (MS), have been widely used to investigate potential sources of bioactive isoquinoline-derived alkaloids.4-6 Mass spectrometry-based approaches constitute the primary strategy for the dereplication of isoquinoline-derived alkaloids and are typically based on diagnostic product ions, making it possible to identify the class and its substitution pattern.7-11 In summary, protonated aporphine alkaloids first exhibit the loss of the amino group and its substituent, represented by the loss of 17 Da (nonsubstituted), 31 Da (N-methyl), 45 Da (N-formyl), or 47 Da (N-methyl N-oxide). Subsequently, losses of peripheral groups are observed, following these rules: if a hydroxyl group (OH) is vicinal to a methoxy group (OCH3) on the aromatic ring, consecutive losses of 32 Da (CH3OH) and 28 Da (CO) are observed. Otherwise, if vicinal methoxy groups are present on the aromatic ring, consecutive losses of 15 Da and competitive losses of 15 and 31 Da are observed. For a methylenedioxy group on the aromatic ring, consecutive losses of 30 Da (formaldehyde) and 28 Da (CO) are observed.7-11 These rules also appear to be valid for proaporphine alkaloids.10-12 Regarding protonated benzylisoquinoline alkaloids, it is possible to determine substitutions in the benzyl and isoquinoline moieties using a set of key ions.7,8,11 On the other hand, for protonated tetrahydroprotoberberine alkaloids, the key ions, formed due to retro-Diels-Alder (RDA) reactions, allow the determination of substitutions on rings A and D.7,8,11 Regarding the analysis, the dereplication of isoquinoline-derived alkaloids is normally based on direct injection (DI) (e.g., DI-ESI-MS)9-12 and hyphenated techniques, such as liquid chromatography coupled to tandem mass spectrometry with electrospray ionization (LC-ESI-MS/MS),4-6 which indeed constitutes a powerful system for the acquisition of chemical data. Although electrospray ionization (ESI) is typically employed as a source due to its ability to analyze polar and ionic compounds, atmospheric pressure chemical ionization (APCI) has also shown significant potential for this approach,7 being a source capable of analyzing both polar and non-polar compounds.13 Among the various families that contain alkaloids in their composition, the Annonaceae family stands out for having a wide variety of isoquinoline-derived alkaloids distributed across various genera.14 An example is the Unonopsis genus, a neotropical genus constituted by approximately fifty species (shrubs and trees) distributed throughout Central America and tropical South America.15 In Brazil, 16 species (8 endemic) are described, with a greater occurrence in the northern region.16 Some Unonopsis species are used in traditional medicine, such as U. floribunda, whose trunk bark is widely used in Peruvian traditional medicine for the treatment of arthritis, bronchitis, rheumatism, diarrhea, and malaria,17,18 as well as U. veneficiorum and U. stipitata, whose leaves are utilized to treat age-related cognitive disorders by indigenous people.19 From the chemical point of view, this genus presents itself as a promising source of isoquinoline derivatives, mainly aporphines, oxoaporphines, proaporphines, benzyltetrahydroisoquinolines, and tetrahydroprotoberberines.9,10,12 Although many species from the Unonopsis genus have been studied from a phytochemical perspective, others remain largely unexplored, such as U. rufescens, which is considered botanically similar to U. floribunda.15 This species is distributed in Guyana, Suriname, French Guiana, and Brazil (Amapá, Amazonas, and Pará).15 To date, isoquinoline-derived alkaloids were detected in its leaves,12 and the lanostane-type triterpene polycarpol was found in its twigs and trunk bark.20 Additionally, the composition of the essential oil from its leaves was reported, with the main compounds identified as the sesquiterpenes spathulenol, caryophyllene oxide, and γ-muurolene.21 No knowledge of the alkaloid composition of its trunk bark was reported. In this context, the present study aims to dereplicate isoquinoline-derived alkaloids from the alkaloid fraction of the trunk bark of U. rufescens. This was stablished through LC-MS/MS analysis and manual interpretation of the fragmentation patterns.
