
 |
 |
 |
 |
 |
Artigo
| Chemical composition and measuring the distribution of haemanthidine by MALDI-TOF MS imaging from Crinum scabrum leaves |
|
Jennifer B. PardoI I. Departamento de Química, Universidade Federal do Espírito Santo (UFES), 29075-910 Vitória - ES, Brasil Received: 02/06/2025 *e-mail: warley.borges@ufes.br Crinum scabrum Herb., a member of the Amaryllidaceae family, is native to Brazil and known for its diverse alkaloid content and biological potential. Previous studies identified crinamine, 6-hydroxycrinamine, and lycorine in this species. In this study, crude methanolic extracts from leaves and bulbs were prepared, followed by alkaloid enrichment via acid-base extraction. Electrospray ionization-linear trap quadrupole-cross-linking tandem mass spectrometry (ESI-LTQ-XL-MS/MS) analysis annotated crinamine, lycorine, and haemanthidine/6-epi-haemanthidine. Further separation using silica gel column chromatography and 1H nuclear magnetic resonance (NMR) analysis revealed the epimers mixture of haemanthidine/6-epi-haemanthidine (C1/2), crinamine (C3), and trisphaeridine (C4). Due to the high yield of C1/2 (24.4%), matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) analysis was performed on the leaves, showing widespread distribution of ion peaks (m/z 318.144) corresponding to C1/2. These results underscore the significance of haemanthidine and its epimer, suggesting that Crinum scabrum leaves are a promising source of these alkaloids and facilitate further investigation of its potential biological activities. INTRODUCTION Alkaloids, nitrogen-containing natural compounds abundant in the plant kingdom, are renowned for their alkaline properties and critical roles in plant survival and ecological interactions.1,2 These compounds frequently act as defense mechanisms against herbivores, pathogens, and competing flora, often through mechanisms such as toxicity, antifeedant properties, or modulation of symbiotic relationships while also contributing to plants' evolutionary adaptability.3,4 Beyond their ecological significance, alkaloids have profoundly influenced human history and culture, serving as the basis for pharmaceuticals ranging from analgesics (e.g., morphine) to anticancer agents (e.g., vinblastine), traditional medicines, and agrochemicals.5 Notably, certain plant families, such as Papaveraceae, Berberidaceae, Fabaceae, Ranunculaceae and Amaryllidaceae, are recognized as rich sources of structurally diverse alkaloids.6 Amaryllidaceae stands out for its unique isoquinoline-type alkaloids, which exhibit diverse bioactivities, including acetylcholinesterase inhibition,7 antiviral effects,8 and cytotoxicity against tumor cells.9 The Amaryllidaceae family comprises approximately 100 genera and 1600 species, primarily found in warm temperate and tropical environments, especially in Southern Africa, the Andes region of South America, and Mediterranean areas.10 Chemotaxonomically, this family is distinguished by its isoquinoline-type alkaloids, with over 600 compounds reported to date.11-17 These alkaloids exhibit broad biological activities, driving their applications in drug discovery and agricultural pest management.18 Alkaloids derived from plants of the Amaryllidaceae family (AA) are well-documented in the literature for their diverse and significant biological activities, which has prompted an ongoing research into new drug discoveries. For instance, galantamine, a clinically approved Alzheimer's drug,19 and lycorine, a potent antiviral agent,20 highlight the pharmaceutical potential of Amaryllidaceae alkaloids. Despite this, numerous species within the family remain underexplored, including Crinum scabrum Herb. Crinum scabrum, sparsely documented in the literature, inhabits tropical and subtropical Africa, including countries such as Nigeria, Ghana, Cameroon21 and South America, particularly in Brazil (Rio de Janeiro, São Paulo and Espírito Santo states).22 However, its current distribution remains unclear due to the lack of reports about this species. This species is notably understudied, with only two reports in the literature: one isolating lycorine23 (1938) and another identifying crinamine and 6-hydroxycrinamine (1985).24 No contemporary studies have investigated its alkaloid profile or ecological adaptations, leaving a critical gap in understanding its chemical ecology and pharmacological potential. This is particularly surprising given the prominence of related Crinum species in traditional medicine and drug discovery. This study detected haemanthidine (5,10b-ethanophenanthridine alkaloid, HA) as the dominant compound in Crinum scabrum leaves, exhibiting pro-apoptotic,25 cytotoxic,26 antiparasitic27 and anti-inflammatory activities.28 Its ecological role may involve allelopathic interactions and symbiotic relationships with endophytes, which enhance alkaloid production via balanced antagonism.29-33 We introduced matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) imaging to spatially resolve HA distribution in leaf tissues, overcoming the limitations of bulk-extract approaches. This technique bridges a critical gap in Amaryllidaceae metabolomics, offering a model for studying alkaloid localization in undercharacterized species.
