
 |
 |
 |
 |
 |
Nota Técnica
| Collision-induced dissociation in ESI-MS/MS of caramboxin isolated from Averrhoa carambola: computational and mass spectrometry studies |
|
Ricardo VessecchiI I. Departamento de Química, Universidade de São Paulo, Av. Bandeirantes, 3900, 14040-901 Ribeirão Preto - SP, Brasil Received: 02/20/2025 *e-mail: npelopes@fcfrp.usp.br Star fruit belongs to the Oxalidaceae family, originally from Asia and widely distributed worldwide as a food source. Traditional knowledge advises patients with kidney problems to avoid consuming this fruit. Bioguided studies have led to the isolation of a toxin with a structural core analogous to the amino acids phenylalanine and tyrosine. Mass spectrometry (MS) is the primary technique used for analyzing biological samples, as understanding the gas-phase decomposition reactions of target analytes enhances the efficiency of these analyses. In this study, we detail the mechanisms involved in the formation of the main ion fragments of caramboxin in electrospray ionization tandem mass spectrometry (ESI-MS/MS), supported by computational chemistry and mass spectral simulations. INTRODUCTION Star fruit, known in Brazil as carambola, belongs to the Oxalidaceae family and originally from Asia, and also it has a wide worldwide distribution as food. Its starry shape and range of flavors from acidic to sweet make it popular in tropical regions.1-4 However, since the 1990s, reports4 have indicated that consuming this fruit can cause toxic effects in patients with chronic kidney disease (CKD). Until relatively recently, the exact cause of these effects was unknown. Later studies5 revealed that the neurotoxicity is not only due to high levels of oxalate but also a toxin with a nucleus similar to the amino acids phenylalanine and tyrosine. This natural toxin, called caramboxin (Figure 1), is a non-proteinogenic amino acid derived from phenylalanine and acts as an agonist of N-methyl-D-aspartate (NMDA) and aminomethylphosphonic acid (AMPA) glutamatergic receptors, is mainly responsible for the toxic effects, inhibiting the gamma-aminobutyric acid (GABAergic) system in the central nervous system (CNS), which can lead to symptoms such as hiccups, mental confusion, seizures, and even death, after consuming star fruit, as described by Garcia-Cairasco et al.5
Metabolic profiling of Averrhoa carambola have been done in specific studies6 utilizing liquid chromatography-tandem mass spectrometry (LC-MS/MS) for the analysis of metabolites. Surely, LC-MS/MS is a powerful analytical technique widely used in the identification and quantification of various compounds, including natural toxins.7 Its application could be beneficial for future research on caramboxin, particularly in understanding its metabolism and distribution within biological systems using MS/MS techniques.8 Additionally, no studies have been conducted on the metabolism, pharmacokinetics, or biosynthetic pathway of this important secondary metabolite. One of the main strategies for such studies is liquid chromatography coupled with electrospray ionization mass spectrometry (LC-ESI-MS/MS), as charge transfer is a highly efficient process that enables the analysis of medium to high polarity substances. This technique is a powerful tool for both the identification and analysis of a wide range of substances or metabolomes and for the quantification of target molecules. Given the capabilities of LC-MS/MS in analyzing complex mixtures and detecting low-abundance compounds, it is promissing for future studies aimed at elucidating the metabolic pathways and potentials of caramboxin. This aspect is fundamental in the structural determination of phase 1 metabolites and biosynthetic pathways, as well as pharmacokinetic protocols. An important step in studies involving sequential mass spectrometry (MS/MS) analyzers is defining the gas-phase ion chemistry involved in analyte fragmentation reactions.9-11 This understanding, achieved through energy-resolved plots,12 helps determine which fragments are most stable at a given collision energy, enabling the development of the optimal quantification protocol.13 Thus, understanding the fragmentation mechanisms in ESI-MS/MS after collisional activation should be a key focus of research aimed at applying analytical methodologies to investigate specific biological responses.11,14,15 Knowledge of these processes opens new perspectives for identifying potential metabolites and understanding the simultaneous effects of caramboxin present in star fruit species within a biological environment.