EXPERIMENTAL General experimental procedures Liquid chromatography coupled to tandem mass spectrometry analysis (LC-MS/MS) was performed using a system comprising an Accela chromatograph coupled with an LCQ ion trap mass spectrometer (Thermo Scientific, Waltham, MA, USA). Semi-preparative HPLC (high-performance liquid chromatography) analysis was carried out on a UFLC system (LC-6 AD pump; DGU-20A5 degasser; SPD-20AV UV detector; rheodyne injector; CBM-20A communication module) (Shimadzu, Columbia, MD, USA). Nuclear magnetic resonance (NMR) spectroscopy analysis was performed with an AVANCE III 600 NMR spectrometers (Bruker, Karlsruhe, Germany), operating at 14.1 T, observing 1H at 600.13 MHz. An analytical mill Q298A (Quimis, Diadema, SP, Brazil) was used to grind the sample. All solvents used for extraction, chromatography, and MS experiments were HPLC grade, purchased from Tedia (Fairfield, OH, USA), and the water was purified by a Milli-Q system. Deuterated methanol (CΔ3OD) (Cambridge Isotope, Tewksbury, USA) was used as solvent for NMR analysis. Authenticated standards of pallidine, stepharine, reticuline, asimilobine, liriodenine, norushinsunine, isopiline, lysicamine, nornuciferine, anonaine, and O-methylisopiline were sourced from the authors' laboratory.11,22 Plant material The trunk bark of U. rufecens was collected in the Esteio Farm (2º23'15.1"S 59º50'53.3"W) of the Projeto Dinâmica Biológica de Fragmentos Florestais (PDBFF), located in the Distrito Agropecuário da SUFRAMA, Manaus, Brazil, from specimen previously identified by Prof. Paul J. M. Maas. A voucher specimen (No. 3767) is deposited in the botany collection of PDBFF. Access to the plant material is registered in the Sistema Nacional de Gestão do Patrimônio Genético e do Conhecimento Tradicional Associado (SisGen) under code AE0F182. Alkaloid extraction and semi-preparative HPLC analysis The alkaloid fraction was obtained according to a previously reported methodology.7 Firstly, the plant material was powdered using an analytical mill. Then, the powdered trunk bark (2 g) was stirred with a vortex mixer with a mixture of 10% ammonium hydroxide (NH4OH) aqueous solution (20 mL) and dichloromethane (CH2Cl2) (20 mL) in a glass container. Subsequently, the organic phase was transferred to another glass container and stirred with a 10% acetic acid (CH3CO2H) aqueous solution (20 mL). Afterward, the aqueous phase was separated and treated with NH4OH until a pH of 10 was achieved. Then, this phase was extracted under vigorous stirring with CH2Cl2 (20 mL). Finally, the organic phase was dried over anhydrous sodium sulfate, transferred to a new container, and the solvent was evaporated to dryness under a nitrogen gas stream, yielding the alkaloid fraction (5.6 mg, 0.28%). The procedure was repeated two more times, yielding a total of 13.4 mg of alkaloid fraction. A part of the alkaloidal fraction (10 mg) was subjected to fractionation by semi-preparative HPLC using 0.01% trifluoroacetic acid aqueous solution (A) and methanol (B) as mobile phases. Elution was performed using a flow of 4.5 mL min-1 and through a gradient of 30% B to 80% B over 20 min. A C18(2) column (250 × 10 mm i.d., 5 