EXPERIMENTAL Plant material The leaves and bulbs of Crinum scabrum were collected at Goiandira, Goiás, Brazil (18º03'34.5" S, 48º07'49.8" W) in October 2016 (SisGen register: A012E5E). The plant was identified by Renata Souza de Oliveira and a voucher specimen (VIES 24824) was deposited at the Herbarium (VIES) from Universidade Federal do Espírito Santo (UFES), Vitória, Espírito Santo, Brazil. Extraction and purification The dried and powered leaves and bulbs of C. scabrum were macerated with methanol at room temperature (24 h; three times). Following filtration and evaporation, the crude extract (E.MeOH; 243 g) underwent acid-base extraction. The methanolic extract was solubilized in 2% (v/v) H2SO4 until the pH reached 2. Then, the acid extract was extracted with successive portions of ethyl acetate to provide fraction I. The aqueous layer was alkalinized with NH4OH 25% (v/v) until pH 10 and extracted with n-hexane (fraction II) and ethyl acetate (fraction III), separately. The fraction III was concentrated (11.6 g) and fractionated by silica-gel column chromatography by eluding with a gradient hexane/EtOAc (from 100:0 to 0:100, v/v) to obtain 6 fractions (III1-6). Of these, III2 (4.76 g) was fractionated on a silica-gel column applying gradient elution with hexane/EtOAc (from 100:0 to 0:100, v/v) and EtOAc/MeOH (from 100:0 to 0:100, v/v) to give 8 fractions (III2-F1-8); III2-F5 yielded compound 1/2 (2.8421 g), III2-F6 compound 3 (0.1413 g) and III2-F8 yielded compound 4 (0.0044 g). Characterization of the compounds by ESI-LTQ-XL-MS/MS The E.EtOAC was analyzed by mass spectrometry using an electrospray ionization-linear trap quadrupole-cross-linking tandem mass spectrometry (ESI-LTQ-XL-MS/MS) system (Thermo Scientific). For sample preparation, 10 μL of the extract was diluted in 900 μL of methanol. Data were acquired in a range of m/z 50 to 1000, with a constant infusion flow of 10 μL min-1. The capillary temperature was set at 380 ºC to optimize ionization and stability of the generated ions. MALDI-TOF MS imaging The leaf cuts were made in a cryostat embedded with TissueTek® (Sakura). The thickness of the cuts was 70 µm (-20 ºC). Subsequently, the α-cyano-4-hydroxycinnamic acid (HCCA) matrix, at a concentration of 7 mg mL-1 and diluted in acetonitrile:water (1:1) containing 0.1% TFA (trifluoroacetic acid), was applied to the sections using ImagePrep (Bruker®). The plate, covered with the HCCA matrix, was then inserted into the MALDI-TOF/TOF Ultraflextreme equipment (Bruker Daltonics). The parameters used to acquire the spectra resulting in the formation of the image of interest were as follows: reflector analysis mode, 500 laser shots per spectrum, PIE (pulsed ion extraction) of 130 ns, positive mode of analysis, and a laser frequency of 1000 Hz. For external calibration of the equipment, a quercetin standard was used. Spectra was acquired within the mass range of 0-1000 Da.
RESULTS AND DISCUSSION Chemical Characterization of the compounds by ESI-LTQ-XL-MS/MS Mass spectra were obtained in positive mode, which is more sensitive for alkaloids analysis (Table 1). The data was processed using Mzmine (version 4.4.0) an open-source software developed by an international community led by researchers from the Okinawa Institute of Science and Technology (OIST), Japan, and the Czech Academy of Sciences, and molecules were annotated by comparison with the GNPS online database (Figure 1).