EXPERIMENTAL Chemicals High-performance liquid chromatography (HPLC)-grade methanol and analytical-grade acetic acid were purchased from J.T. Baker (Phillipsburg, USA), and the water was distilled and purified using a Millipore Milli-Q Plus system (Bedford, USA). Caramboxin was obtained as reported by Garcia-Cairasco et al.5 Mass spectrometry analysis ESI-MS/MS analyses were performed using both low-resolution and high-resolution instruments. Low-resolution experiments were conducted on an AB API 3200 mass spectrometer (Sciex, Bremen, Germany) equipped with a quadrupole analyzer (QqQ). All analyses were carried out in positive ion mode, with the capillary voltage set at 5.5 kV. Fragmentation experiments were performed on the protonated molecule [M + H]+ using collision-induced dissociation (CID), with collision energies ranging from 0 to 50 eV. For high-resolution analysis, a timsTOF Flex MALDI-2 mass spectrometer was used, operating with an ESI source and collision energies ranging from 5 to 30 eV. Computational studies Computational studies were performed using the Gaussian 09 and Gaussview (version 4.1, 2000-2006) programs.16 Equilibrium geometries and harmonic vibrational frequencies were calculated at the B3LYP/6-311++G(d,p) level.17,18 Recently, a benchmark study by Bhatia19 reaffirmed that the B3LYP method is the most suitable for calculating proton affinity (PAs) in compounds containing nitrogen, oxygen, and carbon. Our choice for B3LYP was based on previous studies20,21 with caramboxin (CBX), which have been demonstrated good results for geometric analysis of this natural compound. The most stable conformers of CBX were previously described by using B3LYP/6-31+G(d,p). Based on these findings, our study began with the reoptimization of the two most stable conformers described by Pichierri21 at the B3LYP/6-311++G(d,p) level. The search for the most reactive protonation site was conducted by analyzing atomic polar tensor (APT) and Merz-Singh-Kollman (MK) atomic charges22 and calculating proton affinity (PA) and gas-phase basicities (GB) values for the possible sites.23,24 PA and GB values were calculated using the B3LYP method, following benchmark studies by Vessecchi and Galembeck,25 as well as other studies26-28 involving natural and synthetic products. PA and BG were calculated through a protonation reaction (M + H+ → MH+) by computing the enthalpies and Gibbs energies at 298.15 K, respectively from this reaction.29 Mass spectra were predicted using the web server CFM-ID 4.030-32 and compared with those obtained experimentally via ESI-MS/MS. Fragmentation mechanisms were proposed based on the identified reactivity sites, and the relative energies of the main fragment ions were calculated at the B3LYP/6-311++G(d,p) level.
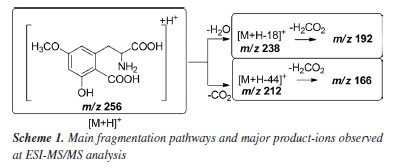
RESULTS AND DISCUSSIONS Mass spectrometry analysis of caramboxin reveals important aspects of the dissociation of protonated molecules. The loss of 18 u was observed as an in-source dissociation event from protonated molecule, [M + H]+, with m/z 256. Additionally, the product ion at m/z 238, [M + H-18]+, was detected in all MS/MS spectra, as shown in Figure 2. The significant intensity of the 18 u elimination observed without applied collision energy supports the data previously reported by Garcia-Cairasco et al.5 In that study, the authors discussed that the ring closure leading to the formation of the m/z 238 ion, [M + H-18]+, is facilitated and may occur through an irreversible process. They stated that, once the lactam ring is formed, the product loses its toxic activity, which explains the absence of the toxin in processed samples. Furthermore, other fragment ions were observed during high-resolution analysis, including m/z 212, 192, and 166, which motivated us to conduct the present study.