μm) (Phenomenex, Torrance, CA, USA) was employed on the fractionation being monitored with UV channels at 250 and 280 nm. A single injection was carried into the column in MeOH (50 μL) affording compound 1 (4.6 mg). LC-MS/MS analysis The alkaloid fraction was diluted to 1 mg mL-1 in MeOH and analyzed (10 µL, injection volume) with an Accela HPLC system (Thermo Scientific) equipped with a Phenomenex Luna C18(2) column (150 mm × 4.6 mm i.d., 5 µm) (Torrance, CA, USA). The mobile phase consisted of 0.01% trifluoroacetic acid aqueous solution (A) and ACN (B). Elution was performed using a flow of 1 mL min-1 and through a gradient of 20% B to 80% B over 15 min, followed by 10 min at 80% B. The outlet from the column was connected through a split valve (flow of 500 µL min-1) to a LCQ fleet mass spectrometer (Thermo Scientific). Ionization was performed with an APCI source operating at positive ion mode. The ionization settings were as follows: positive polarity, source voltage, 6 kV, discharge current, 5 µA; vaporizer temperature, 330 ºC; sheath gas pressure, 35 arbitrary unit (arb); ion sweep gas pressure, 0.0 arb; aux gas pressure, 15 arb; capillary temperature, 250 ºC; tube lens offset, 110 V; skimmer offset, 0 V; mass range, m/z 100 to 800. Helium was used as collision gas, and the MSn spectra were obtained using collision energies ranging from 20 to 27%. Analysis of authenticated standards by DI-MS For direct injection mass spectrometry (DI-MS) analysis, stock solutions (1 mg mL-1) of the authenticated standards of pallidine, stepharine, reticuline, asimilobine, liriodenine, norushinsunine, isopiline, lysicamine, nornuciferine, anonaine, and O-methylisopiline were prepared in HPLC-grade MeOH. Aliquot (10 µL) of the stock solution was subsequently transferred to vials containing 1 mL of MeOH. Finally, the diluted solution (10 ppm) was analyzed by direct injection into a LCQ fleet mass spectrometer (Thermo Scientific) equipped with an APCI source operating in positive polarity mode. The ionization settings were the same as those of the LC-MS/MS analysis.
RESULTS AND DISCUSSION LC-MS analysis The alkaloid fraction of U. rufescens was analyzed using LC-MS/MS in positive ionization mode, and the fragmentation data were subsequently interpreted manually. The tentative identification of compounds 1-16 was based on the previously reported fragmentation pattern knowledge regarding benzyltetrahydroisoquinolines, proaporphine, and aporphinoids in species of the genus Unonopsis and other species of Annonaceae.7-12 These compounds were supposed as alkaloids based on the even m/z ions (Figures 1S-15S, Supplementary Material), indicating protonated structures containing only one nitrogen atom, a characteristic feature of isoquinoline-derived alkaloids.1-3 The LC-MS total ion chromatogram (TIC) of the alkaloid fraction (Figure 1) revealed a major peak (retention time (tR) = 4.23 min) corresponding to a protonated alkaloid at m/z 328, along with other minor peaks between 5.49 and 11.90 min, which contained protonated alkaloids ranging from m/z 266 to 312 (Table 1). The LC-MS/MS or LC-MS2 spectra of these compounds (Figures 16S-31S, Supplementary Material) were then interpreted manually.