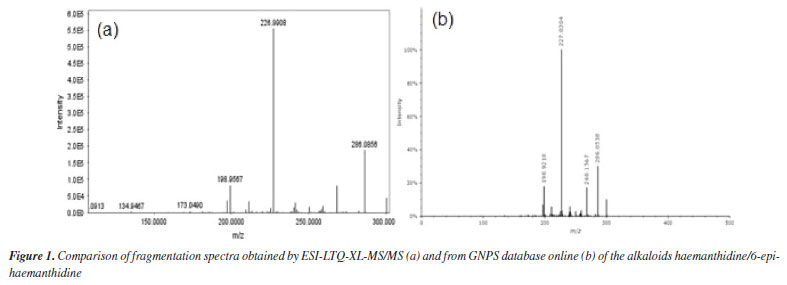
The proposed ESI-MS/MS fragmentation analyses for haemanthidine, crinamine, and lycorine revealed characteristic patterns. In the case of haemanthidine, the precursor ion [M + H]+ was observed at m/z 318, with a main fragment at m/z 227, which is common in Amaryllidaceae alkaloids (AA)34 and suggests the elimination of 59 Da corresponding to a -OCH3 group and a nitrogen-containing bridge, as illustrated in Scheme 1. This fragmentation mechanism highlights the loss of key functional groups associated with the core structure of the alkaloid. For the other analyzed compounds (crinamine and lycorine), the detailed fragmentation pathways are described in the Supplementary Material. Confirmation of isolated compounds Chromatographic purification of the leaves and bulbs of C. scabrum yielded four known alkaloids (Figure 2). Following separation and purification, one-dimensional 1H nuclear magnetic resonance (NMR) spectra were recorded and compared with literature data (see Supplementary Material) to confirm compound identities.
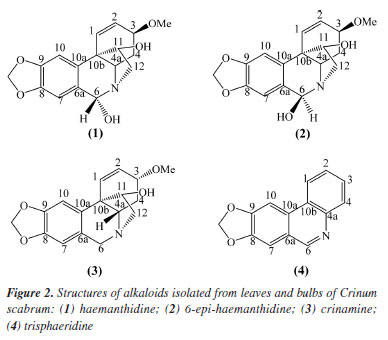
Compounds 1/2 were isolated as a white amorphous solid. The 1H NMR spectrum showed signals for four aromatic protons, indicating a para-orientation characteristic of phenanthridine core. Two singlets at dH 6.98 (1H, s, H-7) and dH 6.82 (1H, s, H-7) corresponded to the C-7 protons of the two epimers, while the C-10 protons resonates as two overlapping singlets at dH 6.77 (1H, s, H-10) and dH 6.75 (1H, s, H-10). These duplications confirmed the presence of two stereoisomers in equilibrium. In the olefin and functional region, the methylenedioxy group (-OCH2O-) protons appeared as two doublets at dH 5.90 (1H, d, J 1.4 Hz) and 5.93 (1H, d, J 1.4 Hz), characteristic of a dioxole ring. Olefinic protons (H1-H2) resonated as a multiplet at dH 6.20-6.25 (4H, m, H-1/2); consistent with the conjugated diene system in the phenanthridine scaffold. A sharp singlet at 3.40 (3H, s, OCH3) confirmed the presence of a methoxy group. For haemanthidine (1), the axial orientation of the C-6 hydroxyl group (6β-OH) was supported by the downfield shift at 5.62 (1H, s, 6β) and the coupling pattern of the C-4 protons: dH 2.17-2.24 (1H, m, 4α) and dH 2.09-2.12 (1H, m, 4β). For 6-epi-haemanthidine (2), the equatorial C-6 hydroxyl (6αOH) resulted in an upfield shift at dH 5.02 (1H, s, 6α) and distinct C-4 proton signals: dH 2.26-2.33 (1H, m, 4α) and dH 2.17-2.24 (1H, m, 4β). Compound 3 was isolated as a white amorphous solid. The 1H NMR spectrum exhibited a phenanthridine-type Amaryllidaceae alkaloid, through the following characteristic signals. Two paraoriented aromatic protons were observed as singlets at dH 6.47 (1H, s, H-7) and dH (1H, s, H-10). This substitution pattern is consistent with the C-7 and C-10 positions of crinamine's aromatic system. Olefinic region showed two hydrogens bonded to sp2 carbon at dH 6.25 (2H, bs, H-1/2), and in functional groups, a characteristic doublet signal of the methylenedioxy at dH 5.89-5.90 (2H, d, OCH2O). Two sets of methine hydrogens dH 3.96-4.02 (1H, m, H-3) and dH 2.08 (1H, m, H-4a), signs of methylene hydrogens at dH 2.04-2.16 (2H, m, H-4), a sharp singlet at dH 3.40 (3H, s, OCH3), indicated a