
 |
 |
 |
 |
 |
Artigo
|
|
| Detecção de flavonoides com propriedades tripanocida e antioxidante nas folhas de Senna multijuga (Fabaceae) Detection of flavonoids with trypanocidal and antioxidant properties in the leaves of Senna multijuga (Fabaceae) |
|
Cássia G. MagalhãesI,II,* I. Departamento de Química, Universidade Estadual de Ponta Grossa, 84030-900 Ponta Grossa - PR, Brasil Recebido: 14/02/2025 *e-mail: cgmagalhaes@uepg.br Senna multijuga (Fabaceae) is a plant species native from Brazil and it has been used to recover degraded areas. There are a few reports about its chemical composition and biological activities. Therefore, this study aimed to conduct preliminary dereplication studies and evaluate the antioxidant activity by chemical and electrochemical methods of the ethanolic extract of S. multijuga (SM) leaves and its trypanocidal activity. High-performance liquid chromatography-electrospray ionization-ion trap-mass spectrometry (HPLC-ESI-IT-MS) based on molecular networking dereplication of the leaves and branches extracts led to annotation of one caffeoylquinic acid, one flavonol, two flavanones, and seven flavonol glycosides. Some of these metabolites are reported for the first time for this species. The leave extract showed significant radical scavenging properties. On the other hand, SM was more cytotoxic to 3T3 fibroblasts than to Trypanosoma cruzi epimastigotes (selectivity index (SI) = 0.23). Results showed a good agreement between the antioxidant power of extracts and their content of phenolic compounds. INTRODUÇÃO A família Fabaceae engloba 795 gêneros e aproximadamente 20.000 espécies endêmicas ou cultivadas mundialmente, devido ao seu alto valor proteico.1 Senna é um dos mais representativos gêneros da família Fabaceae e suas espécies ocorrem como árvores, arbustos ou subarbustos na Ásia, África e América.2 No território brasileiro, ocorrem nos diferentes biomas, onde são bastante utilizadas em paisagismo3 reflorestamento para a prevenção da desertificação. Espécies deste gênero apresentam uso popular diversificado, com destaque para ação tônica e no tratamento de doenças sexualmente transmissíveis, febre tifoide e malária.4 Vários metabólitos secundários foram isolados de Senna spp., sendo a maioria antraquinonas, flavonoides, terpenos, saponinas, alcaloides, entre outros.5 S. multijuga (Rich.) H.S.Irwin & Barneby (syn. Cassia multijuga Rich; Senna multijuga var. verrucosa (Vogel) H.S.Irwin & Barneby) é usada como planta ornamental, bem como na recuperação de áreas degradadas. No Brasil, a espécie ocorre em regiões de Mata Atlântica.6 Estudos anteriores7 realizados com S. multijuga demonstraram a presença de alcaloides piridínicos 7'-multijuguinol e 8'-multijuguinol com fraca atividade inibidora de acetilcolinesterase in vitro quando comparados com o composto padrão fisostigmina. Até o presente momento, são escassos os estudos relacionados à composição química e potencial bioativo dessa espécie. Atualmente, devido à acelerada perda da biodiversidade vegetal,8 a pesquisa em produtos naturais continua sendo um objeto importante de estudo visando a descoberta de princípios biologicamente ativos com potencial para descoberta de novos fármacos, agroquímicos ou produtos tecnológicos cujo processo de obtenção cause menos impacto ambiental. Tendo em vista que alguns antioxidantes sintéticos apresentam potencial citotóxico e carcinogênico em estudos in vivo,9 a busca por antioxidantes que causem menos efeitos colaterais permanece necessária. Além disso, a continuidade da caracterização química e biológica de espécies pouco estudadas evidencia novos conhecimentos voltados para a busca de produtos naturais já conhecidos, mas ainda com bioatividade pouco estudada. O desenvolvimento de protótipos para doenças negligenciadas, como a doença de Chagas, causada pelo protozoário Trypanosoma cruzi, é ainda um grande desafio devido a carência de medicamentos que causem menos efeitos colaterais e que sejam mais eficazes, sobretudo na fase crônica da doença.10-12 Estudos relatam o potencial anti-T. cruzi de compostos extraídos de S. villosa, tanto em estudos in vitro13,14 quanto in vivo.15 Entretanto, uma das limitações é a dificuldade de obter quantidades suficientes do composto porque o rendimento é muito baixo, em torno de 2,7%.16 Atualmente, métodos modernos de metabolômica, combinando técnicas analíticas eficientes e ferramentas computacionais têm sido empregados no mapeamento químico de extratos, viabilizando o cruzamento de resultados de bioatividade de maneira mais dinâmica. Como exemplo, os resultados de uma triagem antiparasitária e do perfil metabolômico de 1.600 extratos vegetais levou a duas classes de metabólitos principais, rotenoides e quinolonas, que poderiam ser responsáveis pela atividade observada contra formas amastigotas de T. cruzi. Naquele estudo,17 observou-se que os rotenoides rotenona e deguelina, encontrados em extratos de Chadsia grevei, Pachyrhizus erosus eDesmodium heterophylum (Fabaceae) foram ativos. Diante do exposto, o objetivo deste trabalho foi investigar os produtos naturais presentes no extrato etanólico das folhas e galhos de S. multijuga, pelo uso de técnicas de desreplicação facilitadas via redes moleculares baseadas em cromatografia líquida de alta eficiência acoplada a espectrômetro de massas com analisador ion trap e ionização por electrospray (HPLC-ESI-IT-MS), bem como avaliar seu potencial antioxidante e tripanocida in vitro.
PARTE EXPERIMENTAL Material vegetal Folhas e galhos de S. multijuga foram coletados no campus Uvaranas da Universidade Estadual de Ponta Grossa (25º5'23"S 50º6'23"O). A espécie foi identificada pela Profa. Dra. Rosângela Tardivo, depositada no herbário da UEPG (HUPG 21655) e registada no Sistema Nacional de Gestão do Patrimônio Genético e Conhecimento Tradicional Associado (SisGen) sob o número A7481FE. Obtenção dos extratos brutos As folhas (6,0 g) e galhos (20 g) de S. multijuga foram secos e triturados em moinho analítico. O pó obtido (5,25 g de cada amostra) foi levado para extração com 125 mL de etanol grau P.A. em banho ultrassônico (Elmasonic S 60 H) com frequência de 37 kHz a 40 ºC por 45 min. Em seguida, os extratos foram filtrados e submetidos à destilação sob pressão reduzida em um evaporador, em banho a 40 ºC. Pré-purificação dos extratos Os extratos foram submetidos a uma pré-purificação no intuito de se remover o excesso de clorofila. Para isso, 5,0 mg dos extratos brutos provenientes de folhas e galhos foram solubilizados em 3,0 mL de metanol (MeOH) grau HPLC e submetidos à extração em fase sólida em cartucho contendo 500 mg de C18, com diâmetro de partícula de 40 mm, marca Phenomenex. O processo de eluição foi realizado com 3,0 mL de uma mistura de água e 0,1% de ácido fórmico (CH2O2) (solvente A) e uma solução de metanol e 0,1% ácido fórmico (solvente B), variando-se a proporção do solvente B em 0, 95 e 100%. Análise por cromatografia líquida acoplada a um analisador de massas de baixa resolução (LC-ESI-IT-MS) Os extratos obtidos foram analisados por meio de cromatógrafo líquido (Shimadzu®) acoplado a um espectrômetro de massas