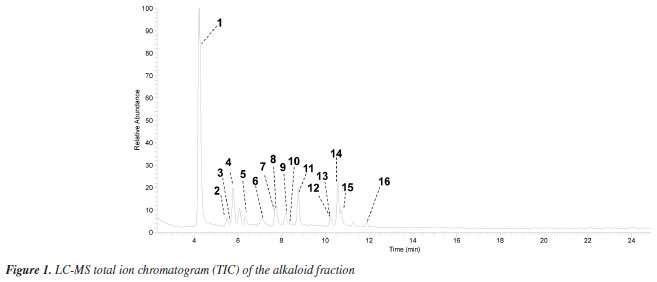
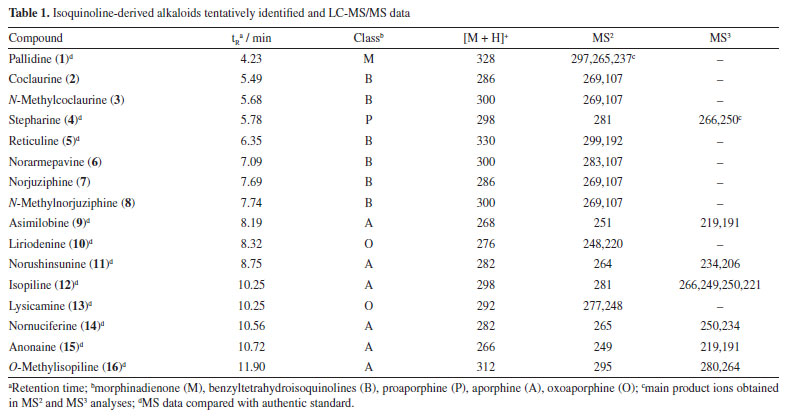
The LC-MS2 spectrum of compound 1 (m/z 328) exhibited an initial loss of 31 Da (-NH2CH3) (m/z 328 → 297), followed by subsequent losses of 32 Da (-CH3OH) (m/z 297 → 265) and 28 Da (-CO) (m/z 265 → 237). Although this fragmentation pattern is consistent with aporphine alkaloids that possess adjacent hydroxyl and methoxyl groups at ring A,7-12 a similar pattern has been previously observed for the morphinadienone alkaloid pallidine (1) (Figure 2), which is also a major compound found in the bark of the related species U. floribunda.22
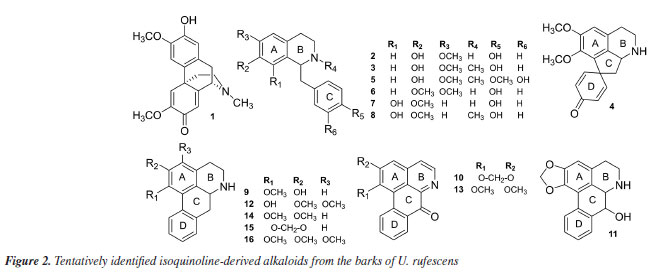
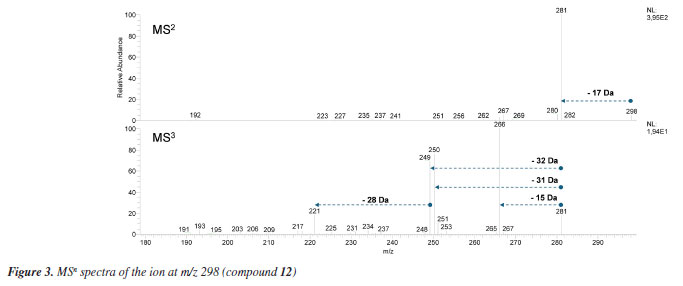
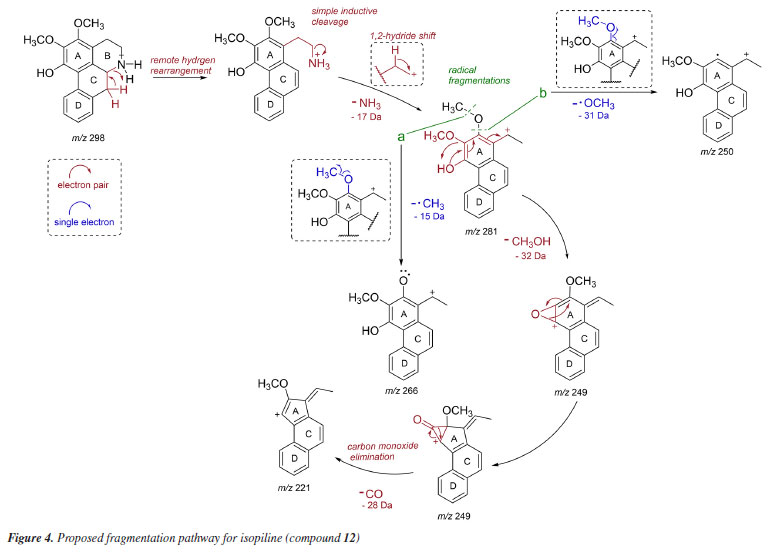
The LC-MS2 spectra of compounds 2 (m/z 286) and 3 (m/z 300) presented an initial loss of 17 Da (-NH3) (m/z 286 → 269) and 31 Da (-NH2CH3) (m/z 300 → 269), respectively, along with a diagnostic ion of benzylisoquinoline skeletons at m/z 107. This evidence has been previously associated with benzylisoquinolines with and without an N-methyl group, and containing hydroxyl and methoxyl groups at ring A, as well as a hydroxyl group at the C ring.7,8,12 The fragmentation of compounds 2 and 3 were consistent with the benzylisoquinoline alkaloids coclaurine (2) and N-methylcoclaurine (3), both previously reported in Annonaceae species.8 The same fragmentation pattern was observed in the LC-MS2 spectra of compounds 7 (m/z 286) and 8 (m/z 300), with these being tentatively identified as norjuziphine (7) and N-methylnorjuziphine (8).7 For these compounds, the elution order was assigned based on the previous behavior of compounds 2 and 7 on C-18 columns.4 Compound 6 (m/z 300), an isomer of compounds 3 and 8, presented in the LC-MS2 spectrum an initial loss of 17 Da (-NH3) (m/z 300 → 283) and the diagnostic ion at m/z 107. This evidence suggests a benzylisoquinoline skeleton without an N-methyl group, containing two methoxyl groups at ring A, along with a hydroxyl group at the C ring. Thus, compound 6 was tentatively identified as norarmepavine (6).23 Finally, the LC-MS2 spectrum of compound 5 (m/z 330) presented a discrete fragment at m/z 299 and a base peak fragment ion at m/z 192 (m/z 300 → 192). This fragmentation pattern is consistent with the benzylisoquinoline alkaloid reticuline (5).7,8,11 The LC-MS2 spectra of compounds 10 (m/z 276) and 13 (m/z 292) presented evidence of oxoaporphine alkaloids, such as a loss of -28 Da (m/z 276 → 248) and -15 Da (m/z 292 → 277), and were tentatively identified as liriodenine (10) and lysicamine (13) through comparison of their MS2 spectra with those reported in the literature.24 On the other hand, the LC-MS2 mass spectra of compounds 4, 9, 11, 12, 14, 15, and 