methoxy group. Compound 4 was isolated as a yellow amorphous solid. The 1H NMR spectrum showed a significantly unshielded signal, a sharp singlet at dH 9.11 (1H, s, H-6) arises from the strong electron-withdrawing effect of the adjacent nitrogen atom in the phenanthridine core. The signal is diagnostic for trisphaeridine and similar N-containing heterocycles. Two para-oriented aromatic protons at dH 7.36 (1H, s, H-7) and dH 7.93 (1H, s, H-10) are consistent with the substitution pattern at C-7 and C-10. It was also observed four sets of aromatic protons at dH 7.66 (1H, m, H-2), dH 7.71 (1H, m, H-3), dH 8.39 (1H, d, H-1) and dH 8.21 (1H, d, H-4) and a characteristic signal of the methylenedioxy group at dH 6.19 (2H, s, OCH2O). Distribution of haemanthidine The chemical profiling and spatial distribution analysis of alkaloids in Crinum scabrum leaves revealed HA as the dominant metabolite, aligning with the study's primary aim to elucidate the species' phytochemical potential and ecological adaptations. The high yield of HA (24.4%) underscores its significance as a major secondary metabolite in C. scabrum, contrasting with prior studies on this species,23,24 which reported only lycorine, crinamine, and 6-hydroxycrinamine. This discrepancy highlights a critical advancement: HA's abundance suggests species-specific biosynthesis pathways, potentially linked to environmental pressures or symbiotic interactions in its native habitats. Analyses by MALDI-TOF/TOF The spatial distribution of HA in Crinum scabrum leaf tissues, as mapped by MALDI-TOF MS imaging (Figure 3), supports its proposed role in plant defense. Accumulation in epidermal and vascular tissues aligns with strategies to deter herbivory and pathogen invasion, consistent with findings in Lycoris radiata, where alkaloid localization correlates with biotic stress responses.31,32 However, unlike Lycoris, C. scabrum exhibits HA as the principal alkaloid rather than lycorine, suggesting divergent evolutionary adaptations within Amaryllidaceae. This distinction may reflect niche-specific ecological pressures, such as competition with invasive flora in its tropical habitats.
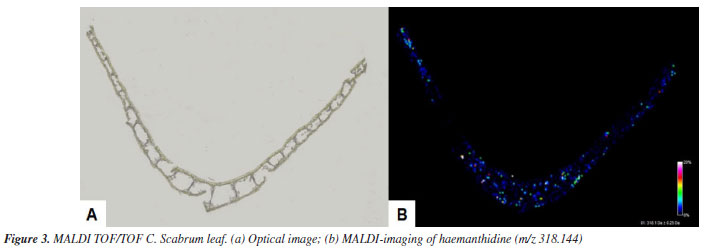
The ion peak at m/z 318.144 [C17H19NO5] was detected throughout the leaf tissue, with intensity gradients (black = absent, white = maximum) indicating HA's broad but heterogeneous accumulation. The experimental mass of the ion peak was compared with literature data.34 This widespread distribution suggests HA may play a systemic role in C. scabrum's ecological interactions, such as deterring herbivores or pathogens across vulnerable tissues, a hypothesis supported by similar alkaloid localization patterns in Amaryllidaceae species. Notably, the HA mass yield in C. scabrum exceeds reported values for other Crinum species, including C. graminicola (mass yield = 0.071%),35 C. variabile (mass yield = 0.057%),36 and Crinum × amabile Donn ex Ker Gawl (mass yield = 0.043%).37 Compared with the mass yield of HA obtained in this study, C. scabrum is a potential HA-producing species. HA's dominance positions C. scabrum as a promising candidate for alkaloid sourcing. HA derivatives are reported to exhibit antiproliferative activity against cancer cell lines38 