AmaZon-SL (Bruker Daltonics®). O sistema de cromatografia líquida operante nos seguintes módulos: bombas (LC-20AD), forno (CTO-20A), degaseificador (DGU-20A3R), módulo de comunicação (CBM-20A) e detetor de arranjo de diodos (SPD-M20A). A injeção de amostra (2 μL) foi realizada por meio de um injetor automático (SIL-20A HT). Os solventes utilizados nas análises foram de grau HPLC. Os extratos secos foram solubilizados em MeOH grau HPLC a 1 mg mL-1 e analisados por cromatografia líquida em fase reversa à temperatura de 35 ºC, com uma coluna Phenomenex Luna C18, de dimensões de 250 × 4,6 mm, partículas de 5 μm e poros de 100 Å como fase estacionária, e a fase móvel foi constituída por solução A: H2O (0,10% CH2O2) e solução B: ACN (0,10% CH2O2). A eluição foi realizada com um gradiente exploratório de 5-100% de solvente B com vazão de 1 mL min-1 em 45 min. Os espectros de massas foram adquiridos em modo positivo na faixa de aquisição de m/z 50-1500. Os parâmetros da fonte de ionização por electrospray (ESI) do espectrômetro de massas foram ajustados com voltagem de saída do capilar 140 V, voltagem do capilar de -3,5 kV, e temperatura de dessolvatação 300 ºC. Utilizou-se nitrogênio como gás de secagem e dessolvatação (7 psi, 4 L min-1). Os dados foram processados com o auxílio do software Data Analysis, versão 4.2 (Bruker, EUA, 2013). Processamento dos dados na plataforma Global Natural Product Social Molecular Networking (GNPS) Os dados brutos adquiridos no formato ".d" em modo positivo foram convertidos para ".mzML" utilizando o software Data Analysis, versão 4.2, e utilizados para gerar as redes moleculares. As redes moleculares foram criadas a fim de se verificar o perfil químico dos extratos etanólicos das folhas (SM) e galhos (SG) de S. multijuga, usando o fluxo de trabalho online no site do GNPS.18 Os dados foram filtrados removendo-se todos os íons fragmentos MS/MS dentro de +/-17 Da do precursor m/z. Os espectros de MS/MS foram filtrados por janela, escolhendo-se apenas os 6 principais íons fragmento na janela de +/-50 Da em todo o espectro. A tolerância de massa do íon precursor e dos íons fragmentos MS/MS foram ajustadas para 0,5 Da. Uma rede foi então criada, onde as arestas foram filtradas para se ter um valor de cosseno acima de 0,7 e mais de 6 sinais correspondentes. Finalmente, o tamanho máximo de uma família molecular foi definido como 100. Os espectros foram então pesquisados nas bibliotecas espectrais do GNPS. Todas as correspondências mantidas entre espectros de rede e espectros de biblioteca precisavam ter pontuação acima de 0,7 e pelo menos 6 sinais correspondentes. Os resultados obtidos podem ser acessados na plataforma do GNPS2.19 Atividade tripanocida O extrato etanólico das folhas de S. multijuga (SM) foi solubilizado em dimetilsulfóxido (DMSO) a 100 mg mL-1. Nos ensaios biológicos, a concentração final máxima de DMSO foi de 0,5%. Formas epimastigotas de Trypanosoma cruzi (cepa Y) foram mantidas a 27 ºC em meio LIT (liver infusion tryptose), suplementado com 10% de soro fetal bovino (SFB), através de dois repiques semanais. Todos os testes com epimastigotas foram realizados com parasitos coletados no quarto dia de cultivo. Para os ensaios de atividade tripanocida, 180 μL poço-1 de uma suspensão contendo 5 × 106 epimastigotas de T.cruzi mL-1 foram semeados em placas de 96 poços e incubados com 20 µL de SM nas concentrações de 5, 25, 50 e 100 µg mL-1 por 48 h a 27 ºC. Como controle, utilizou-se DMSO 0,5%. O percentual de inibição sobre o parasita foi estimado em microscópio invertido através da observação da redução da motilidade, mudança na morfologia e integridade dos parasitas, seguido da técnica colorimétrica do MTT (brometo de 3-(4,5-dimetiltiazol-2-il)-2,5-difenil tetrazólio).20,21 Brevemente, a placa foi centrifugada por 5 min a 4000 rpm, o sobrenadante foi descartado e, em seguida, foram adicionados 50 μL da solução 2 mg mL-1 de MTT em cada poço. Após 3 h de incubação a 37 ºC, a placa foi centrifugada a 4000 rpm por 5 min. O sobrenadante foi removido e o sedimento de parasitas foi suspenso em 20 μL de dodecilsulfato de sódio (SDS) 10% em HCl 0,01 M e 80 μL de DMSO por poço para análise dos parasitas e solubilização dos cristais de formazan. Em seguida, a densidade óptica foi determinada a 570 nm em leitor de microplacas, modelo Biotek EL 800. As densidades ópticas foram convertidas em percentual de viabilidade em relação ao controle não tratado e utilizadas para o cálculo da CI50 (concentração inibitória de 50%). Ensaios de citotoxidade e determinação do índice de seletividade (IS) Fibroblastos da linhagem 3T3/A31 (ATCC-CCL163TM) (2 × 104 células poço-1) foram incubados a 37 ºC e 5% de CO2 com diferentes concentrações (25, 50, 100 e 200 µg mL-1) de SM em placas de 96 poços, em um volume final de 200 μL de meio Roswell Park Memorial Institute (RPMI) suplementado com 10% SFB. Após 48 h de incubação, a integridade do tapete celular foi observada em microscópio invertido, seguida do ensaio colorimétrico do MTT.21 Em seguida, a densidade óptica foi determinada em comprimento de onda de 570 nm, utilizando-se leitor de microplacas Biotek EL 800. As densidades ópticas foram convertidas em percentual de viabilidade em relação ao controle não tratado e utilizadas para o cálculo da CC50 (concentração citotóxica de 50%). O índice de seletividade (IS) foi calculado através da relação da CC50/CI50. Valores de IS > 1 são indicativos que o extrato testado é seleletivo contra epimastigotas de T. cruzi, enquanto que IS < 1 indica que o extrato foi mais citotóxico para fibloblastos 3T3 do que ativos para T. cruzi.22 Ensaios de atividade antioxidante Determinação do conteúdo de compostos fenólicos totais (CFT) O conteúdo de compostos fenólicos totais (CFT) foi realizado seguindo o protocolo proposto por Singleton et al.,23 com adaptações em relação ao volume e obedecendo a estequiometria da reação. Preparou-se uma curva de calibração com ácido gálico (100 mg L-1) como padrão. Em uma placa de 96 poços, foram adicionados 100 μL de SM, 100 μL de reagente Folin-Ciocalteu, 10% (v v-1) e 100 μL de solução aquosa de carbonato de sódio (Na2CO3) 7,5% (m v-1). Para a construção da curva analítica, foi realizado o mesmo procedimento porém, adicionando-se ácido gálico, variando-se a concentração de 100 a 10 μg mL-1, no lugar da amostra. Logo após a adição do reagente Folin-Ciocalteu, aguardou-se 5 min e depois adicionou-se a solução de carbonato de sódio. Logo em seguida, a placa foi incubada à temperatura ambiente por aproximadamente 30 min no escuro. Realizou-se a leitura em um espectrofotômetro no comprimento de onda de 765 nm. A partir dos valores de absorbância obtidos, foi calculado o CFT das amostras de acordo com a curva de calibração, sendo o resultado expresso em mg equivalente de ácido gálico por grama de extrato (mg Ag g-1 extrato). Todas as análises foram realizadas em triplicata. A leitura de absorbância foi realizada em uma leitora de placas ELISA Biotek μQuant® (Synergy H1 Hybrid Reader) para esse experimento e para os demais dessa sessão. Determinação do conteúdo de flavonoides totais (FT) A determinação do