16 presented primarily only the base peak as useful information, providing no additional data to assist in their tentative identifications. Although the m/z information of these compounds and the initial losses provided strong evidence of aporphine alkaloids previously reported in species of the Unonopsis group, such as U. duckei,10,12 U. floribunda,22 U. guatterioides,9,12 U. stipitata,25 O. amazonicum,7 and Bocageopsis pleiosperma,11 an additional MS3 analysis was conducted (Figures 32S-38S, Supplementary Material). The MS2 and MS3 spectra of compounds 4 (m/z 298), 14 (m/z 282), and 16 (m/z 312) presented fragmentation pathways consistent with proaporphine and aporphine alkaloids that have adjacent methoxyl groups at ring A and an absence of N-methyl groups.7-12 Their losses were assigned as 17 Da (-NH3) (m/z 298 → 281; m/z 282 → 265; m/z 312 → 295), followed by competitive losses of 15 Da (-∙CH3) (m/z 281 → 266; m/z 265 → 250; m/z 295 → 280) and 31 Da (-∙OCH3) (m/z 281 → 250; m/z 265 → 234; m/z 295 → 264). Therefore, the fragmentation patterns observed for compounds 4, 14, and 16 were consistent with the proaporphine alkaloid stepharine (4)11 and the aporphine alkaloids nornuciferine (14)7-11 and O-methylisopiline (16),7 respectively. On the other hand, the MS2 and MS3 spectra of compound 9 (m/z 268) presented a fragmentation pathway consistent with aporphine alkaloids that have adjacent hydroxyl and methoxyl groups at ring A and the absence of N-methyl groups.7-12 Thus, an initial loss of 17 Da (-NH3) was observed (m/z 268 → 251), followed by subsequent losses of 32 Da (-CH3OH) (m/z 251 → 219) and 28 Da (-CO) (m/z 219 → 191). This fragmentation pattern is consistent with the aporphine alkaloid asimilobine (9).7-12 The MS2 and MS3 spectra of compound 11 (m/z 282) presented a fragmentation pathway consistent with a 7-hydroxyaporphine alkaloid containing a methylenedioxy group at ring A and the absence of an N-methyl group.7 For this compound, an initial loss of 18 Da (-H2O) was observed (m/z 282 → 264), followed by subsequent losses of 30 Da (-CH2O) (m/z 264 → 234) and 28 Da (-CO) (m/z 234 → 206). This fragmentation is coherent with the aporphine alkaloid norushinsunine (11).7 Moreover, compound 15 (m/z 266) presented MS2 and MS3 spectra similar to compound 11, with only the first loss being different (17 Da instead of 18 Da). The subsequent losses of 30 Da (-CH2O) (m/z 249 → 219) and 28 Da (-CO) (m/z 219 → 191) confirmed compound 15 as anonaine.7-12 Finally, the MS2 and MS3 spectra (Figure 3) of compound 12 (m/z 298) presented a mixed fragmentation pathway consistent with an aporphine alkaloid that has adjacent hydroxyl and methoxyl groups, as well as adjacent methoxyl groups at ring A, in addition to the absence of