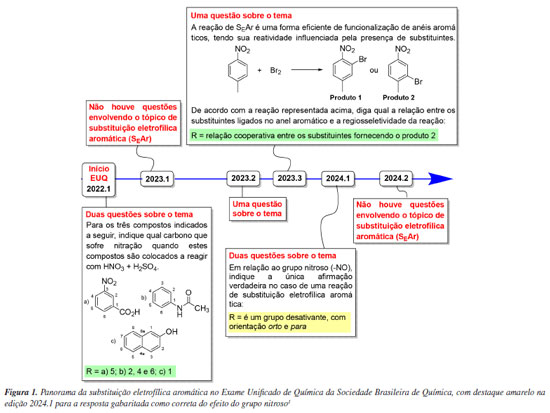
INTRODUÇÃO
A reação de substituição eletrofílica aromática (SEAr) é um tema importante no ensino de química orgânica em função do seu emprego na pesquisa e na indústria. Por este motivo, está presente de forma recorrente no Exame Unificado de Química da Sociedade Brasileira de Química (EUQ-SBQ), Figura 1.1
Na primeira edição de 2024 do EUQ-SBQ, uma questão inqueriu sobre a reatividade do nitrosobenzeno e o efeito dirigente do grupo nitroso (-N=O ou NO) na reação de SEAr, Figura 1.
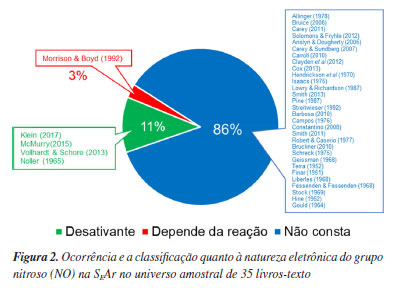
A inserção do grupo nitroso no EUQ-SBQ sugere sua relevância como grupo funcional na formação dos químicos, sendo esperado que os livros-texto abordem o tema de forma adequada. No entanto, a realidade nos manuais de ensino é distinta, uma vez que a influência do grupo NO na reação de SEAr é raramente discutida nos cursos de química orgânica de graduação e pós-graduação e, quando apresentada (Figura 2), não é condizente com a literatura científica primária sobre o tema, como será discutido adiante. Por causa dessa inconsistência, a questão envolvendo o grupo nitroso na SEAr, indicada na Figura 1, foi gabaritada de forma errada no EUQ-SBQ.1
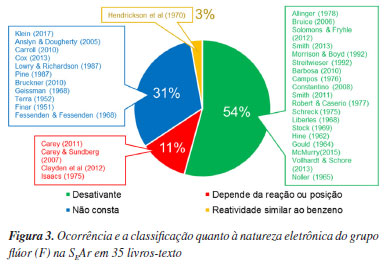
Assim como o grupo nitroso, é raro encontrar nos livros-texto uma explicação detalhada sobre o efeito do átomo de flúor na SEAr e, da mesma forma, as informações fornecidas são conflitantes com a literatura científica primária. Em algumas fontes, o flúor é classificado como desativante, enquanto em outras sua classificação varia conforme a reação, Figura 3. Essa dualidade de explicações gera insegurança no estudante quanto ao efeito do flúor na SEAr.
Em função da ênfase da SEAr na formação dos profissionais da Química e dos cenários acima descritos, bem como da nossa continuada contribuição ao ensino teórico de Química Orgânica,2-5 o presente trabalho organiza as informações que explicam o comportamento dos grupos funcionais nitroso e flúor, corrigindo erros e imprecisões contidos em alguns livros de graduação. Além disso, sugere que a explicação completa da química desses grupos seja abordada apenas em programas de pós-graduação, devido à complexidade desses grupos na SEAr em comparação com os substituintes tradicionalmente estudados na graduação.
OCORRÊNCIA E CLASSIFICAÇÃO DOS GRUPOS NITROSO E FLÚOR NOS LIVROS DIDÁTICOS DE QUÍMICA ORGÂNICA
Para avaliar o cenário do ensino da reação de SEAr e compreender a origem do erro no gabarito da questão do EUQ-SBQ 2024.1, foram analisados trinta e cinco livros de química orgânica para graduação e pós-graduação6-40 (detalhes na Tabela 1S, Material Suplementar), envolvendo o período entre 1951-2017, e os resultados são apresentados nas Figuras 2 e 3 para os grupos NO e F, respectivamente.
Nos poucos livros que abordam o efeito do grupo NO,10,19,21,28,37 as explicações contêm equívocos tanto em relação aos fatos experimentais quanto teóricos. A literatura especializada sobre o grupo NO41-50 revela uma reatividade distinta e mais complexa do que a apresentada nesses livros, evidenciando a necessidade de uma revisão no ensino simplificado deste grupo funcional nos textos didáticos.
De maneira similar, a maioria dos livros consultados de graduação e avançados mostram o flúor como um grupo desativante na SEAr (Figura 3), análogo aos halogênios cloro e bromo. Apesar deste fato, analisando a literatura primária sobre a influência do átomo de flúor em reações de SEAr,51-63 nota-se que o flúor apresenta reatividade particular não consistente com os demais halogênios.
DEFINIÇÕES DE TERMOS ESPECÍFICOS E O CONCEITO DE REATIVIDADE QUÍMICA NO CONTEXTO DA SEAR
Para facilitar a compreensão aos estudantes que se iniciam no estudo da SEAr e subsidiar a discussão feita neste trabalho, são apresentadas definições de termos relacionados à reatividade química no contexto da SEAr, com argumentação centrada no mecanismo iônico. Contudo, a substituição eletrofílica aromática também pode ocorrer por outros mecanismos de reação, como o SET (single electron transfer),12-14,22 e os argumentos apresentados ainda continuam válidos sob esse prisma.