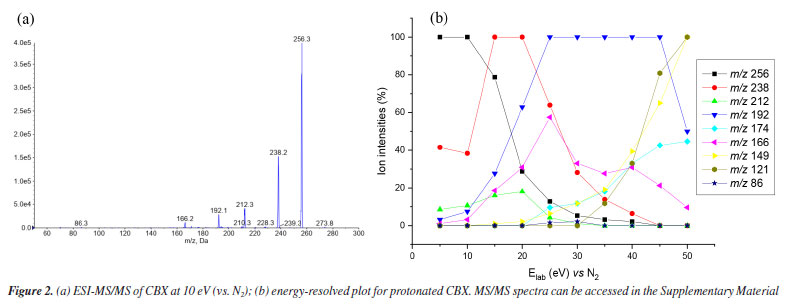
Low-resolution analysis using a QqQ analyzer yielded the same product ions previously described by Garcia-Cairasco et al.5 In this study, experiments were conducted at higher collision energies (Figure 3), as shown in the energy-resolved plot (Figure 2b). A complete collection of spectra is available in the Supplementary Material. Once again, the same product ions were observed in the mass spectra. Additionally, other product ions were detected at low intensity, appearing at higher collision energies (up to 30 eV). In high-resolution mass spectrometry (HRMS) analysis using the MALDI-2 timsTOF equipment, the spectra recorded between 5-30 eV exhibited a similar pattern, with high mass accuracy (see Figure 2S in the Supplementary Material).
A summary of the main product ions is depicted in Scheme 1. Two alternative pathways for the fragmentation of [M + H]+ can be observed: one involving the elimination of 18 u and the other via direct loss of CO2. The high occurrence of [M + H-18]+ in the MS/MS spectra suggests that the elimination of H2O is a favored process.
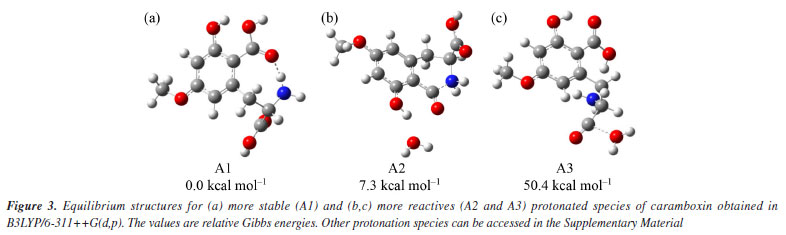
For this reason, we will focus on discussing the formation of the main product ions, which can result from the neutral elimination of H2O, CO2, and H2CO2. In the gas-phase, this reaction was confirmed being highly favored, further corroborating the findings previously discussed. Predicted mass spectra were carried out in competitive fragmentation model, CFM-ID,31 with the objective of testing this methodology and verifying the possibility of this tool to be used contributing to ESI-MS/MS studies. Simulated spectrum at 10 eV versus N2, as suggested by neural network of CFM-ID, shows similar ions observed at experimental spectrum in ESI-MS/MS. The suggested product-ions are: m/z 256, 238, 210, 169, and 167. Only the fragment-ion of m/z 238 was observed using this model, when compared to experimental ones (see Figure 2). On the other hand, the suggested m/z 210 and 167 have similar ions in experimental spectra, m/z 212 and 166, respectively (see Supplementary Material). To explore the fragmentation mechanisms and the possible eliminations of H2O and CO2 from the protonated molecule, we calculated key descriptors to better understand the behavior of this analyte during electrospray ionization and the formation of major ions in the mass spectra. Analyzing the APT atomic charges (Figure 3S, Supplementary Material), we identified the oxygen atoms of the methoxy and hydroxyl groups as the most nucleophilic sites. Although protonation at the methoxy group may occur, no evidence was found in the mass spectra, as no fragment ions of the [M + H-32]+ type were observed (see Figure 2). However, these protonation sites were considered in the reactivity analysis by evaluating relative energy values. The most stable protonation sites are described when proton takes place at carboxylate (aromatic), with the proton migration for N atom. This result is due to the hydrogen bond between the N atom and C=O (see structure A1 in Figure 3a). This bond is also observed in other protonated forms of CBX, which is an indication of ring cyclization, as previously justified by the authors who isolated this natural product.5 For the fragmentation studies, the most stable ions (highest PA value) and the most reactive ions (lowest PA value) were obtained using quantum chemical calculations. Protonation at carbonyl sites led to proton migration to other sites during optimization. Here, we have considered two approaches: one based on more reactive ions and dissociative protonations,33 and another following the recommendations of studies carried out by Bouchoux,34 where proton migration was a possible hypothesis. Thus, our initial discussion of dissociation mechanisms will follow these two possibilities. More recently, Lee et al.35 demonstrated the impact of protonation on CID spectra, showing that protonation sites can influence fragmentation pathways. Therefore, we will discuss the fragmentation of [CBX + H]+ from this perspective. The protonations at -OH provide an ion-neutral complex, with the direct elimination of H2O during geometry optimization (see structures A2 and A3, Figures 3b and 3c, respectively). Thus, the elimination of H2O observed in the MS/MS spectrum can be evidenced from the formation of these ions, when proton take place on OH, what can indicate the in-source elimination of 18 Da and formation of m/z 238. However, it is necessary to approach the intensity of this ion-fragment, since these more reactive protonation sites (dissociative protonation) may occur in a smaller proportion when compared to other sites of reactivity to the proton accommodation. From these results, the fragmentation mechanisms were proposed and the relative Gibbs energies and enthalpies for pathways were calculated from these species (Figure 3 and Scheme 2).