and antiparasitic effects against Plasmodium falciparum.39 Compared to other Crinum species such as C. asiaticum, rich in crinamine,40,41 or C. jagus, a lycorine producer,42 C. scabrum's HA-rich profile offers a unique chemical resource. This distinction is pharmacologically advantageous, as HA's 5,10b-ethanophenanthridine scaffold is less explored than lycorine's phenanthridine core, opening avenues for structure-activity relationship (SAR) studies.43 While earlier studies on C. scabrum focused on whole-plant extracts,23,24 our tissue-specific approach using MALDI-TOF MS provides unprecedented resolution into HA's biosynthesis and storage, as this technique allows to eliminate matrix interference, facilitating research into the spatial distribution of plant tissues.44 The ecological and pharmacological implications of HA warrant further investigation. For instance, HA's interaction with endophytes in C. scabrum could be probed to assess its role in "balanced antagonism",31 while in vitro assays could validate its bioactivity against neglected tropical disease targets. Additionally, comparative transcriptomics of HA-rich versus HA-poor Crinum species may unravel genetic drivers of alkaloid diversification.
CONCLUSIONS This study addresses the limited chemical characterization of Crinum scabrum by combining phytochemical profiling with MALDI-TOF MS imaging to characterize its major alkaloids and resolve the spatial distribution of haemanthidine (HA) within leaf tissues. Four Amaryllidaceae alkaloids haemanthidine/6-epi-haemanthidine (C1/2), crinamine (C3), and trisphaeridine (C4) were isolated from an alkaloid-enriched extract. Notably, C1/2 exhibited an atypical mass yield, and MALDI-TOF MS imaging revealed the spatial distribution of HA across leaf tissues. By correlating HA's localization with its biosynthetic and ecological roles, we provide critical insights into its adaptive functions in plant defense and resource allocation. Furthermore, this work highlights HA's pharmacological potential, positioning C. scabrum as a promising source for natural product discovery. The novel application of MALDI-TOF MS imaging advances methodologies for studying alkaloid dynamics in undercharacterized plant species.
SUPPLEMENTARY MATERIAL Some general remarks and illustrations regarding compound annotation, as well as comparison with the literature, are available at: http://quimicanova.sbq.org.br, in the form of a PDF file, with free access.
DATA AVAILABILITY STATEMENT All data generated or analyzed during this study are included in this published article (and its Supplementary Material).
ACKNOWLEDGMENTS This study was funded by the Coordenação de Aperfeiçoamento de Pessoal de Nivel Superior - Brasil (CAPES) - Finance Code 001, the National Council of Scientific and Technological Development (CNPq, process 305190/20172 and JE grant, 312505/2021-3) and the Foundation of Support to Research and Innovation of Espirito Santo (FAPES PPE-Agro No. 76418880/16, 76419363/16 and 308/2022). We would also like to acknowledge LabPetro (UFES, Brazil) for performing ESI-LTQ-XL-MS/MS measurements and the NMR analysis. We also acknowledge INCTBioNat (CNPq 465637/2014-0, FAPESP 2015/17177-6, 2014/50926-0, 2017/15840-0, and 2021/10066-5).