an N-methyl group. This hypothesis was considered due to the competitive losses of 15 Da (-∙CH3) (m/z 281 → 266), 31 Da (-∙OCH3) (m/z 281 → 250), and 32 Da (-CH3OH) (m/z 281 → 249), which occurred after the initial loss of 17 Da (-NH3) (m/z 298 → 281) (Figure 3). Another evidence was the loss of 28 Da (-CO) (m/z 249 → 221) occurring after the loss of 32 Da (-CH3OH). This fragmentation pattern is consistent with the aporphine alkaloid isopiline (12).7 Since mixed fragmentation pathway for aporphine alkaloids is uncommon, a proposed fragmentation pathway for this type of aporphine skeleton is presented in Figure 4.
Although the fragmentation pattern observed for the tentatively identified compounds 1-16 is in agreement with previously published data based on ion trap (IT), triple quadrupole (QqQ), and quadrupole time-of-flight (Q-TOF) analyzers,7,8 some differences in the data contained within the spectra can be noted (e.g., the presence or absence of certain fragment ions, differing relative intensities of fragment ions, etc.). The differences between mass spectra of natural products obtained using different analyzers or instrumental conditions is a well-established issue26 and the reason why studies on the fragmentation patterns of a given class of natural products should be continuously encouraged, and databases should be used with caution in the dereplication process. Therefore, in order to validate the dereplication of the compounds presented in this study, the MS2 spectra of compounds 1, 5, 10, and 13, and MS2 and MS3 spectra of compounds 4, 9, 11, 12, 14, 15, and 16 were also directly compared with authentic standards analyzed on the same instrument. This comparative analysis showed high spectral similarity between the tentatively identified compounds and authentic standards, therefore, endorsing the dereplication presented in this study.
CONCLUSIONS Through the LC-MS/MS analysis conducted in this study, it was possible to dereplicate several isoquinoline-derived alkaloids from the trunk bark of Unonopsis rufescens, such as pallidine (1), a morphinadienone alkaloid previously reported as the major alkaloid also in the closely related species U. floribunda, whose trunk bark is widely used in traditional medicine. Therefore, in addition to expanding the knowledge of this yet underexplored species, this study also supports future biological investigations. Furthermore, the observation of a mixed fragmentation pathway for isopiline contributes to the research on the fragmentation patterns of isoquinoline-derived alkaloids. Thus, this study not only contributes to the chemical knowledge of U. rufescens but also presents a useful strategy for the dereplication of alkaloids widely distributed in the Annonaceae family.