conteúdo de flavonoides totais (FT) foi realizada de acordo com o protocolo de Hacke et al.24 O extrato hidroalcoólico foi preparado na concentração de 500 μg mL-1 para este ensaio. Foram adicionados 100 μL de SM em microplacas de 96 poços, após a mistura de 100 μL de solução aquosa de AlCl3 (10% m v-1). Esta mistura foi incubada por 30 min e submetida à análise espectroscópica a 415 nm. Uma curva padrão foi realizada com quercetina em uma faixa de concentração de 0 a 100 μg mL-1. Os resultados foram expressos como mg QER (equivalente de quercetina) g-1 de extrato. As análises foram realizadas em triplicata. Atividade de captura do radical DPPH• (2,2-difenil-1-picrilhidrazil) A determinação da captura do radical DPPH• foi realizada conforme descrito por Brand-Williams et al.,25 com modificações. As amostras dos extratos hidroalcoólicos foram inicialmente preparadas como uma solução (1000 μg mL-1) em EtOH e diluídas a 100 μg mL-1. Para determinar a concentração inibitória de 50% (CI50), as concentrações foram variadas de forma decrescente, de 100 a 3,125 μg mL-1 de ácido ascórbico (AA) como controle positivo. Em uma microplaca de 96 poços, foram adicionados, em cada poço, 100 μL de solução de amostra, 80 μL de etanol e 100 μL de solução DPPH• (120 μmol L-1). Para o controle do branco, foram adicionados 180 μL de etanol e 120 μL da solução de amostra. Para o controle negativo, foram adicionados 100 μL de etanol e 100 μL de solução DPPH•. A mistura de reação foi incubada por 30 min no escuro. A absorbância foi medida em um leitor de microplacas no comprimento de onda de 517 nm. A inibição foi calculada empregando-se a Equação 1: 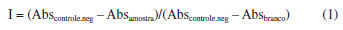 onde a ABSamostra é a absorbância medida das amostras, Abscontrole.neg é a absorbância do controle negativo e Absbranco é a absorbância do branco. O experimento foi realizado em triplicata nos poços das placas (n = 3) e realizadas três repetições. O CI50 foi expresso em μg mL-1. Atividade de captura do radical ABTS•+ Avaliou-se a atividade antioxidante também pela captura do radical ABTS•+ (2,2''-azinobis ácido 3-etilbenzotiazolina-6-sulfônico), baseando-se no protocolo de Xiao et al.,26 com modificações. Para este ensaio, 5 mL de solução estoque de ABTS (7 mM) foram misturados com 88 μL de peroxidisulfato de potássio (140 mM, K2S2O6). Em seguida, a solução foi armazenada à temperatura ambiente com ausência de luz por 16 h antes do uso. A solução de ABTS•+ foi diluída com tampão fosfato pH 7,4 (10 mM) até se obter o valor de absorbância de 0,7, em comprimento de onda de 734 nm. A medição foi realizada no equipamento de leitura de placas ELISA. Para a avaliação, foram adicionados a uma microplaca de 96 poços, em triplicata, 20 μL da amostra em diferentes concentrações (decrescente, variando de 100 a 3,125 μg mL-1). Depois disso, adicionou-se 80 μL de tampão fosfato pH 7,4 (10 mM) e 100 μL de solução de trabalho ABTS•+. Então, a placa foi incubada por 30 min na ausência de luz. O branco foi preparado com 100 μL de amostra e 100 μL de tampão fosfato. O controle positivo foi preparado usando ácido ascórbico no lugar das amostras. A leitura da placa foi realizada em um espectrofotômetro no comprimento de onda de 734 nm. Os resultados foram expressos como CI50 (μg mL-1), como na Equação 1. Avaliação do poder redutor de íons férrico O poder redutor pelo método de poder antioxidante de redução de ferro, do inglês ferric-reducing antioxidant power (FRAP), foi avaliado conforme proposto por Berker et al.27 Preparou-se uma solução de complexo Fe-o-fenantrolina 1,10 × 10-2 mol L-1 a partir de sulfato férrico amoniacal dodecahidratado (NH4Fe(SO4)2.12H2O) dissolvido em ácido clorídrico (1 mol L-1) e misturado com solução de o-fenantrolina. Em uma placa de 96 poços foram adicionados 150 µL do extrato hidroalcoólico (500 µg mL-1) de SM e 150 µL da solução complexo de o-fenantrolina. Incubou-se no escuro por 30 min até finalização da reação. Para o branco, adicionou-se 150 µL de amostra e 150 µL de etanol. O ácido ascórbico foi utilizado como controle positivo e construção da curva analítica. As leituras da absorbância foram medidas no comprimento de onda de 510 nm. O aumento da coloração vermelho-alaranjada indicou a presença do complexo ferroso-fenantrolina (Fe2+-o-fen), avaliado pela medida da absorbância em 510 nm. Os resultados foram expressos em mg AA g-1 e os experimentos foram realizados em triplicada. Avaliação eletroquímica Os estudos eletroquímicos à temperatura ambiente foram realizados utilizando-se um potenciostato/galvanostato Metrohm® Autolab (PGSTAT204), gerenciado pelo software NOVA, versão 2.1.7 (Methrom, Suiça, 2022). As medições foram feitas a partir de um sistema padrão de três eletrodos, onde o eletrodo de carbono vítreo (ECV) atuou como eletrodo de trabalho, um eletrodo Ag/AgCl 3 mol L-1 serviu como referência e um fio de platina funcionou como eletrodo auxiliar. A solução eletrolítica consistiu em um tampão Britton-Robinson (BR) 0,04 mol L-1 em níveis de pH de 2,0, 4,0, 7,0 e 10,0, preparado com 0,04 mol L-1 H3PO4, 0,04 mol L-1 H3BO3 e 0,04 mol L-1 CH3COOH. Ajustes de pH foram feitos usando 0,5 mol L-1 NaOH, conforme necessário. Uma solução de etanol e tampão BR (1:1 v v-1) foi utilizada, incorporando-se os extratos à concentração final de 5 mg mL-1. Voltamogramas cíclicos foram obtidos na faixa de potencial de -0,2 a +1,4 V (vs. Ag/AgCl) a uma velocidade de varredura de 0,1 V s-1. Antes das medições, o ECV foi polido com pasta de alumina de 0,3 μm em uma almofada de polimento, limpa em banho ultrassônico em etanol por 5 min. Análise estatística Os valores de CI50 (concentração inibitória de 50%) foram estimados a partir da triplicata de dois experimentos independentes, utilizando os programas GraphPad Prism Instat3®, versão 3.10 (GraphPad Software Inc., EUA, 2003), e Origin, versão 2022b (OriginLab Corporation, Northampton, EUA, 2022). Os resultados foram submetidos à análise de variância (ANOVA) pelo teste de Tukey e expressos como média e desvio padrão. Os valores de p < 0,05 foram considerados significantes.
RESULTADOS E DISCUSSÃO Desreplicação dos extratos e anotação dos metabólitos A análise das redes moleculares possibilitou o mapeamento químico dos extratos, facilitando o processo de anotação dos metabólitos secundários por meio da observação dos padrões de fragmentação e da correlação com as bibliotecas espectrais. O processamento na plataforma GNPS resultou em 47 correspondências com as bibliotecas espectrais e permitiu a anotação de sete compostos: um flavonol (quercetina), quatro flavonóis O-glicosilados (quercetina-O-glicosídeo, kaempferol-O-rutinosídeo, quercetina-O-rutinosídeo e quercetina-O-malonilglicosídeo) e duas flavanonas (naringenina e eriodictiol) (Figura 1). Todos os dados moleculares foram inspecionados manualmente, a fim de se excluir possíveis anotações presentes no branco (MeOH) e/ou características de compostos sintéticos e/ou também advindas de outros tipos de organismos. Além disso, ao verificar que as fragmentações não eram compatíveis com as bibliotecas espectrais, essas correspondências espectrais foram descartadas.