REFERENCES 1. Setyorini, D.; Antarlina, S. S.; Food Sci. Technol. 2022, 42, e49822. [Crossref] 2. Ferreira, M.-J. U.; Molecules 2022, 27, 1347. [Crossref] 3. Goyal, S. In Natural Products: Phytochemistry, Botany and Metabolism of Alkaloids, Phenolics and Terpenes; Goyal, S.; Merillón, J., eds.; Springer: Berlin, Heidelberg, 2013, ch. 3. 4. Debnath, B.; Singh, W. S.; Das, M.; Goswami, S.; Singh, M. K.; Maiti, D.; Manna, K.; Mater. Today Chem. 2018, 9, 56. [Crossref] 5. Aniszewski, T. In Alkaloids - Secrets of Life; Aniszewski, T., ed.; Elsevier, 2007, ch. 3. 6. Wink, M. In Encyclopedia of Food and Health; Caballero, B.; Finglas, P.; Toldrá, F., eds.; Academic Press: Heidelberg, 2015, p. 97. 7. Sibanyoni, M. N.; Chaudhary, S. K.; Chen, W.; Adhami, H.-R.; Combrinck, S.; Maharaj, V.; Schuster, D.; Viljoen, A.; Fitoterapia 2020, 146, 104650. [Crossref] 8. Nair, J. J.; van Staden, J.; Phytomedicine 2023, 108, 154480. [Crossref] 9. Doskočil, I.; Hošťálková, A.; Šafratová, M.; Benešová, N.; Havlík, J.; Havelek, R.; Kuneš, J.; Královec, K.; Chlebek, J.; Cahlíková, L.; Phytochem. Lett. 2015, 13, 394. [Crossref] 10. Koutová, D.; Maafi, N.; Havelek, R.; Opletal, L.; Blunden, G.; Řezáčová, M.; Cahlíková, L.; Molecules 2020, 25, 2337. [Crossref] 11. Herbário da Universidade de Coimbra, Amaryllidaceae, https://www.uc.pt/herbario_digital/learn_botany/Familias/Amaryllidaceae, accessed in July 2025. 12. Nair, J. J.; van Staden, J.; S. Afr. J. Bot. 2021, 142, 467. [Crossref] 13. Flora Campestre, Família Amaryllidaceae, https://www.ufrgs.br/floracampestre/familia-amaryllidaceae/, accessed in July 2025. 14. Presley, C. C.; Krai, P.; Dalal, S.; Su, Q.; Cassera, M.; Goetz, M.; Kingston, D. G. I.; Bioorg. Med. Chem. 2016, 24, 5418. [Crossref] 15. Chauhan, N. S.; Sharma, V.; Dixit, V. K.; Thakur, M.; BioMed Res. Int. 2014, 2014, 868062. [Crossref] 16. Nair, J. J.; van Staden, J.; Food Chem. Toxicol. 2013, 62, 262. [Crossref] 17. Cahlíková, L.; Vrabec, R.; Pidaný, F.; Peřinová, R.; Maafi, N.; Al Mamun, A.; Ritomská, A.; Wijaya, V.; Blunden, G.; Molecules 2021, 26, 5240. [Crossref] 18. Berkov, S.; Osorio, E.; Viladomat, F.; Bastida, J.; The Alkaloids: Chemistry and Biology 2020, 83, 113. [Crossref] 19. Burns, A.; Bernabei, R.; Bullock, R.; Jentoft, A. J. C.; Frölich, L.; Hock, C.; Raivio, M.; Triau, E.; Vandewoude, M.; Wimo, A.; Came, E.; Van Baelen, B.; Hammond, G. L.; van Oene, J. C.; Schwalen, S.; Lancet Neurol. 2009, 8, 39. [Crossref] 20. Chen, H.; Lao, Z.; Xu, J.; Li, Z.; Long, H.; Li, D.; Lin, L.; Liu, X.; Yu, L.; Liu, W.; Li, G.; Wu, J.; Virology 2020, 546, 88. [Crossref] 21. Nair, J. J.; van Staden, J.; Bonnet, S. L.; Wilhelm, A.; Nat. Prod. Commun. 2017, 12, 635. [Crossref] 22. Flora do Brasil, Crinum scabrum Herb., https://floradobrasil2020.jbrj.gov.br/reflora/floradobrasil/FB116471, accessed in July 2025. 23. Reichert, B.; Arch. Pharm. Ber. Dtsch. Pharm. Ges. 1938, 276, 328 (CA 32:48797). 