SUPPLEMENTARY MATERIAL Complementary material for this work is available at http://quimicanova.sbq.org.br/, as a PDF file, with free access.
DATA AVAILABILITY STATEMENT All data are avaliable in the text.
ACKNOWLEDGMENTS We would like to thank Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação de Amparo à Pesquisa do Estado do Amazonas (FAPEAM), Financiadora de Estudos e Projetos (FINEP), and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) (Finance Code 001) for financial support.
AUTHOR CONTRIBUTIONS LAFA was responsible for investigation, formal analysis, data curation, methodology, and writing the original draft; BRL, ABBS, FASF, BRCL, APAC, and RSF for formal analysis, data curation; HHFK and ADLS for data curation, and writing the original draft; FMAS and MLBP for conceptualization, data curation, funding acquisition, project administration, resources, and writing the original draft.
REFERENCES 1. Shang, X.-F.; Yang, C.-J.; Morris ‐ Natschke, S. L.; Li, J.-C.; Yin, X.-D.; Liu, Y.-Q.; Guo, X.; Peng, J.-W.; Goto, M.; Zhang, J.-Y.; Lee, K. H.; Med. Res. Rev. 2020, 40, 2212. [Crossref] 2. Plazas, E.; Avila M, M. C.; Muñoz, D. R.; Cuca S, L. E.; Pharmacol. Res. 2022, 177, 106126. [Crossref] 3. Guinaudeau, H.; Lebœuf, M.; Cavé, A.; J. Nat. Prod. 1994, 57, 1033. [Crossref] 4. Lima, J. M.; Leme, G. M.; Costa, E. V.; Cass, Q. B.; J. Chromatogr. B 2021, 1164, 122493. [Crossref] 5. Dantas, E. P.; Monteiro, J.; de Medeiros, L. S.; Romanelli, M. M.; Amaral, M.; Tempone, A. G.; Lago, J. H. G.; Soares, M. G.; Sartorelli, P.; J. Braz. Chem. Soc. 2020, 31, 1908. [Crossref] 6. Justino, A. B.; Florentino, R. M.; França, A.; Filho, A. C. M. L.; Franco, R. R.; Saraiva, A. L.; Fonseca, M. C.; Leite, M. F.; Espindola, F. S.; PLoS One 2021, 16, e0250394. [Crossref] 7. de Lima, B. R.; da Silva, F. M. A.; Soares, E. R.; de Almeida, R. A.; da Silva-Filho, F. A.; Barison, A.; Costa, E. V.; Koolen, H. H. F.; de Souza, A. D. L.; Pinheiro, M. L. B.; J. Braz. Chem. Soc. 2020, 31, 79. [Crossref] 8. Paz, W. H. P.; de Oliveira, R. N.; Heerdt, G.; Angolini, C. F. F.; de Medeiros, L. S.; Silva, V. R.; Santos, L. S.; Soares, M. B. P.; Bezerra, D. P.; Morgon, N. H.; Almeida, J. R. G. S.; da Silva, F. M. A.; Costa, E. V.; Koolen, H. H. F.; J. Nat. Prod. 2019, 82, 2220. [Crossref] 9. da Silva, F. M. A.; Koolen, H. H. F.; de Almeida, R. A.; de Souza, A. D. L.; Pinheiro, M. L. B.; Costa, E. V.; Quim. Nova 2012, 35, 944. [Crossref] 10. da Silva, F. M. A.; de Souza, A. D. L.; Koolen, H. H. F.; Barison, A.; Vendramin, M. E.; Costa, E. V.; Ferreira, A. G.; Pinheiro, M. L. B.; Phytochem. Anal. 2014, 25, 45. [Crossref] 11. Soares, E. R.; da