A reatividade de uma substância é sua tendência a reagir com outras substâncias, sendo mais reativo aquela que participa de reações com maior rapidez ou facilidade,6,7 desde que a comparação seja realizada por meio de uma reação sob condições reacionais comuns.
A natureza eletrônica de um grupo funcional (ativante ou desativante) no contexto de SEAr associa diretamente a classificação do substituinte com a reatividade do anel aromático que o contém,7 onde grupos funcionais que tornam um composto aromático monossubstituído mais reativo que o benzeno são denominados ativantes e, por consequência, substituintes que tornam um composto aromático menos reativo que o benzeno são denominados desativantes, Figura 4a.
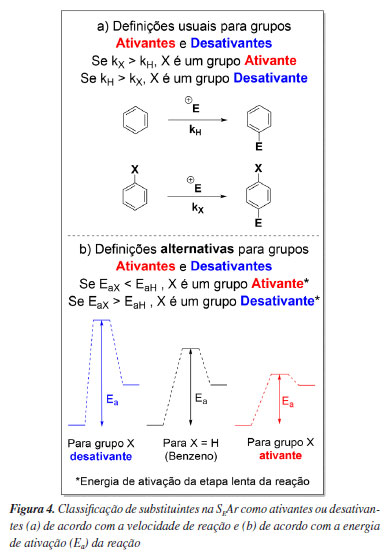
Embora esta definição seja bem disseminada, a constante da velocidade de reação (k) não é um bom parâmetro para estabelecer se um grupo é ativante ou desativante, pois esta é a consequência, e não a causa, de dois outros fatores que influenciam a cinética da reação, um fator energético associado à energia de ativação e um fator estatístico associado à variação de entropia na formação do estado de transição (ΔS‡). Além disso, uma velocidade de reação maior não necessariamente está associada a uma energia de ativação mais baixa, devido à existência do termo estatístico que pode mascarar os resultados.64-67 Com base nisso, propomos que definir substituintes como ativantes e desativantes seja feita com cuidado. Uma forma mais assertiva é analisar os "fatores de velocidade parciais", que serão definidos adiante. De forma simples, estes fatores refletem a velocidade de reação normalizada para o número de posições reativas, sendo que se for maior que 1, o grupo funcional é ativante, caso contrário, é desativante. Para as discussões subsequentes (como no tópico sobre o efeito do flúor na SEAr, onde se faz necessário ponderar termos energéticos e entrópicos), sugerimos que este conceito de ativação e desativação revisto seja adotado, associando essas propriedades sempre aos fatores de velocidade parciais (fo, fm, fp), e não às constantes velocidades de reação dos substratos (k).
Embora estas definições sejam simples e efetivas para classificar o substituinte do anel benzênico do reagente como ativantes ou desativantes quando estes apresentam estas naturezas eletrônicas bem pronunciadas, requerem um rigor maior ao analisar dados cinéticos para inferir sobre grupos substituintes nos casos limítrofes, como quando os efeitos de ressonância e indutivo são ambos pronunciados e concorrentes (Figura 5). Vale a pena mencionar que os efeitos indutivos (ou de campo) e de ressonância estão relacionados à seletividade de posição por meio de seu impacto no estado de transição, e não no reagente. Como o estado de transição é estruturalmente semelhante ao complexo sigma (ou íon arênio), seu efeito é amplificado nesse intermediário.
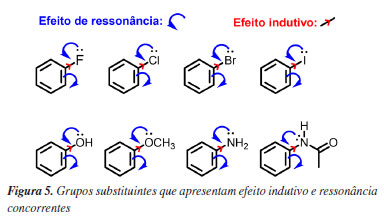
Quanto à orientação e ao efeito diretor (ou dirigente), os substituintes podem também ser classificados como orto, para ou meta dirigentes, em relação às posições preferenciais de substituição na SEAr para um substrato, já que são dependentes do substituinte nele presente. Quanto à orientação, os substituintes mais comumente conhecidos estão contidos em apenas duas categorias, os que dirigem simultaneamente para as posições orto e para e os que dirigem preferencialmente para a posição meta, que em conjunto com a classificação como ativantes ou desativantes leva à existência de três categorias globais, Figura 6.
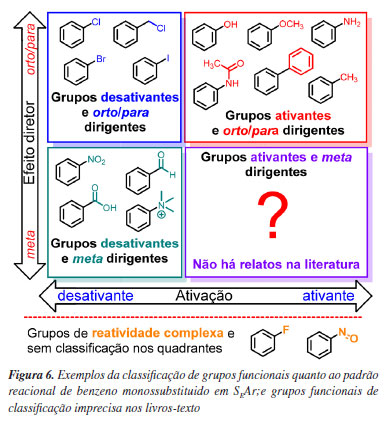
A classificação dos grupos funcionais como mostrada na Figura 6 é bem descrita e recorrente nos livros-texto. Entretanto, os grupos nitroso e flúor, que aparecem menos frequentemente nos livros e/ou apresentam inconsistências quando confrontados com dados da literatura científica primária, encontram-se situados fora destes quadrantes, de forma que sua reatividade complexa deve ser analisada com cuidado, caso se opte em ensiná-la no contexto da SEAr, Figura 6. Os tópicos seguintes apresentam material didático para o ensino adequado do efeito desses dois substituintes.