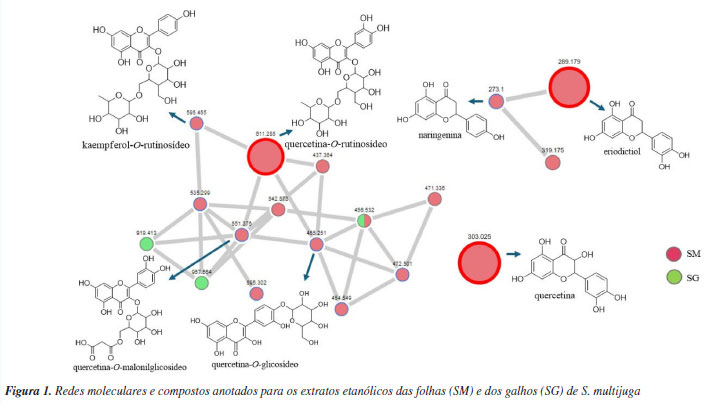
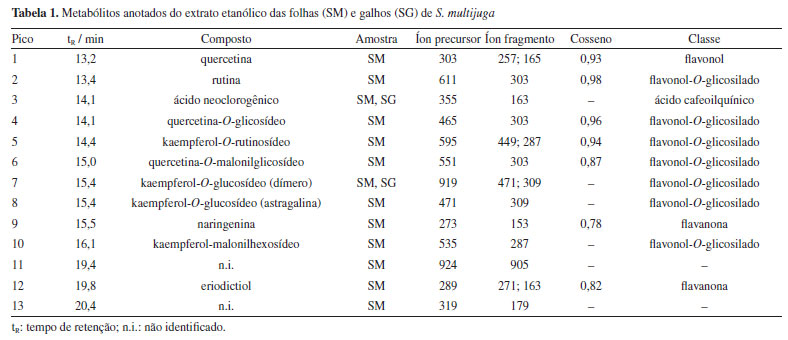
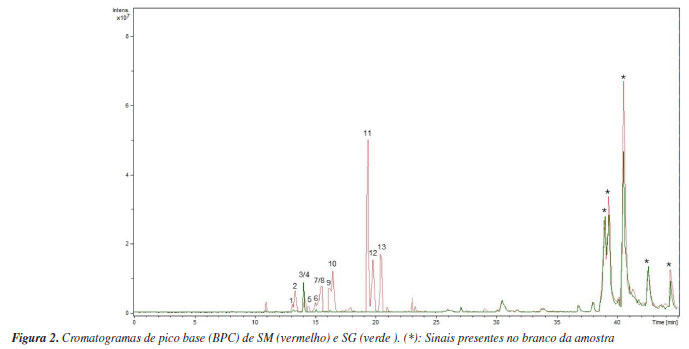
Foram anotados compostos cujo valor de cosseno fosse maior ou igual a 0,7 (Tabela 1), conforme definido nos parâmetros da análise. O parâmetro representa a similaridade espectral de MS/MS entre o espectro da amostra e os espectros presentes na biblioteca. Quanto mais próximo de 1, mais similares são os espectros.28 Foram anotados derivados O-glicosilados de quercetina, caracterizados pela presença do íon fragmento m/z 303, correspondente à sua aglicona. Para o íon precursor de m/z 465, sugere-se o composto quercetina-O-glicosídeo, cuja fragmentação evidencia a perda de uma unidade de hexose (162 Da), indicada pelo íon fragmento de m/z 303. Para a quercetina-O-rutinosídeo (m/z 611), foi observada a perda do dissacarídeo (hexose mais deoxihexose, 308 Da), gerando o íon fragmento m/z 303, característico da aglicona. Essa mesma perda (308 Da) foi verificada para o kaempferol-O-rutinosídeo, gerando então o íon fragmento de m/z 287, confirmando a presença deste flavonoide.29 Para a quercetina-O-malonilglicosídeo, a perda característica de malonilexose (-248 Da) originou o íon fragmento m/z 303, atribuído ao núcleo característico de um flavonol. Adicionalmente, a fragmentação dessa aglicona resultou na formação do íon m/z 257, relativo à perda de água e CO, um padrão típico desses flavonoides. Em seguida, observou-se o sinal de m/z 165, característico da reação retro Diels-Alder, devido à clivagem do anel C da quercetina.30 O íon precursor de m/z 289 foi anotado como eriodictiol, com base em suas fragmentações características, como a formação do íon fragmento de m/z 271 que ocorre devido à perda de água, enquanto o íon fragmento de m/z 163 resulta da retrociclização e clivagem do anel A, um comportamento típico dessa flavanona em espectrometria de massas.31 Para a naringenina (m/z 273), a fragmentação gerou o íon fragmento de m/z 153, que corresponde à perda da unidade C8H8O, relativa ao anel B, conforme descrito na literatura.32 Alguns compostos presentes nas redes moleculares não tiveram correspondência com aqueles disponíveis na plataforma GNPS. No entanto, por se tratarem de metabólitos já reportados na literatura, foi possível atribuir o padrão de fragmentação desses compostos a derivados do kaempferol, como a astragalina, cujo íon precursor [M + Na]+ foi observado em m/z 471. A perda da unidade glicosídica da astragalina gerou o íon fragmento em m/z 309 [M + Na]+. Foi possível atribuir o sinal em m/z 919 [2M + Na]+ ao íon precursor do dímero desse flavonol glicosilado, sendo possível observar os íons framentos em m/z 471, relativo ao monômero, e em m/z 309, atribuído a [M + Na]+ relativo à aglicona.33 O íon precursor de m/z 535 foi anotado como kaempferol-malonilhexosídeo, onde a perda da unidade malonilexose (-248 Da) originou o íon fragmento m/z 287, atribuído ao kaempferol.34 O sinal em m/z 355 [M + H]+ foi atribuído ao íon precursor do ácido neoclorogênico, onde a clivagem do éster leva ao cátion relativo ao ácido quínico em m/z 193.35 Com exceção da rutina, os demais metabólitos são mencionados pela primeira vez para S. multijuga. Na Figura 2 estão representados os cromatogramas de pico base (BPC) de SM e SG. No entanto, para que haja determinação inequívoca da estrutura dos compostos, são necessárias técnicas espectroscópicas e espectrométricas complementares para a caracterização química, como a ressonância magnética nuclear de 1H e 13C. Além disso, tendo em vista que não foi possível determinar a estrutura química relativa a dois sinais intensos observados no BPC de SM, o fracionamento desse extrato, visando elucidar a estrutura dos metabólitos que geraram os respectivos sinais, é importante.