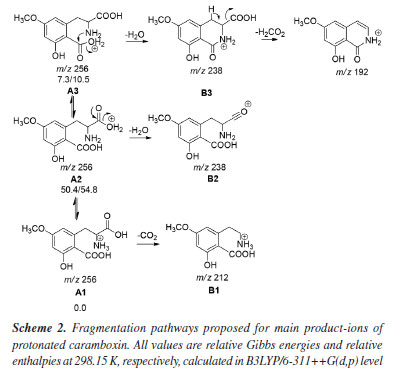
Formation of the m/z 238, which is the most important fragment-ion observed in the MS/MS spectra was proposed from two alternative ways: by protonation in carboxylate from amide bond, which generates an ion-neutral complex; or via protonation on carboxilate at aromatic ring. Analyzing Figure 3 and Scheme 2, it is possible to conclude that the formation of the m/z 192 depends on the dissociation of the m/z 238. Thus, the proposed fragmentation pathway for the formation of the m/z 192 ion takes into account previously published5 mechanisms, where potential cyclization and lactam formation should be considered. Therefore, identifying the possible protonation site as the starting point of fragmentation must also account for the potential fragments generated from this precursor ion.33,36,37 Another relevant point to discuss is that, unlike most protonated substances with methoxy substituents on the aromatic ring,38-40 the radical elimination of 15 u from [M + H]+ does not occur. The unique radical fragment observed (m/z 177) appeared at highest collision energies above 35 eV, in low-resolution studies at QqQ, see energy-resolved plot, Figure 2, and ESI-MS/MS spectra in Supplementary Material. These findings are consistent with those reported by Garcia-Cairasco et al.,5 who observed that the presence of two carboxylic acid groups promotes the rapid elimination of CO2 and H2O, thereby reducing the internal energy of the ion and the likelihood of this pathway occurring.
CONCLUSIONS The findings of this study provide a comprehensive understanding of the gas-phase dissociation mechanisms of caramboxin, particularly the favored elimination pathways of CO2 and H2O. The detailed characterization of the m/z 238 ion, along with other key fragment ions, not only corroborates previous reports on the irreversible lactam formation but also highlights crucial fragmentation routes. These insights have a direct impact on future spatial imaging and pharmacokinetic studies, as they offer a deeper understanding of the structural stability and ionization behavior of caramboxin. By elucidating its fragmentation patterns, this work paves the way for improved analytical approaches in mass spectrometry-based imaging, enabling precise localization of caramboxin in biological tissues. Furthermore, the identification of its major ions and their formation mechanisms will enhance pharmacokinetic studies by refining detection strategies and contributing to a more accurate assessment of its bioavailability and metabolism.
SUPPLEMENTARY MATERIAL Complementary material for this work is available at http://quimicanova.sbq.org.br/, as a PDF file, with free access.
DATA AVAILABILITY STATEMENT All data are available in the text and Supplementary Material. Other data can be requested from the corresponding author.