REATIVIDADE E ORIENTAÇÃO DO GRUPO NITROSO NA SUBSTITUIÇÃO ELETROFÍLICA AROMÁTICA
O grupo nitroso é relatado como grupo desativante nos livros didáticos, porém imprecisamente como orto e para dirigente, comportamento que se assemelharia aos halogênios cloro e bromo. São poucos os livros didáticos que exemplificam o grupo nitroso em SEAr e, quando explicitam o grupo NO, as explicações estão em desacordos com fatos experimentais, além de representarem de maneira imprecisa estruturas químicas do nitrosobenzeno, ferindo princípios da teoria da ligação de valência (TLV) e da teoria de repulsão de pares de elétrons na camada de valência (VSEPR). Por exemplo, na segunda edição do livro de Klein (2013),10 a justificativa para a regiosseletividade em orto e para ocorre através da ressonância do par de elétrons não ligantes do nitrogênio do NO para explicar a estabilidade do intermediário cátion arênio formado, onde o nitrosobenzeno e os cátion arênios são desenhados com um ângulo de 180º entre o carbono ipso do anel e o grupo NO (átomos C1-N=O lineares sugerindo a hibridização sp para o N), Esquema 1a.
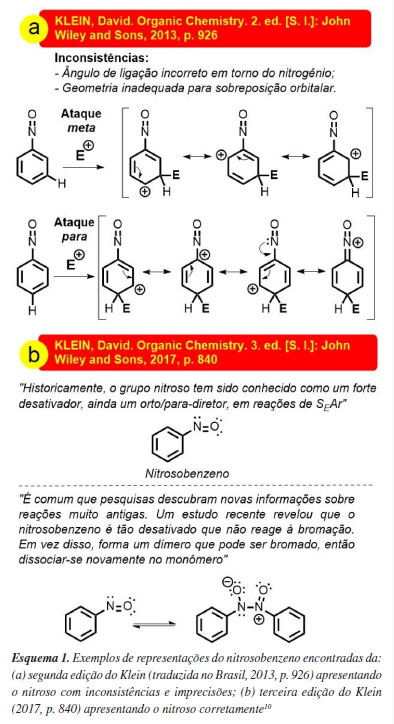
As estruturas de ressonância no Esquema 1a são dependentes da conformação onde o par de elétrons não ligante do nitrogênio do NO fica paralelo à nuvem π do sistema aromático que, ao ser deslocalizado para o anel, acaba ferindo alguns princípios de geometria e ângulo das ligações nas estruturas representadas, não considerando a natureza da hibridização do orbital sp2 do N onde se encontra o par de elétrons não ligante, ignorando o ângulo das ligações envolvidas, lançando mão de estruturas de ressonância às quais se faz necessário que o par de elétrons esteja localizado em um orbital p e tenha simetria π, assim ferindo os princípios da TLV.
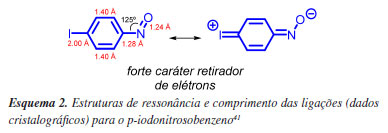
A natureza da hibridização sp2 do nitrogênio do grupo nitroso é evidenciada através da cristalografia de raio-X do p-iodonitrosobenzeno, onde o ângulo de ligação entre os átomos C1-N=O é 125º, com o grupo NO e o anel coplanares. Ademais, o comprimento da ligação C1-N 1.28 Å é próximo da ligação NO 1.24 Å, evidenciando o forte caráter retirador de elétrons do grupo nitroso, Esquema 2.41
É bem conhecido que arilnitrosos dimerizam levando à formação da espécie azodióxi através de uma reação de adição nucleofílica (Esquema 3),42-44 sendo que arilnitrosos substituídos por grupos retiradores de densidade eletrônica tendem a dimerizar, enquanto que os substituídos por grupos doadores tendem a permanecer na forma monomérica.41 Ao contrário do grupo -N=O, o grupo azodióxi possui o par de elétrons não ligante localizado no orbital p do nitrogênio (em azul no Esquema 3) com geometria adequada para entrar em ressonância com o anel, o que justifica a estabilidade do intermediário cátion arênio formado pelo ataque do eletrófilo nas posições orto e para, sendo o grupo diretor azodióxi o responsável pela reatividade e regiosseletividade observada nas reações de SEAr envolvendo arilnitrosos. Ou seja, a espécie que reage com o eletrófilo não é o arilnitroso, pois este dimeriza antes de reagir com o eletrófilo, e sim o dímero azodióxi que, após a reação de SEAr, sofre dissociação aos respectivos monômeros, com indicado no Esquema 4.
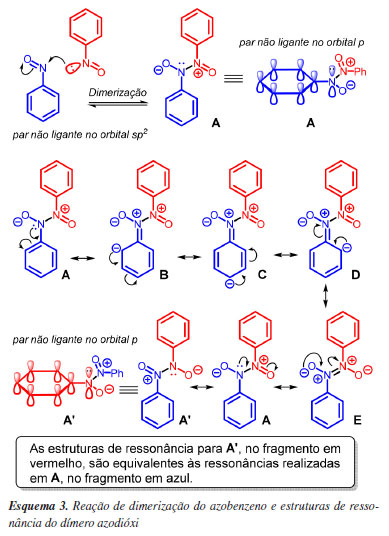
De todos os livros consultados que mencionam o grupo nitroso, apenas o Klein, em sua terceira edição (2017),10 corrige a imprecisão e menciona a participação da espécie azodióxi nas reações de SEAr (Esquema 1B), mas ainda assim não apresenta o mecanismo completo, como indicado no Esquema 4.