Flavonoides são fitoconstituintes abundantes em espécies do gênero Senna, podendo ser isolados de diferentes partes do vegetal.5 A rutina foi previamente isolada dessa espécie6 e foi o componente majoritário do extrato metanólico das folhas S. italica e S. alexandrina.36 Quercetina-O-glicosídeo e naringenina foram isoladas de S. macranthera e S. siamea, respectivamente.5 Mohamed et al.37 descreveram um efeito radioprotetor da combinação de rutina e naringenina, atribuído ao efeito de sequestro de radicais livres já descritos para essas substâncias. Visando aumentar sua biodisponibilidade, vários estudos demonstram a eficácia de nanoformulações de naringenina em estudos in vivo.38 Tendo em vista que os compostos anotados foram majoritariamente atribuídos ao extrato das folhas (SM), os ensaios de atividade tripanocida e antioxidante foram realizados com essa amostra. Atividade tripanocida e citotóxica SM não apresentou atividade tripanocida in vitro, sendo que as formas epimastigotas de T. cruzi (cepa Y) mantiveram taxas médias de viabilidade acima de 90% (Figura 3). Por outro lado, os fibroblastos da linhagem 3T3 foram mais sensíveis (CC50 = 122,91 µg mL-1), resultando em um índice de seletividade abaixo de 1, o que indica que SM foi mais citotóxico do que ativo contra o parasita (Figura 3). Contrariamente, o extrato hidroalcoólico de S. occidentalis não exibiu citotoxicidade significativa frente a esses fibroblastos, indicando a possibilidade de seu uso em formulações tópicas.39
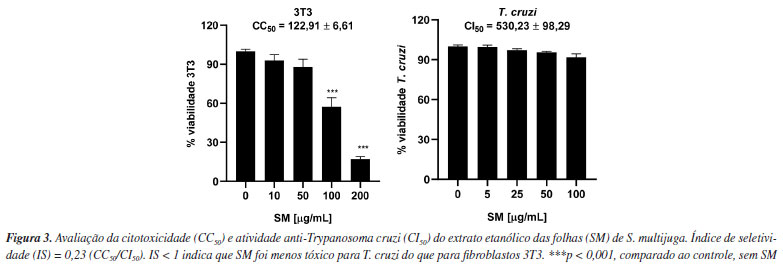
Considerando o potencial tripanocida de outras espécies da família Fabaceae, verificou-se que a biochanina A, uma isoflavona isolada dos frutos de Cassia fistula, exibiu promissora atividade contra T. cruzi (concentração efetiva (CE50) = 18.32 μg mL-1), sendo 24 vezes mais ativa que benzonidazol.40 Essa constatação abre perspectivas para a continuidade da avaliação do potencial tripanocida de extratos obtidos de outras partes de S. multijuga, bem como de outras espécies da família Fabaceae. Em se tratando do efeito tripanocida dos compostos isolados, a astragalina apresentou baixo CI50 (15,7 ± 11,1 μg mL-1) quando comparado ao benzonidazol (15,8 ± 0,9 μg mL-1).41 O kaempferol, por sua vez, exibiu efeito fraco (CI50 23,9 ± 1,6 μg mL-1) frente ao parasita, enquanto seus análogos glicosilados (kaempferol-3-O-glicosídeo e kaempferol-3-O-rutinosídeo) e a quercetina foram inativos.42 Tais evidências indicam que a busca por moléculas ativas contra T. cruzi permanece desafiadora. Essa constatação abre perspectivas para a continuidade da avaliação do potencial tripanocida de extratos obtidos de outras partes de S. multijuga, no entanto, com o uso de solventes de outra natureza, visando obter classes químicas que se apresentassem mais promissoras, como foi observado para o extrato clorofórmico das folhas de S. villosa.13,14 Alguns dos metabólitos anotados no presente estudo já tiveram seu potencial citotóxico avaliado. Demonstrou-se o efeito citotóxico da quercetina sobre fibroblastos 3T3, que foi constatado quando utilizadas concentrações superiores a 50 mM, ao passo que a rutina isolada não exibiu citotoxicidade (CI > 200 mM).43 A atividade citotóxica do eriodictiol já foi também reportada44 para diferentes linhagens tumorais, com destaque para o carcinoma hepatocelular (HepG2), onde esse metabólito foi capaz de suprimir a motilidade e crescimento celular pela desativação da inflamassoma NLRP3. O kaempferol exibiu atividade anti-proliferativa de células tumorais de bexiga por apoptose, suprimindo a função de algumas enzimas.45 Dessa forma, a avaliação do potencial citotóxico de extratos contendo esses metabólitos permanece relevante. Atividade antioxidante A concentração de compostos fenólicos totais (CFT) foi determinada utilizando-se ácido gálico como padrão para construção da curva analítica. O resultado foi expresso como mg AG g-1. O teor de flavonoides totais (FT) foi determinado utilizando-se quercetina como padrão para construção da curva analítica. O resultado foi expresso como mg QER g-1. Para SM, observou-se que CFT (Abs = 0,0223[amostra] + 0,0916, R2 = 0,9945) foi de 177,25 ± 1,3 mg AG g-1, bastante superior ao reportado para para o extrato metanólico das folhas de S. alexandrina (22,25 ± 0,29 mg QER g-1) e S. italica (21.54 ± 0.28 mg QER g-1).30 O teor de FT (Abs = 0,0287[amostra] +0,063, R2 = 0,9981) de SM foi de 40,81 ± 0,88 mg QER g-1, valor quase 25% menor que o descrito para o extrato metanólico das folhas de S. auriculata (156,64 ± 0,97 mg QER g-1).46 Essas observações indicam a influência do solvente sobre a extração. A espécie estudada no presente trabalho é rica em CFT e FT. Compostos fenólicos e flavonóides são de ampla ocorrência nos mais diversos tipos de extratos de Senna spp., entre os quais destacam-se catequinas, proantocianidinas, rutina, quercimeritrina, glicosídeos de kaempferol e crisofanol.4 A presença de compostos fenólicos em geral e de flavonoides no extrato SM justifica a atividade antioxidante, pois tais metabólitos são capazes de neutralizar espécies reativas de oxigênio (EROs), tais como oxigênio singleto, radical hidroxila, superóxido, radicais peroxila, alcoxila e outras espécies reativas radicalares ou não, de outros elementos.47,48 Essa ação antioxidante é classificada em dois distintos mecanismos: transferência de átomos de hidrogênio (TAH) e transferência de elétron singleto, ou simplesmente, transferência de elétron (TE). Ensaios baseados em TAH consistem na habilidade do antioxidante em doar e transferir átomos de hidrogênio para estabilizar radicais livres ou espécies reativas não radicalares. Os ensaios baseados em TE, por sua vez, consistem na transferência de um ou mais elétrons para reduzir e estabilizar o alvo oxidante. Ensaios como capacidade de absorção de radicais de oxigênio (ORAC) e parâmetros de captura de radicais totais são exemplos de ensaios que avaliam a atividade antioxidante por mecanismo de TAH, enquanto ensaios de captura de radicais como DPPH•, ABTS+• e poder redutor de íons férrico (FRAP) são exemplos de ensaios que avaliam por meio de mecanismo de TE.27,49,50 Os resultados obtidos de atividade antioxidante estão apresentados na Tabela 2. SM exibiu CI50 de 128,81 ± 0,36 μg mL-1 frente ao radical DPPH•, sendo considerado um potencial antioxidante moderado se comparado com o controle positivo ácido ascórbico (AA), cujo CI50 foi de 6,79 ± 0,51 μg mL-1. Outro fator que pode ter contribuído para essa inibição moderada está no fato de que os flavonoides glicosilados apresentam menor potencial antioxidante em relação à sua respectiva aglicona. A coplanaridade de um flavonoide, como no caso da quercetina, favorece a propriedade antioxidante do extrato, mas, por outro lado, as suas respectivas formas glicosiladas têm a coplanaridade comprometida. Flavonóis não glicosilados que são coplanares apresentam deslocalização de carga pelo alinhamento dos orbitais, o que favorece a doação e transferência de átomos de hidrogênio e elétrons da hidroxila fenólica.51 Portanto, a presença de flavonóis glicosilados pode ser um fator que justifica a atividade antioxidante moderada.
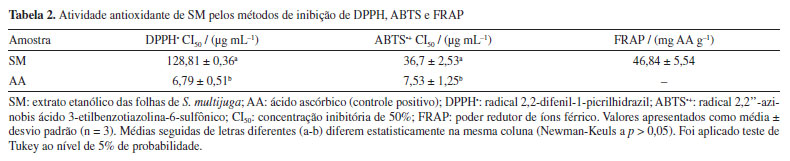
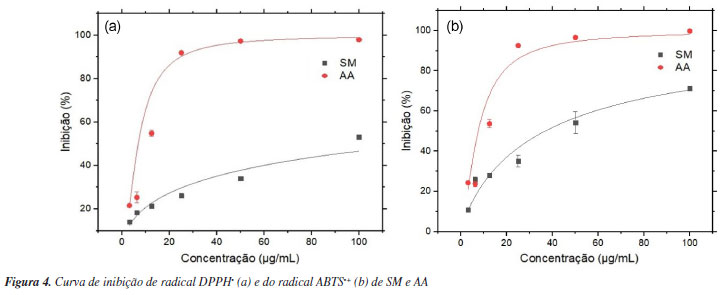
Os ensaios de captura de radical DPPH• podem ter mecanismos antioxidantes diferentes, dependendo de fatores como efeito do solvente e pH, por exemplo. Etanol e metanol são parcialmente ionizantes e, por esse meio, a inibição pode ocorrer tanto por transferência de átomos de hidrogênio como por transferência de elétrons. Portanto, o radical DPPH• reage com compostos antioxidantes por meio de transferência de elétrons com perda de prótons sequencial. Por outro lado, o etanol pode causar desaceleração na reação por causa do aumento da viscosidade, o que dificulta a difusão e interação dos antioxidantes com o radical.52 Tais evidências, além do aumento da interferência estérica, sugerem o incremento da dificuldade de acesso da hidroxila fenólica ao sítio radicalar do DPPH•, justificando a diferença nos valores obtidos de CI50 entre os ensaios de captura do radical DPPH• (128,81 ± 0,36 μg mL-1) e ABTS•+ (36,7 ± 2,53 μg mL-1) por SM. O radical ABTS•+ é menos impedido estericamente em comparação ao DPPH• e é produzido através da oxidação com persulfato de potássio. SM apresentou inibição de 71% do radical ABTS•+ com 100 μg mL-1, enquanto que para o DPPH•, foi capaz de inibir 53% nessa mesma concentração (Figura 4). O método de captura do radical ABTS•+ é mais versátil e possui reação mais rápida em comparação ao DPPH•. O uso de tampão pH 7,4 proporciona um ambiente mais controlado para a reação, que pode ocorrer através dos mecanismos de TAH e TE. Além disso, a reação com o radical ABTS•+ pode ocorrer tanto em meios lipofílicos quanto hidrofílicos,49 ampliando a gama de substâncias a terem seu potencial antioxidante avaliado.