ACKNOWLEDGMENTS The authors thank Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, grants 2020/02207-5 and 2022/11454-1) for financial support, Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
REFERENCES 1. de Araújo, P. S. R.; Minami, K.; Sci. Agric. 2001, 58, 91. [Crossref] 2. de Oliveira, M. T. R.; Berbert, P. A.; Pereira, R. C.; Vieira, H. D.; Carlesso, V. O.; Revista Brasileira de Sementes 2011, 33, 251. [Crossref] 3. Prati, P.; Nogueira, J. N.; Dias, C. T. S.; Boletim do Centro Pesquisa de Processamento de Alimentos 2002, 20, 221. [Crossref] 4. Muthu, N.; Lee, S. Y.; Phua, K. K.; Bhore, S. J.; Bioinformation 2016, 12, 420. [Crossref] 5. Garcia-Cairasco, N.; Moyses-Neto, M.; Del Vecchio, F.; Oliveira, J. A. C.; dos Santos, F. L.; Castro, O. W.; Arisi, G. M.; Dantas, M.; Carolino, R. O. G.; Coutinho-Netto, J.; Dagostin, A. L. A.; Rodrigues, M. C. A.; Leão, R. M.; Quintiliano, S. A. P.; Silva, L. F.; Gobbo-Neto, L.; Lopes, N. P.; Angew. Chem. 2013, 52, 13067. [Crossref] 6. Biswal, R. P.; Patnana, D. P.; Vutukuri, V. N. R. K.; Anal. Chem. Lett. 2022, 12, 505. [Crossref] 7. Cubbon, S.; Antonio, C.; Wilson, J.; Thomas-Oates, J.; Mass Spectrom. Rev. 2010, 29, 671. [Crossref] 8. Schmid, R.; Petras, D.; Nothias, L. F.; Wang, M.; Aron, A. T.; Jagels, A.; Tsugawa, H.; Rainer, J.; Garcia-Aloy, M.; Dührkop, K.; Korf, A.; Pluskal, T.; Kameník, Z.; Jarmusch, A. K.; Caraballo-Rodríguez, A. M.; Weldon, K. C.; Nothias-Esposito, M.; Aksenov, A. A.; Bauermeister, A.; Albarracin Orio, A.; Grundmann, C. O.; Vargas, F.; Koester, I.; Gauglitz, J. M.; Gentry, E. C.; Hövelmann, Y.; Kalinina, S. A.; Pendergraft, M. A.; Panitchpakdi, M.; Tehan, R.; Le Gouellec, A.; Aleti, G.; Mannochio Russo, H.; Arndt, B.; Hübner, F.; Hayen, H.; Zhi, H.; Raffatellu, M.; Prather, K. A.; Aluwihare, L. I.; Böcker, S.; McPhail, K. L.; Humpf, H. U.; Karst, U.; Dorrestein, P. C.; Nat. Commun. 2021, 12, 3832. [Crossref] 9. Bouchoux, G.; Eckert-Maksić, M.; Mass Spectrom. Rev. 2018, 37, 139. [Crossref] 10. Amad, M. H.; Cech, N. B.; Jackson, G. S.; Enke, C. G.; J. Mass Spectrom. 2000, 35, 784. [Crossref] 11. Demarque, D. P.; Crotti, A. E. M.; Vessecchi, R.; Lopes, J. L. C.; Lopes, N. P.; Nat. Prod. Rep. 2016, 33, 432. [Crossref] 12. Harrison, A. G.; J. Mass Spectrom. 1999, 34, 1253. [Crossref] 13. Borges, L. S.; Batista, J. H. C.; Bozzini, L.; Lourenço, C. D.; Lopes, N. P.; Clososki, G. C.; Vessecchi, R.; Rapid Commun. Mass Spectrom. 2023, 37, 1. [Crossref] 14. Kind, T.; Fiehn, O.; Bioanalytical Reviews 2010, 2, 23. [Crossref] 15. Holcapek, M.; Jirásko, R.; Lísa, M.; J. Chromatogr. A 2010, 1217, 3908. [Crossref] 16. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A. V.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H. P.; Ortiz, J. V.; Izmaylov, A. F.; Sonnenberg, J. L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery Jr., J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M. J.; Heyd, J. J.; Brothers, E. N.; Kudin, K. N.; Staroverov, V. N.; Keith, T. A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A. P.