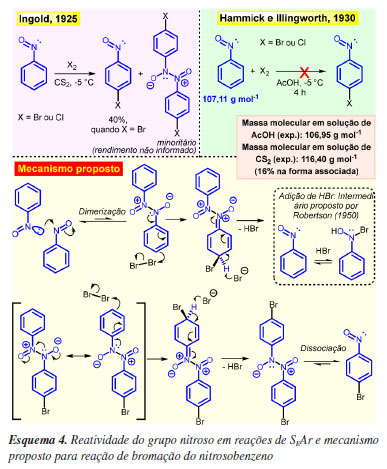
São várias as evidências experimentais que sustentam a dimerização e o mecanismo proposto. Os estudos realizados por Ingold45 para a reação de bromação e cloração do nitrosobenzeno em dissulfeto de carbono (CS2) descrevem produtos halogenados para substituídos, como o p-bromonitrosobenzeno obtido em 40% e o pp'-dibromoazodioxibenzeno obtido em pequena quantidade, através de uma reação rápida em que é possível observar o consumo dos reagentes orgânicos logo após a adição dos reagentes de halogenação, Esquema 4.
Posteriormente, Hammick e Illingworth46 reinvestigaram as reações de halogenação do nitrosobenzeno, obtendo os mesmos resultados descritos por Ingold ao empregar CS2. Todavia, na tentativa de alterar o solvente da reação para ácido acético (AcOH) não houve transformações mesmo após quatro horas. Além disso, foram conduzidos experimentos para determinar a massa molar do nitrosobenzeno (107,11 g mol-1) em ambos os solventes, CS2 e AcOH. A massa molecular do nitrosobenzeno em solução de AcOH foi mensurada por crioscopia, resultando em um valor médio de 106,35 g mol-1, próximo do valor esperado de 107,11 g mol-1. Por outro lado, a massa molecular mensurada em solução em CS2, através do aumento do ponto de ebulição da solução pelo método de Landsberger-Walker, apresentou valor médio de 116,40 g mol-1. Ambos os valores mensurados, quando comparados ao valor esperado para a massa molecular do nitrosobenzeno (107,11 g mol-1), revelam que, em solução de AcOH, este prevalece exclusivamente em sua forma monomérica, enquanto que, em CS2, o valor encontrado acima do esperado revela-se como uma mistura do nitrosobenzeno e seu dímero (presente em 16%), Esquema 4.
Esses resultados racionalizados em conjunto reforçam que a reatividade do nitrosobenzeno nas reações de halogenação depende da presença do dímero azodióxi, de modo que, no solvente que suprime esse equilíbrio não há reação, a exemplo do AcOH, enquanto quando constatada sua presença, mesmo em pequenas quantidades, em um solvente o qual favorece o equilíbrio de dimerização, é o suficiente para iniciar a reação e deslocar o equilíbrio para a formação dos produtos. Dessa maneira, o dímero é o responsável pela reatividade e regiosseletividade observada nas reações.
Anos depois, Robertson et al.47 revisitou a reação de bromação do nitrosobenzeno, empregando tetracloreto de carbono (CCl4) como solvente e ácido bromídrico como catalisador, realizando um estudo cinético. A adição de HBr ao sistema, além de acelerar a reação de bromação e torná-la reprodutível, revelou coeficientes de uma reação bimolecular nos instantes iniciais, indicando uma reação de adição do HBr ao grupo -N=O, saturando-o, em um processo autocatalítico, levando a formação de um intermediário mais reativo (com par de elétron não ligante no orbital p) responsável pela reatividade e regiosseletividade na SEAr (caixa pontilhada em destaque no Esquema 4).
Construindo um paralelo direto entre a estrutura do azodióxi e o intermediário de Robertson, em termos de estruturas de ressonância e deslocalização eletrônica, a presença do par de elétron não ligante no orbital p do intermediário de Robertson pode gerar estruturas de ressonância similares ao azodióxi (como demonstrado no Esquema 3) com carga negativa deslocalizada em orto e para, sem ferir nenhum dos princípios da química e com possibilidade de também orientar nessas posições.
Assim, um possível mecanismo para bromação pode ser proposto tanto com participação do dímero azodióxi quanto com o intermediário de Robertson, uma vez que ao longo da reação de SEAr com Br2 há a formação de HBr à medida que a reação avança e, ao final, há formação do mesmo produto por caminhos complementares que justificam os fatos experimentais, Esquema 4. Qualquer que seja a real espécie reativa, a determinada por Ingold ou por Robertson, não é diretamente o grupo funcional -N=O o responsável pela reatividade e regiosseletividade observadas.
Com o objetivo de melhor compreender eletronicamente a racionalização proposta nos Esquemas 3 e 4 acerca do nitroso e seu dímero, foi realizada uma busca na literatura acerca de sua distribuição de orbitais moleculares de fronteira. Devido à inexistência destes dados, as duas estruturas foram calculadas usando o pacote de química computacional Orca 4.2.1 (Max-Planck-Institut für Kohlenforschung, Alemanha, 2019)68 e os resultados estão apresentados na Figura 7, com as topologias e energias dos orbitais moleculares de fronteira para o nitrosobenzeno e para o seu dímero, calculados usando o nível de teoria e base M06/def2-TZVP (na Figura 1S, Material Suplementar, são apresentados os dados calculados também via B3LYP/def2-SVP e RHF/def2-SVP, e os resultados são essencialmente análogos entre si e aos da Figura 7 na localização da densidade eletrônica que explica a regiosseletividade aqui apresentada, quando se analisa os orbitais HOMO (orbital molecular ocupado de mais alta energia), HOMO-1 e HOMO-2).