O poder redutor de íons férrico (Fe3+) (FRAP) foi avaliado partindo da curva analítica (y = 0,035x + 0,464, R2 = 0,9845) (Tabela 2) utilizando AA como padrão, obtendo-se 46,84 ± 5,54 mg AA g-1. A importância de se avaliar o efeito antioxidante de extratos na redução de íons férrico (Fe3+) a íons ferrosos (Fe2+) reside na presença da reação de Foto-Fenton que ocorre em organismos aeróbicos. O Fe atua como catalisador quando reage com o radical superóxido (O2•-) produzindo íon hidroxila e radical hidroxila através das reações de Foto-Fenton e Haber-Weiis. O radical hidroxila, por sua vez, é altamente reativo com curto período de vida e não há enzima antioxidante para neutralizá-lo.47,53,54 Considerando que SM apresentou significativo poder antioxidante na redução de íons férrico, torna-se um candidato capaz de prevenir a geração do radical hidroxila, evitando danos oxidativos a biomoléculas e organelas. Além de técnicas espectrofotométricas comumente utilizadas para a determinação do potencial antioxidante de extratos vegetais, métodos eletroquímicos se configuram como técnicas alternativas para se quantificar essa propriedade.55 Os métodos eletroquímicos permitem a obtenção de dados valiosos sobre reações de transferência de elétrons, sem a necessidade de etapas prévias de tratamento do analito, garantindo respostas rápidas com alta sensibilidade.56 A análise da atividade antioxidante é realizada por meio da avaliação do potencial de oxidação (Epa), sendo que valores mais baixos indicam maior facilidade na doação de elétrons e, consequentemente, maior atividade antioxidante.57 Na análise feita com SM, os resultados obtidos a partir dos voltamogramas cíclicos (Figura 5) indicaram redução acentuada do Epa à medida que o pH aumentava (Tabela 3), sugerindo que o ambiente alcalino facilita a doação de elétrons pelos compostos antioxidantes presentes em SM. Observou-se também a formação de duas correntes de pico anódicas (Ipa), sendo o primeiro pico um indicativo da presença de rutina no extrato, que apresenta potenciais próximos a 0,23 V em pH 7.58 Já o segundo pico visualizado é um indicativo de compostos nitrogenados, podendo apresentar potenciais próximos a 1,0 V,59 sendo, possivelmente, referentes aos alcaloides que possam estar presentes no extrato. Alguns alcaloides descritos na literatura,60-62 como as β-carbolinas harmano, harmina, harmalina e harmalol e também a quinina, apresentam potenciais próximos aos valores obtidos nos voltamogramas, variando de 0,4 até 1,5 V, a depender do pH.
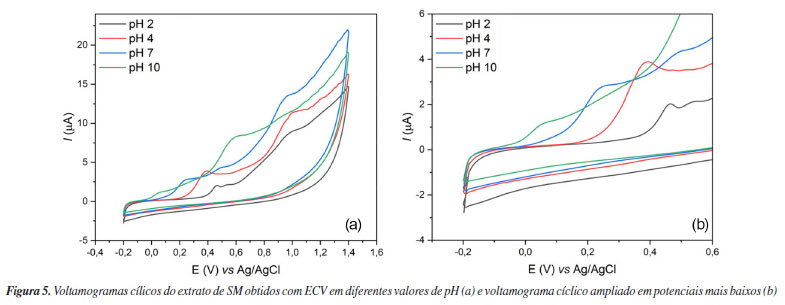
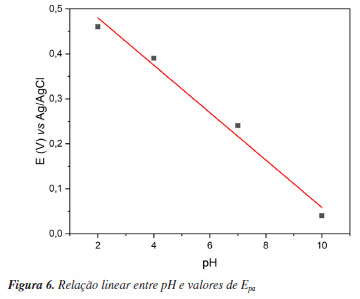
Esse comportamento é habitual para antioxidantes fenólicos, pois o aumento do pH pode desprotonar os grupos -OH, resultando na formação de fenolatos. Estes fenolatos são melhores nucleófilos em comparação aos fenóis, doando com facilidade seus elétrons.63 Além disso, foi identificada uma relação linear entre o pH e o Epa do primeiro pico (Figura 6), evidenciando que, em todos os casos, o processo de transferência de elétrons está acoplado à transferência de prótons (proton-coupled electron transfer, PCET), um mecanismo frequentemente observado na oxidação de compostos fenólicos. Esse resultado sugere que a reação envolve a transferência equimolar de prótons e elétrons.64
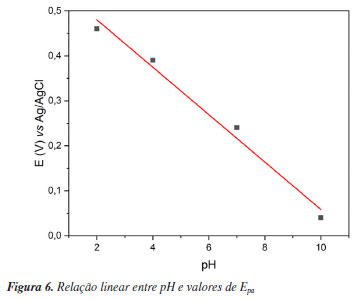
Esta conclusão é suportada pelas equações experimentais (-0,053pH + 0,58, R2 = 0,99), que apresentaram valores de inclinação próximos ao teórico de 0,059 V por unidade de pH, conforme previsto pela equação de Nernst (Equações 2 e 3).  A 25 ºC e com as constantes resolvidas:  Esse é o primeiro relato sobre o potencial antioxidante de S. multijuga por meio de processos eletroquímicos.
CONCLUSÕES O estudo do perfil químico dos extratos de folhas e galhos de Senna multijuga levou à anotação de flavonóis glicosilados e flavanonas, o que justifica a atividade antioxidante observada. A ausência de citotoxicidade frente a células saudáveis estimula a continuidade da avaliação do potencial dessa espécie frente a formas amastigotas intracelulares de T. cruzi.
DECLARAÇÃO DE DISPONIBILIDADE DE DADOS Todos os dados estão disponíveis no texto. Em caso de dúvida ou necessidade de algum esclarecimento adicional, entrar em contato com o autor de correspondência.
AGRADECIMENTOS Os autores agradecem à Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), processos No. 2013/07600-3 (CIBFar-CEPID) e No. 2014/50926-0 (INCT BioNat CNPq/FAPESP), ao Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) e Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). Os autores agradecem o suporte financeiro das bolsas de A. L. P. S., E. K. e A. V. B. da CAPES (No. 88887.373581/2019-00, No. 88887.753099/2022-00, 88887.821103/2023-00) e M. S. R. da FAPESP (2024/02602-2).
CONTRIBUIÇÕES DO AUTOR Cássia G. Magalhães foi responsável por conceituação, curadoria de dados, análise formal, investigação, redação (rascunho original, revisão e edição); Ana Letícia P. dos Santos por investigação, redação rascunho original; Marcelino S. do Rosário por investigação, redação rascunho original; Elton Kazmierczak por investigação, redação rascunho original; André V. Bassani por investigação, redação rascunho original; Christiana A. Pessoa por conceituação, curadoria de dados, análise formal, validação, redação (revisão e edição); Jorge Iarmul por curadoria de dados, validação; Iriane Eger por conceituação, curadoria de dados, análise formal, investigação, validação, redação (rascunho original, revisão e edição); Vanderlan S. Bolzani por conceituação, curadoria de dados, análise formal, validação, redação (revisão e edição).