Silva, F. M. A.; de Almeida, R. A.; de Lima, B. R.; da Silva Filho, F. A.; Barison, A.; Koolen, H. H. F.; Pinheiro, M. L. B.; de Souza, A. D. L.; Phytochem. Anal. 2015, 26, 339. [Crossref] 12. Silva, F. M. A.; Silva Filho, F. A.; Lima, B. R.; Almeida, R. A.; Soares, E. R.; Koolen, H. H. F.; Souza, A. D. L.; Pinheiro, M. L. B.; J. Braz. Chem. Soc. 2016, 27, 599. [Crossref] 13. Albert, A.; Engelhard, C.; Anal. Chem. 2012, 84, 10657. [Crossref] 14. Lúcio, A. S. S. C.; Almeida, J. R. G. S.; da-Cunha, E. V. L.; Tavares, J. F.; Barbosa Filho, J. M.; The Alkaloids: Chemistry and Biology 2015, 74, 233. [Crossref] 15. Maas, P. J. M.; Westra, L. Y. Th.; Vermeer, M.; Blumea - Biodiversity, Evolution and Biogeography of Plants 2007, 52, 413. [Crossref] 16. https://floradobrasil2015.jbrj.gov.br/FB110538, accessed in June 2025. 17. Jovel, E. M.; Cabanillas, J.; Towers, G. H. N.; J. Ethnopharmacol. 1996, 53, 149. [Crossref] 18. Kvist, L. P.; Christensen, S. B.; Rasmussen, H. B.; Mejia, K.; Gonzalez, A.; J. Ethnopharmacol. 2006, 106, 390. [Crossref] 19. Adams, M.; Gmünder, F.; Hamburger, M.; J. Ethnopharmacol. 2007, 113, 363. [Crossref] 20. da Silva, F. M. A.; de Lima, B. R.; Soares, E. R.; de Almeida, R. A.; da Silva Filho, F. A.; Corrêa, W. R.; Salvador, M. J.; de Souza, A. Q. L.; Koolen, H. H. F.; de Souza, A. D. L.; Pinheiro, M. L. B.; Rev. Bras. Farmacogn. 2015, 25, 11. [Crossref] 21. da Silva, F. M. A.; Koolen, H. H. F.; Pereira, J. L. S.; Flach, A.; da Costa, L. A. M. A.; de Souza, A. D. L.; Pinheiro, M. L. B.; Rec. Nat. Prod. 2015, 9, 530. [Link] accessed in June 2025 22. da Silva, F. M. A.; da Silva Filho, F. A.; de Souza, C. A. S.; Maciel, J. B.; Costa, E. V.; Barison, A.; Koolen, H. H. F.; de Souza, A. D. L.; Pinheiro, M. L. B.; Biochem. Syst. Ecol. 2018, 79, 12. [Crossref] 23. Zhang, L.; Shi, J.; Tang, J.; Cheng, Z.; Lu, X.; Kong, Y.; Wu, T.; Anal. Chim. Acta 2017, 967, 52. [Crossref] 24. da Silva, F. M. A.; Bataglion, G. A.; de Almeida, R. A.; Heerdt, G.; Sousa, I. L.; da Silva Filho, F. A.; de Alencar, D. C.; Costa, E. V.; de Souza, A. D. L.; Pinheiro, M. L. B.; Morgon, N. H.; Koolen, H. H.; Int. J. Mass Spectrom. 2017, 418, 30. [Crossref] 25. da Silva, F. M. A.; da Silva-Filho, F. A.; de Souza, C. A. S.; Maciel, J. B.; Costa, E. V.; Barison, A.; Koolen, H. H. F.; de Souza, A. D. L.; Pinheiro, M. L. B.; Biochem. Syst. Ecol. 2018, 79, 69. [Crossref] 26. Demarque, D. P.; Crotti, A. E. M.; Vessecchi, R.; Lopes, J. L. C.; Lopes, N. P.; Nat. Prod. Rep. 2016, 33, 432. [Crossref]
Guest Editor handled this article: Antonio E. M. Crotti |

On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access