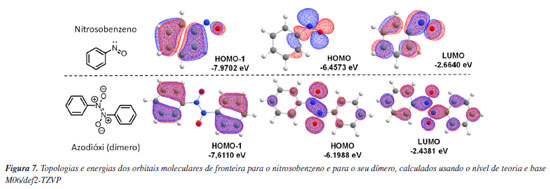
O HOMO do nitrosobenzeno possui os maiores coeficientes localizados sobre o grupo -N=O, o que justifica o nitrogênio desta porção atuar como nucleófilo na reação de dimerização, e não há contribuição relevante de coeficientes nos átomos de carbono do anel aromático. De forma similar, o LUMO (orbital molecular desocupado de mais baixa energia) do nitrosobenzeno possui os maiores coeficientes localizados também sobre o grupo N=O, com simetria π, que corrobora com sua reatividade já conhecida em reações do tipo nitroso aldol, análoga à de compostos carbonílicos, e em reações de cicloadição do tipo nitroso Diels-Alder.48 Assim, ao avaliar o HOMO e o LUMO do nitrosobenzeno é possível racionalizar a sua baixa reatividade em SEAr e corroborar que a reação de dimerização é favorecida pelo controle de orbitais moleculares.
Não é incomum que sistemas instaurados contendo heteroátomos apresentem seu orbital HOMO centrado nos elétrons não ligantes e em ligações sigma, que é o caso do nitrosobenzeno e seu dímero. Por este motivo, a análise da reatividade destes compostos com eletrófilos via SEAr pode ser feita analisando o orbital HOMO-1, que é o orbital mais alto em energia que descreve os elétrons de natureza pi. Para sistemas insaturados contendo maiores quantidades de heteroátomos, pode ser necessária também a análise de outros orbitais ocupados de natureza π, como HOMO-2 ou em diante.69 Assim, ao analisar o HOMO-1 do nitrosobenzeno, nota-se uma distribuição homogênea dos coeficientes sobre o anel aromático, que sugere a inexistência de um efeito diretor pronunciado caso este composto fosse reativo em SEAr. Por outro lado, ao analisar o HOMO do dímero do nitrosobenzeno, se observa que a região do grupo azodióxi apresenta os maiores valores dos coeficientes. Contudo, no anel aromático há contribuição relevante dos coeficientes nos carbonos orto e para, o que explica a regiosseletividade observada nas SEAr. Estendendo a análise para o HOMO-1, também de simetria π, nota-se o reforço de coeficientes em todas as posições do anel.
Figuras dos orbitais HOMO-2, HOMO-1, HOMO e LUMO para os dois compostos estão disponíveis no Material Suplementar para maiores análises, entretanto, salientamos que, embora a análise computacional possa corroborar os dados experimentais e permitir o acesso a propriedades que seriam inacessíveis ou de difícil obtenção por via experimental, os dados verdadeiros, reais e, portanto, confiáveis são os obtidos pela experimentação.
Além disso, os deslocamentos de ressonância magnética nuclear de 13C de benzenos monossubstituídos, quando comparados os efeitos de proteção e desproteção e a regiosseletividade em SEAr para cada um, sugerem que o grupo nitroso seja um grupo desativante e meta dirigente,49 mesmo sem evidências de SEAr com esta orientação. O grupo nitroso também é relatado como um desativante mais forte que o grupo nitro (-NO2) ao comparar a constante de Hammett para ambos, sendo σp de 0,91 para o NO, valor maior quando comparado ao de 0,79 para o NO2.49,50
O conjunto dos cálculos dos orbitais moleculares de fronteira realizados, associado aos fatos experimentais já conhecidos e aqui destacados, norteiam a natureza e o comportamento do nitrosobenzeno e seu respectivo dímero frente às reações SEAr, indicando que o grupo nitroso é desativante e não possui reatividade significativa frente as reações de SEAr, enquanto que a espécie responsável pela reatividade e regiosseletividade é o dímero com o grupo azodióxi, dissociando-se ao final da reação ao monômero nitroso substituído. Portanto, simplificar a explicação do comportamento do grupo nitroso como sendo desativante, porém orto e para dirigente, é de um reducionismo exagerado que não é compatível com o ensino científico, por não levar em consideração o conjunto dos fatos experimentais disponíveis na literatura, e causa insegurança ao estudante por não ser possível explicar esse comportamento por meio da estrutura de Lewis do grupo nitroso e suas implicações no efeito indutivo e de ressonância, como é para os outros grupos funcionais no benzeno monossubstituído.
REATIVIDADE E ORIENTAÇÃO DO ÁTOMO DE FLÚOR NA SUBSTITUIÇÃO ELETROFÍLICA AROMÁTICA
A reatividade e orientação do átomo de flúor na substituição eletrofílica aromática também não são adequadamente explicadas nos textos didáticos. Como apresentado na Figura 3, a maioria dos livros de graduação e da pós-graduação indicam o flúor como desativante na SEAr.
Estudos experimentais e teóricos indicam que o flúor apresenta reatividade particular não análoga ao comportamento do cloro e do bromo em reações de SEAr.51-63 Os que aprofundam a análise51-56 mostram que o grupo flúor difere dos demais grupos funcionais por alterar de forma não homogênea a eletrônica do anel aromático, comportando-se como um grupo ativante em para e desativante nas posições orto e meta em reações de SEAr.