REFERÊNCIAS 1. Usman, M.; Khan, W.R.; Yousaf, N.; Akram, S.; Murtaza, G.; Kudus, K. A.; Ditta, A.; Rosli, Z.; Rajpar, M. N.; Nazre, M.; Molecules 2022, 27, 3863. [Crossref] 2. Lum Nde, A.; Chukwuma, C. I.; Erukainure, O. L.; Chukwuma, M. S.; Matsabisa, M. G.; J. Ethnopharmacol. 2022, 283, 114663. [Crossref] 3. de Macedo, E. M. S.; Silva, J. G. A.; Silva, M. G. V.; Rev. Virtual Quim. 2016, 8, 169. [Crossref] 4. Alshehri, M. M.; Quispe, C.; Herrera-Bravo, J.; Sharifi-Rad, J.; Tutuncu, S.; Aydar, E. F.; Topkaya, C.; Mertdinc, Z.; Ozcelik, B.; Aital, M.; Kumar, N. V. A.; Lapava, N.; Rajkovic, J.; Ertani, A.; Nicola, S.; Semwal, P.; Painuli, S.; González-Contreras, C.; Martorell, M.; Butnariu, M.; Bagiu, I. C.; Bagiu, R. V.; Barbhai, M. D.; Kumar, M.; Daştan, S. D.; Calina, D.; Cho, W. C.; Oxid. Med. Cell. Longevity 2022, 2022, 6025900. [Crossref] 5. Oladeji, O. S.; Adelowo, F. E.; Oluyori, A. P.; S. Afr. J. Bot. 2021, 138, 1. [Crossref] 6. Serrano, M. A. R.; Pivatto, M.; Francisco, W.; Danuello, A.; Regasini, L. O.; Lopes, E. M. C.; Lopes, M. N.; Young, M. C. M.; Bolzani, V. S.; J. Nat. Prod. 2010, 73, 482. [Crossref] 7. Francisco, W.; Pivatto, M.; Danuello, A.; Regasini, L. O.; Baccini, L. R.; Young, M. C. M.; Lopes, N. P.; Lopes, J. L. C.; Bolzani, V. S.; J. Nat. Prod. 2012, 75, 408. [Crossref] 8. Ferreira, P. M. P.; Arcanjo, D. D. R.; Peron, A. P.; J. Toxicol. Environ. Health, Part B 2023, 26, 257. [Crossref] 9. Silvestrini, A.; Meucci, E.; Ricerca, B. M.; Int. J. Mol. Sci. 2023, 24, 10978. [Crossref] 10. García-Huertas, P.; Cardona-Castro, N.; Biomed. Pharmacother. 2021, 142, 112020. [Crossref] 11. Lazarin-Bidóia, D.; Garcia, F. P.; Ueda-Nakamura, T.; Silva, S. O.; Nakamura, C. V.; Mem. Inst. Oswaldo Cruz 2022, 117, 1. [Crossref] 12. Martínez-Peinado, N.; Cortes-Serra, N.; Tallini, L. R.; Pinazo, M. J.; Gascon, J.; Bastida, J.; Alonso-Padilla, J.; Parasites Vectors 2021, 14, 337. [Crossref] 13. Guzmán, E.; González, R.; Flores, S.; Zavala, J.; Rosado, M.; Pérez, S.; Pharm. Biol. 2004, 42, 504. [Crossref] 14. Guzmán, E.; Perez, C.; Zavala, M. A.; Acosta-Viana, K. Y.; Perez, S.; Phytomedicine 2008, 15, 892. [Crossref] 15. Jimenez-Coello, M.; Acosta-Viana, K. Y.; Guzman-Marin, E.; Perez, G. C.; Perez, G. M. S.; Pharm. Biol. 2010, 48, 666. [Crossref] 16. Jimenez-Coello, M.; Guzman-Marin, E.; Perez-Gutierrez, E.; Polanco-Hernandez, G. M.; Acosta-Viana, K. Y.; Afr. J. Tradit., Complementary Altern. Med. 2011, 8, 164. [Crossref] 17. Gaudry, A.; Marcourt, L.; Kaiser, M.; Flückiger, J.; David, B.; Grondin, A.; Wolfender, J. L.; RSC Adv. 2025, 15, 15240. [Crossref] 18. Wang, M.; Carver, J. J.; Phelan, V. V.; Sanchez, L. M.; Garg, N.; Peng, Y.; Nguyen, D. D.; Watrous, J.; Kapono, C. A.; Luzzatto-Knaan, T.; Porto, C.; Bouslimani, A.; Melnik, A. V.; Meehan, M. J.; Liu, W. T.; Crüsemann, M.; Boudreau, P. D.; Esquenazi, E.; Sandoval-Calderón, M.; Kersten, R. D.; Pace, L. A.; Quinn, R. A.; Duncan, K. R.; Hsu, C. C.; Floros, D. J.; Gavilan, R. G.; Kleigrewe, K.; Northen, T.; Dutton, R. J.; Parrot, D.; Carlson, E. E.; Aigle, B.; Michelsen, C. F.; Jelsbak, L.; Sohlenkamp, C.; Pevzner, P.; Edlund, A.; McLean, J.; Piel, J.; Murphy, B. T.; Gerwick, L.; Liaw, C. C.; Yang, Y. L.; Humpf, H. U.; Maansson, M.; Keyzers, R. A.; Sims, A. C.; Johnson, A. R.; Sidebottom, A. M.; Sedio, B. E.; Klitgaard, A.; Larson, C. B.; Boya, C. A. P.; Torres-Mendoza, D.; Gonzalez, D. J.; Silva, D. B.; Marques, L. M.; Demarque, D. P.; Pociute, E.; O'Neill, E. C.; Briand, E.; Helfrich, E. J. N.; Granatosky, E. A.; Glukhov, E.; Ryffel, F.; Houson, H.; Mohimani, H.; Kharbush, J. J.; Zeng, Y.; Vorholt, J. A.; Kurita, K. L.; Charusanti, P.; McPhail, K. L.; Nielsen, K. F.; Vuong, L.; Elfeki, M.; Traxler, M. F.; Engene, N.; Koyama, N.; Vining, O. B.; Baric, R.; Silva, R. R.; Mascuch, S. J.; Tomasi, S.; Jenkins, S.; Macherla, V.; Hoffman, T.; Agarwal, V.; Williams, P. G.; Dai, J.; Neupane, R.; Gurr, J.; Rodríguez, A. M. C.; Lamsa, A.; Zhang, C.; Dorrestein, K.; Duggan, B. M.; Almaliti, J.; Allard, P. M.; Phapale, P.; Nothias, L. F.; Alexandrov, T.; Litaudon, M.; Wolfender, J. L.; Kyle, J. E.; Metz, T. O.; Peryea, T.; Nguyen, D. T.; VanLeer, D.; Shinn, P.; Jadhav, A.; Müller, R.; Waters, K. M.; Shi, W.; Liu, X.; Zhang, L.; Knight, R.; Jensen, P. R.; Palsson, B.; Pogliano, K.; Linington, R. G.; Gutiérrez, M.; Lopes, N. P.; Gerwick, W. H.; Moore, B. S.; Dorrestein, P. C.; Bandeira, N.; Nat. Biotechnol. 2016, 34, 828 [Crossref]; GNSP Documentation, https://ccms-ucsd.github.io/GNPSDocumentation/, acessado em julho 2025. 19. GNPS2, https://www.gnps2.org/status?task=c3159a 62d3f84cd898c8b747915ca36a, acessado em julho 2025. 20. da Silva, L. E.; Joussef, A. C.; Pacheco, L. K.; da Silva, D. G.; Steindel, M.; Rebelo, R. A.; Bioorg. Med. Chem. 2007, 15, 7553. [Crossref] 21. Sieuwerts, A. M.; Klijn, J. G. M.; Peters, H. A.; Foekens, J. A. T.; Eur. J. Clin. Chem. Clin. Biochem. 1995, 33, 813. [Crossref] 22. Tronina, T.; Bartmańska, A.; Popłoński, J.; Rychlicka, M.; Sordon, S.; Filip-Psurska, B.; Huszcza, E.; Int. J. Mol. Sci. 2023, 24, 7408. [Crossref] 23. Singleton, V. D.; Orthofer, R.; Lamuela-Ravento, R. M.; Methods Enzymol. 