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Millam, J. M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Farkas, O.; Foresman, J. B.; Fox, D. J.; Gaussian 09, revision D.01; Gaussian, Inc., Wallingford, CT, 2013. 17. Becke, A. D.; Phys. Rev. 1988, 38, 3098. [Crossref] 18. Lee, C.; Yang, W.; Parr, R. G.; Phys. Rev. B 1988, 37, 785. [Crossref] 19. Bhatia, M.; Comput. Theor. Chem. 2023, 1223, 114101. [Crossref] 20. Gobato, R.; Parana Journal of Science and Education 2017, 3, 1. [Link] accessed in June 2025 21. Pichierri, F.; J. Mol. Struct. 2015, 1079, 274. [Crossref] 22. Singh, U. C.; Kollman, P. A.; J. Comput. Chem. 1984, 5, 129. [Crossref] 23. Deakyne, C. A.; Int. J. Mass Spectrom. 2003, 227, 601. [Crossref] 24. Range, K.; Riccardi, D.; Cui, Q.; Elstner, M.; York, D. M.; Phys. Chem. Chem. Phys. 2005, 7, 3070. [Crossref] 25. Vessecchi, R.; Galembeck, S. E.; J. Phys. Chem. A 2008, 112, 4060. [Crossref] 26. Cardozo, K. H. M.; Vessecchi, R.; Carvalho, V. M.; Pinto, E.; Gates, P. J.; Colepicolo, P.; Galembeck, S. E.; Lopes, N. P.; Int. J. Mass Spectrom. 2008, 273, 11. [Crossref] 27. Cardozo, K. H. M.; Vessecchi, R.; Galembeck, S. E.; Guaratini, T.; Gates, P. J.; Pinto, E.; Lopes, N. P.; Colepicolo, P.; J. Braz. Chem. Soc. 2009, 20, 1625. [Crossref] 28. Modesto-Costa, L.; Martinez, S. T.; Pinto, A. C.; Vessecchi, R.; Borges, I.; J. Mass Spectrom. 2018, 53, 934. [Crossref] 29. Berruyer-Penaud, F.; Bouchoux, G.; Payen, O.; Sablier, M.; J. Mass Spectrom. 2004, 39, 613. [Crossref] 30. Wang, F.; Liigand, J.; Tian, S.; Arndt, D.; Greiner, R.; Wishart, D. S.; Anal. Chem. 2021, 93, 11692. [Crossref] 31. Allen, F.; Greiner, R.; Wishart, D.; Metabolomics 2015, 11, 98 [Crossref]; CFM-ID, https://cfmid.wishartlab.com/, accessed in June 2025. 32. Allen, F.; Pon, A.; Wilson, M.; Greiner, R.; Wishart, D.; Nucleic Acids Res. 2014, 42, 94. [Crossref] 33. Tu, Y. P.; J. Org. Chem. 2006, 71, 5482. [Crossref] 34. Bouchoux, G.; J. Mass Spectrom. 2013, 48, 505. [Crossref] 35. Lee, J.; Tantillo, D. J.; Wang, L. P.; Fiehn, O.; J. Chem. Inf. Model. 2024, 64, 7457. [Crossref] 36. Hu, N.; Tu, Y. P.; Liu, Y.; Jiang, K.; Pan, Y.; J. Org. Chem. 2008, 73, 3369. [Crossref] 37. Vessecchi, R.; Borges, L. S.; Emery, F. S.; Lopes, N. P.; Int. J. Mass Spectrom. 2017, 418, 92. [Crossref] 38. Carnevale Neto, F.; Andréo, M. A.; Raftery, D.; Lopes, J. L. C.; Lopes, N. P.; Castro-Gamboa, I.; Maia, B. H. L. N. S.; Costa, E. V.; Vessecchi, R.; Rapid Commun. Mass Spectrom. 2020, 34, e8533. [Crossref] 39. Vieira, T. M.; Orenha, R. P.; Crevelin, E. J.; Furtado, S. S. P.; Vessecchi, R.; Parreira, R. L. T.; Crotti, A. E. M.; Rapid Commun. Mass Spectrom. 2020, 34, e8699. [Crossref] 40. Vessecchi, R.; Julião Zocolo, G.; Rubio Gouvea, D.; Hubner, F.; Cramer, B.; de Marchi, M. R. R.; Humpf, H. U.; Lopes, N. P.; Rapid Commun. Mass Spectrom. 2011, 25, 2020. [Crossref]
|

On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access