Mais da metade dos livros-textos consultados apresentam erroneamente o flúor apenas como desativante, em detrimento aos de trabalhos da literatura científica primária. Nestes livros, a interpretação equivocada que leva à incoerência surge por não levarem em conta o fator estatístico que pode mascarar os resultados de velocidade de reação.
Como o conceito de ativação/desativação visa analisar o efeito intrínseco do substituinte, e não a facilidade estatística de colisões efetivas entre os reagentes, é necessário normalizar os dados para a análise da reatividade, Figura 8a.
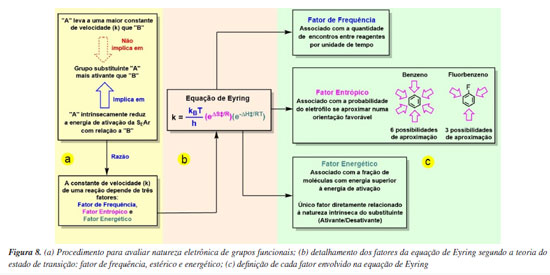
Para normalizar os dados experimentais avaliando apenas o efeito energético do substituinte no anel aromático, deve-se levar em consideração os fatores que impactam na velocidade das reações químicas e removê-los da comparação. Neste sentido, empregando a teoria do estado de transição, a constante de velocidade (k) com que as reações químicas ocorrem depende de três fatores, um de frequência, um estérico e um energético (Figura 8b).64-67 O primeiro é relativo à quantidade de encontros (ou choques) entre os reagentes por unidade de tempo, sendo proporcional a 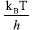 (onde kII é a constante de Boltzmann, T é a temperatura e h é a constante de Planck). O segundo é relativo à probabilidade de os reagentes entrarem em contato com orientação espacial favorável para romper e formar as ligações corretas no estado de transição (choques efetivos), proporcional a
(onde kII é a constante de Boltzmann, T é a temperatura e h é a constante de Planck). O segundo é relativo à probabilidade de os reagentes entrarem em contato com orientação espacial favorável para romper e formar as ligações corretas no estado de transição (choques efetivos), proporcional a 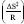 (onde ΔS‡ é a entropia de ativação e R é constante dos gases ideais), de modo que quanto menos provável um estado de transição for, mais o termo entrópico contribui para uma velocidade da reação mais baixa. O terceiro termo é relativo à fração de moléculas com energia suficiente para alcançar o estado de transição, proporcional a e
(onde ΔS‡ é a entropia de ativação e R é constante dos gases ideais), de modo que quanto menos provável um estado de transição for, mais o termo entrópico contribui para uma velocidade da reação mais baixa. O terceiro termo é relativo à fração de moléculas com energia suficiente para alcançar o estado de transição, proporcional a e 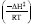 pela distribuição de Maxwell-Boltzmann (onde ΔH‡ é a entalpia), de modo que quanto mais alta a energia de ativação for, mais o termo energético contribui para uma velocidade de reação mais baixa.
pela distribuição de Maxwell-Boltzmann (onde ΔH‡ é a entalpia), de modo que quanto mais alta a energia de ativação for, mais o termo energético contribui para uma velocidade de reação mais baixa.
Nos casos em que os fatores energéticos e estérico apontam em sentidos contrários, o fator estérico pode acabar mascarando o efeito do fator energético na velocidade, por exemplo, superestimando dados de velocidade de reação, quando há quantidades distintas de posições reativas entre dois compostos sob comparação. Uma forma de evidenciar isso é empregar o fluorbenzeno como exemplo de que não é adequado inferir ativação/desativação via velocidades de reação, pois reações com este substrato ocorrem quase em sua totalidade nas posições orto e para em relação ao flúor, havendo praticamente três posições livres passíveis de aproximação do eletrófilo que levam ao avanço da reação, contra seis do benzeno utilizado como comparação. Assim, de forma qualitativa, ao comparar dados de constante de velocidade desses dois, a do benzeno é superestimada por um fator de aproximadamente duas vezes (Figura 8c). Levar em conta o número de posições reativas ao avaliar dados cinéticos ao invés de pura comparação das constantes de velocidade já é algo conhecido, como discutido por Nigst et al.,70 em que hidrazina (H2N-NH2) tem sua constante de velocidade de reação de adição nucleofílica dividida por dois para comparar com a de monoaminas (R-NH2), pois H2N-NH2 apresenta dois centros reativos nitrogenados, enquanto que R-NH2 contém um. Assim, afirmações acerca de ativação e desativação baseados apenas na constante de velocidade de reação devem ser normalizados.