1999, 299, 152. [Crossref] 24. Hacke, A. C. M.; Marques, J. A.; Vellosa, J. C. R.; Boligon, A. A.; da Silva, F. D. A.; de Souza, D.; Bonini, J. S.; Rocha, J. B. T.; Pereira, R. P.; New J. Chem. 2018, 42, 3642. [Crossref] 25. Brand-Williams, W.; Cuvelier, M. E.; Berset, C.; Food Sci. Technol. 1995, 28, 25. [Crossref] 26. Xiao, F.; Xu, T.; Lu, B.; Liu, R.; Food Front. 2020, 1, 60. [Crossref] 27. Berker, K. I.; Güçlü, K.; Tor, I.; Apak, R.; Talanta 2007, 72, 1157. [Crossref] 28. Moura, M. S.; Bellete, B. S.; Vieira, L. C. C.; Sampaio, O. M.; Rev. Virtual Quim. 2022, 14, 214. [Crossref] 29. Zou, X. Y.; He, Y. J.; Yang, Y. H.; Yan, X. P.; Li, Z. B.; Yang, H.; Separations 2022, 9, 76. [Crossref] 30. Lin, L. Z.; Harnly, J. M.; J. Agric. Food Chem. 2007, 55, 1084. [Crossref] 31. Vigbedor, B. Y.; Akoto, C. O.; Neglo, D.; J. Evidence-Based Complementary Altern. Med. 2023, 2023, 9345047. [Crossref] 32. Wang, H. P.; Lin, Z. Z.; Wang, H.; Yang, X.; Niu, N.; Sci. Rep. 2024, 14, 9679. [Crossref] 33. Imperato. F.; Am. Fern. J. 2000, 90, 141. [Crossref] 34. Liu, P.; Kallio, H.; Yang, B.; Food Chem. 2014, 160, 180. [Crossref] 35. Willems, J. L.; Khamis, M. M.; Saeid, W. M.; Purves, R. W.; Katselis, G.; Low, N. H.; El-Aneed, A.; Anal. Chim. Acta 2016, 933, 164. [Crossref] 36. Omer, H. A. A.; Caprioli, G.; Abouelenein, D.; Mustafa, A. M.; Uba, A. I.; Ak, G.; Ozturk, R. B.; Zengin, G.; Yagi, S.; Molecules 2022, 27, 5590. [Crossref] 37. Mohamed, T. A.; Ali, S. K.; Elshamy, A. I.; Saleh, I. A.; Ibrahim, M. A. A.; Atia, M. A. M.; Alshammari, S. O.; Mohamed, A. E. H. H.; Hussien, T. A.; Hamed, A. R.; Saedi, H. R. E.; Abdel-Azim, N. S.; Shams, K. A.; Efferth, T.; Saker, M.; Paré, P. W.; Hegazy, M. E. F.; Phytomedicine 2022, 100, 54019. [Crossref] 38. Sharma, S.; Hafeez, A.; Usmani, S. A.; J. Drug Delivery Sci. Techol. 2022, 76, 103724. [Crossref] 39. Lombardo, M.; Kiyota, S.; Kato, E. T. M.; Mathor, M. B.; Pinto, T. J. A.; Kaneko, T. M.; Acta Sci., Biol. Sci. 2015, 37, 9. [Crossref] 40. Sharma, A.; Kumar, A.; Jaitak, V.; Journal of Herbal Medicine 2021, 26, 100407. [Crossref] 41. Marín, C.; Ramírez-Macías, I.; López-Céspedes, A.; Olmo, F.; Villegas, N.; Díaz, J. G.; Sánchez-Moreno, M.; J. Nat. Prod. 2011, 74, 744. [Crossref] 42. Tasdemir, D.; Kaiser, M.; Brun, R.; Yardley, V.; Schmidt, T. J.; Tosun, F.; Rüedi, P.; Antimicrob. Agents Chemother. 2006, 50, 1352. [Crossref] 43. Başaran, E. B. R. U.; Öztürk, A. A.; Şenel, B.; Demirel, M.; Sarica, Ş.; Eur. J. Pharm. Sci. 2022, 172, 106153. [Crossref] 44. Yin, H.; Li, Y.; Feng, Y.; Tian, L.; Li, Y.; Nutrients 2024, 16, 4237. [Crossref] 45. Parveen, S.; Bhat, I. U. H.; Bhat, R.; Asian Pac. J. Trop. Biomed. 2023, 13, 411. [Crossref] 46. Prasathkumar, M.; Raja, K.; Vasanth, K.; Khusro, A.; Sadhasivam, S.; Sahibzada, M. U. K.; Gawwad, M. R. A.; Al Farraj, D. A.; Elshikh, M. S.; Arabian J. Chem. 2021, 14, 103345. [Crossref] 47. Sharifi-Rad, M.; Kumar, N. V. A.; Zucca, P.; Varoni, E. M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Fokou, P. V. T.; Azzini, E.; Peluso, I.; Mishra, A. P.; Nigam, M.; El Rayess, Y.; Beyrouthy, M.; El Polito, L.; Iriti, M.; Martins, N.; Martorell, M.; Docea, A. O.; Setzer, W. N.; Calina, D.; Cho, W. C.; Sharifi-Rad, J.; Frontiers in Physiology 2020, 11, 1. [Crossref] 48. Phaniendra, A.; Jestadi, D. B.; Periyasamy, L.; Indian J. Clin. Biochem. 2015, 30, 11. [Crossref] 49. Schaich, K. M.; Tian, X.; Xie, J.; J. Funct. Foods 2015, 14, 111. [Crossref] 50. Shahidi, F.; Zhong, Y.; J. Funct. Foods 2015, 18, 757. [Crossref] 51. Kumar, S.; Pandey, A. K.; Sci. World J. 2013, 2013, 6025900. [Crossref] 52. Xie, J.; Schaich, K. M.; J. Agric. Food Chem. 2014, 62, 4251. [Crossref] 53. Kazmierczak, E.; Magalhães, C. G.; Pereira, R. P.; Ecletica Quim. 2023, 48, 41. [Crossref] 54. Gulcin, I.; Alwasel, S. H.; Processes 2022, 10, 132. [Crossref] 55. Alam, M. W.; Najeeb, J.; Naeem, S.; Usman, S. M.; Nahvi, I.; Alismail, F.; Abuzir, A.; Farhan, M.; Nawaz, A.; Antioxidants 2022, 11, 1205. [Crossref] 56. Yan, F.; Su, B.; Anal. Chem. 2016, 88, 11001. [Crossref] 57. Jara-Palacios, M. J.; Begines, E.; Heredia, F. J.; Escudero-Gilete, M. L.; Hernanz, D.; Foods 2024, 13, 906. [Crossref] 58. Simić, A.; Manojlović, D.; Segan, D.; Todorović, M.; Molecules 2007, 12, 2327. [Crossref] 59. Adenier, A.; Chehimi, M. M.; Gallardo, I.; Pinson, J.; Vilà, N.; Langmuir 2004, 20, 8243. [Crossref] 60. Agüí, L.; Peña-Farfal, C.; Yáñez-Sedeño, P.; Pingarrón, J. M.; Electroanalysis 2007, 19, 237. [Crossref] 61. Dushna, O.; Dubenska, L.; Marton, M.; Hatala, M.; Vojs, M.; Microchem. J. 2023, 191, 108639. [Crossref] 62. Ali, H. S.; Yardım, Y.; Food Chem. 2024, 441, 138262. [Crossref] 63. Evans, C. M.; Kirby, A. J.; J. Chem. Soc., Perkin Trans. 2 1984, 7, 1269. [Crossref] 64. Tyburski, R.; Liu, T.; Glover, S. D.; Hammarström, L.; J. Am. Chem. Soc. 2021, 143, 560. [Crossref]
Editor Convidado responsável pelo artigo: Lucas S. Abreu |

On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access