24. Jeffs, P. W.; Abou-Donia, A.; Campau, D.; Staiger, D.; J. Org. Chem. 1985, 50, 1732. [Crossref] 25. Havelek, R.; Seifrtova, M.; Kralovec, K.; Bruckova, L.; Cahlikova, L.; Dalecka, M.; Vavrova, J.; Rezacova, M.; Opletal, L.; Bilkova, Z.; Phytomedicine 2014, 21, 479. [Crossref] 26. Hohmann, J.; Forgo, P.; Molnár, J.; Wolfard, K.; Molnár, A.; Thalhammer, T.; Máthé, I.; Sharples, D.; Planta Med. 2002, 68, 454. [Crossref] 27. Şener, B.; Orhan, I.; Satayavivad, J.; Phytother. Res. 2003, 17, 1220. [Crossref] 28. Çitoǧlu, G.; Tanker, M.; Gümüşel, B.; Phytother. Res. 1998, 12, 205. [Crossref] 29. Joosten, L.; van Veen, J. A.; Phytochem. Rev. 2011, 10, 127. [Crossref] 30. Matsuura, H. N.; Fett-Neto, A. G. In Plant Alkaloids: Main Features, Toxicity, and Mechanisms of Action; Carlini, C.; Ligabue-Braun, R., eds.; Springer: Dordrecht, 2017, ch. 2. 31. Liu, Z.; Zhou, J.; Li, Y.; Wen, J.; Wang, R.; Microbiol. Res. 2020, 239, 126501. [Crossref] 32. Zhou, J.; Liu, Z.; Wang, S.; Li, J.; Li, Y.; Chen, W.-K.; Wang, R.; Environ. Microbiol. 2020, 22, 1421. [Crossref] 33. Schulz, B.; Boyle, C.; Mycol. Res. 2005, 109, 661. [Crossref] 34. Torras-Claveria, L.; Berkov, S.; Viladomat, F.; Bastida, J.; S. Afr. J. Bot. 2021, 136, 81. [Crossref] 35. Masi, M.; Mubaiwa, B.; Mabank, T.; Karakoyun, Ç.; Cimmino, A.; Van Otterlo, W. A. L.; Green, I. R.; Evidente, A.; S. Afr. J. Bot. 2018, 118, 188. [Crossref] 36. Steyn, K. H.; de Villiers, A.; van Otterlo, W. A. L.; Green, I. R.; S. Afr. J. Bot. 2022, 146, 503. [Crossref] 37. Chaichompoo, W.; Rojsitthisak, P.; Pabuprapap, W.; Siriwattanasathien, Y.; Yotmanee, P.; Suksamrarn, A.; Phytochemistry 2023, 205, 113473. [Crossref] 38. Luchetti, G.; Johnston, R.; Mathieu, V.; Lefranc, F.; Hayden, K.; Andolfi, A.; Lamoral-Theys, D.; Reisenauer, M. R.; Champion, C.; Pelly, S. C.; vanOtterlo, W. A. L.; Magedov, I. V.; Kiss, R.; Evidente, A.; Rogelj, S.; Kornienko, A.; ChemMedChem 2012, 7, 815. [Crossref] 39. Herrera, M. R.; Machocho, A. K.; Nair, J. J.; Campbell, W. E.; Brun, R.; Viladomat, F.; Codina, C.; Bastida, J.; Fitoterapia 2001, 72, 444. [Crossref] 40. Sun, Q.; Shen, Y.-H.; Tian, J.-M.; Tang, J.; Su, J.; Liu, R.-H.; Li, H.-L.; Xu, X.-K.; Zhang, W.-D.; Chem. Biodiversity 2009, 6, 1751, [Crossref] 41. Arai, M. A.; Akamine, R.; Sadhu, S. K.; Ahmed, F.; Ishibashi, M.; J. Nat. Med. 2015, 69, 538. [Crossref] 42. Jayawardena, T. U.; Merindol, N.; Liyanage, N. S.; Gélinas, S.-E.; Lionel, B.; Seydou, K.; Seck, M.; Evidente, A.; Desgagné-Penix, I.; Heliyon 2025, 11, e42580. [Crossref] 43. Lamoral-Theys, D.; Andolfi, A.; Van Goietsenoven, G.; Cimmino, A.; Le Calvé, B.; Wauthoz, N.; Mégalizzi, V.; Gras, T.; Bruyère, C.; Dubois, J.; Mathieu, V.; Kornienko, A.; Kiss, R.; Evidente, A.; J. Med. Chem. 2009, 52, 6244. [Crossref] 44. Zhan, L.; Huang, X.; Xue, J.; Liu, H.; Xiong, C.; Wang, J.; Nie, Z.; Food Chem. 2021, 338, 127984. [Crossref]
Guest Editor handled this article: Antonio E. M. Crotti |

On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access