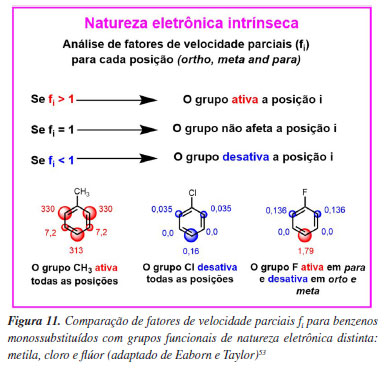
Para analisar o efeito do substituinte na SEAr e afirmar acerca de sua natureza ativante/desativante, a alternativa é normalizar os dados mediante o uso de fatores de velocidade parciais) para cada posição do anel do benzeno monossubstituído (forto, fmeta e fpara), ajustando os dados cinéticos para refletir a velocidade da reação por posição reativa no substrato, removendo assim o efeito do fator estatístico e permitindo a análise apenas pelo fator energético. Essa forma de análise reflete efetivamente o potencial ativante ou desativante do grupo funcional em SEAr, Figura 9.11-18
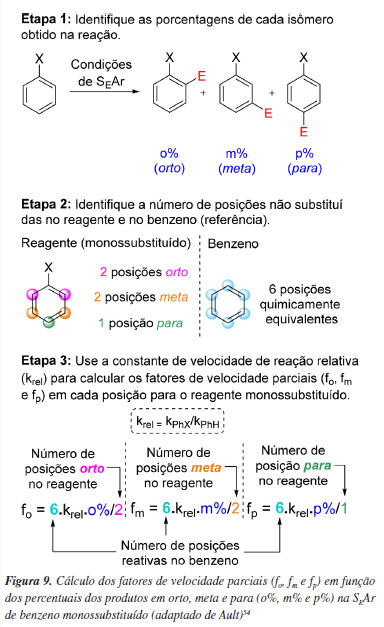
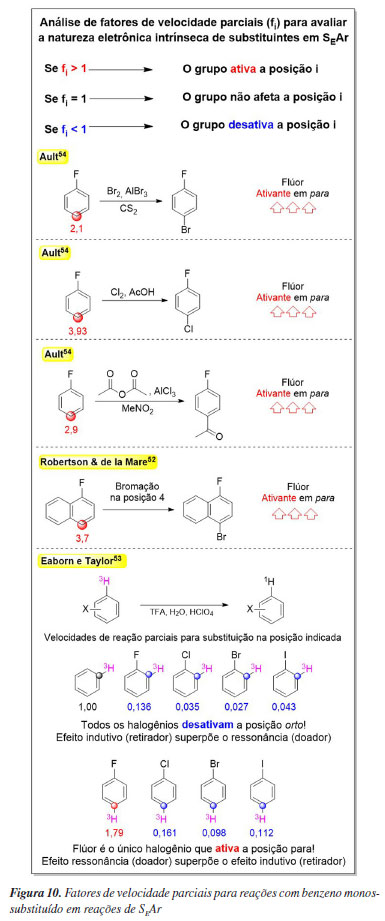
Na literatura,52-54 há exemplos de reações em que foram avaliados os fatores de velocidade parciais de reação de anéis aromáticos substituídos por flúor para analisar o efeito eletrônico por posição reativa, Figura 10. Estes estudos mostram fatores de velocidade parciais < 1 para a posição orto e fatores de velocidade parciais > 1 para a posição para, demonstrando que o grupo funcional flúor, que normaliza os dados cinéticos é um desativante em orto e ativante em para frente a reações de SEAr (Figura 11) e o diferencia dos grupos funcionais clássicos (e de seus análogos Cl e Br) que, ou apenas ativam ou apenas desativam um anel benzênico frente a esse tipo de reação (Figura 12).
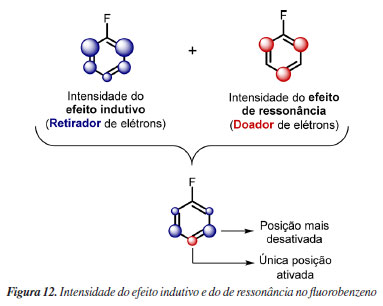
A razão pela qual a reatividade do flúor é tão distinta da de outros grupos funcionais se deve a três fatores. O primeiro decorre de sua alta eletronegatividade, tornando seu efeito indutivo retirador de elétrons extremamente pronunciado. O segundo fator está relacionado aos seus orbitais p de valência serem do nível 2, assim como o carbono, que possibilita uma interação eficiente por ressonância com o anel aromático, favorecendo a doação de elétrons.
Por fim, é possível ponderar as influências do efeito indutivo e de ressonância ou, de forma algo mais precisa, o efeito de campo e o efeito de ressonância para o substituinte do anel benzênico. Como o emprego do efeito indutivo ou o de campo leva a argumentos igualmente válidos e similares, será empregado aqui o efeito indutivo pois, na graduação, o ensino do efeito de campo no contexto da SEAr não é usual, enquanto o efeito indutivo é recorrente. Para cursos avançados na pós-graduação, é recomendável o emprego do efeito de campo.26 Assim, enquanto o efeito indutivo se propaga pelas ligações sigma, diminuindo ao longo de cada ligação percorrido, sendo máximo em orto e praticamente nulo em para, a ressonância ocorre por deslocalização eletrônica em átomos alternados, sendo muito efetiva na posição para. Assim, em orto, o forte efeito indutivo retirador não é compensado pela ressonância, desativando essa posição, enquanto em para o efeito indutivo é mínimo e a ressonância mantém sua contribuição doadora de elétrons, ativando essa posição. Já a posição meta, além de ainda sentir o efeito indutivo do flúor, não recebe contribuição doadora por ressonância devido à alternância eletrônica imposta por esse efeito. Como resultado, essa posição permanece desativada, sendo até mais desativada que a orto, Figura 10.
Como fator adicional à complexidade do padrão de reatividade de aromáticos contendo flúor, certas reações com eletrófilos de alta densidade de carga positiva (eletrófilos duros), como o íon nitrônio em nitrações, mostram o flúor atuando como desativante até mesmo na posição para. Esse efeito foi racionalizado como resultado da repulsão entre o dipolo molecular e o cátion nitrônio. Além disso, há indícios de que, nessas condições, a SEAr possa não ocorrer via um intermediário clássico do tipo complexo sigma (intermediário de Wheland), mas sim por um complexo π. Consequentemente, os fatores parciais de reação não podem ser utilizados diretamente, pois diferentes mecanismos estão envolvidos na reação. A discussão mais detalhada sobre essa particularidade, observada principalmente em nitrações, pode ser encontrada na literatura especializada.52,54,55
O conjunto de informações aqui organizadas amplia outro esforço para ensinar o comportamento do átomo de flúor em química orgânica,56 detalhando as informações de forma condizente com os dados da literatura científica.
CONCLUSÕES
Os dados experimentais da literatura e cálculos dos orbitais moleculares para o nitrosobenzeno, e seu dímero, indicam que o grupo nitroso é desativante e não possui reatividade significativa nas reações de SEAr. Quando o nitrosobenzeno sofre transformação nas condições de reações de SEAr, a espécie responsável pela reatividade e regiosseletividade é o seu dímero com o grupo azodióxi, que é orientador orto/para e se dissocia no final da reação formando o monômero nitroso substituído. Portanto, é errado simplificar a explicação do comportamento do grupo nitroso como sendo desativante, mas orto/para dirigente.
O efeito do átomo de flúor no fluorobenzeno é bem mais complexo, sendo ativante na posição para por efeito de ressonância, e desativante nas posições orto e meta por efeito indutivo. Portanto, é errado simplificar a explicação do comportamento do flúor como sendo desativante e orto/para dirigente.
Para racionalizar o comportamento experimental do fluorobenzeno é necessário considerar os fatores de velocidade parciais para cada posição do benzeno monossubstituído (forto/fmeta/fpara), que normalizam os dados cinéticos para refletir a velocidade da reação em cada posição, suprimindo o efeito estatístico, e permite a análise apenas pelo termo energético da equação de Eyring.
A complexidade dos grupos funcionais nitroso e flúor na SEAr, aliada à divergência entre dados experimentais e informações nos livros didáticos, comprometem o ensino da reatividade e regiosseletividade do nitrosobenzeno e do fluorobenzeno e seus derivados na graduação. É recomendável que os efeitos dos grupos NO e F sejam discutidos em cursos avançados, como os de mecanismos em química orgânica ou de química orgânica avançada, para evitar a propagação de equívocos conceituais presentes nos livros didáticos.
MATERIAL SUPLEMENTAR
Tabela 1S contendo a relação dos livros consultados com a indicação da página onde ocorre a citação aos grupos flúor e nitroso em SEAr, Tabela 2S com detalhes do cálculo computacional para nitrosobenzeno e seu dímero, e Figura 1S com a topologia e energia dos orbitais calculados estão disponíveis em http://quimicanova.sbq.org.br, na forma de arquivo PDF, com acesso livre.
DECLARAÇÃO DE DISPONIBILIDADE DE DADOS
Todos os dados estão disponíveis no texto do artigo e no material suplementar.
AGRADECIMENTOS
Os autores agradecem o suporte financeiro do Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), da Fundação de Amparo à Pesquisa do Estado da Bahia (FAPESB) e da Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) - Código de Financiamento 001. Agradecemos as bolsas de pós-graduação da CAPES para I. Sande e L. Antonelli, a bolsa PIBIC-FAPESB de M. A. R. de Jesus, e a bolsa de produtividade em pesquisa de S. Cunha.
REFERÊNCIAS
1. Exame Unificado de Química, https://euq.sbq.org.br/, acessado em julho 2025.
2. Cunha, S.; Quim. Nova 2018, 41, 825. [Crossref]
3. Cunha, S.; Quim. Nova 2018, 41, 699. [Crossref]
4. Cunha, S.; Quim. Nova 2003, 26, 948. [Crossref]
5. Antonelli, L.; De Paula, R.; Cunha, S.; Quim. Nova 2024, 47, e-20230093 [Crossref]; Antonelli, L.; Nascimento, T. S.; Cunha, S.; Rev. Virtual Quim. 2024, 16, 494. [Crossref]
6. Barbosa, L. C. A.; Introdução à Química Orgânica, 2ª ed.; Pearson: São Paulo, 2010.
7. Constantino, M. G.; Química Orgânica: Curso Básico Universitário, vol. 1, 1ª ed.; LTC: Rio de Janeiro, 2008.
8. Allinger, N. L.; Cava, M. P.; de Jongh, D. C.; Jonhson, C. R.; Lebel, N. A.; Stevens, C. L.; Química Orgânica, 2ª ed.; Guanabara Dois: Rio de Janeiro, 1978.
9. Bruice, P. Y.; Química Orgânica, vol. 1, 4ª ed.; Pearson Prentice Hall: São Paulo, 2006.
10. Klein, D.; Organic Chemistry, 3rd ed.; John Wiley and Sons: New Jersey, 2017; Klein, D.; Organic Chemistry, 2nd ed.; John Wiley and Sons: New Jersey, 2013 (existe a tradução desta edição no Brasil).
11. Carey, F. A.; Química Orgânica, vol. 1, 7ª ed.; McGraw-Hill-Bookmann: São Paulo, 2011.
12. Carey, F. A.; Sundberg, R. J.; Advanced Organic Chemistry Part A: Structure and Mechanisms, 5th ed.; Springer: New York, 2007.
13. Carroll, F. A.; Perspectives on Structure and Mechanism in Organic Chemistry, 2nd ed.; Wiley: New Jersey, 2010.
14. Isaacs, N. S.; Reactive Intermediates in Organic Chemistry, 2nd ed.; Wiley-Blackwell: New Jersey, 1975.
15. Pine, S. H.; Organic Chemistry, 5th ed.; Mcgraw-Hill: New York, Montréal, 1987.
16. Campos, M. M.; Química Orgânica, vol. 1, 1ª ed.; Edgard Blücher: São Paulo, 1976.
17. Liberles, A.; Introduction to Theoretical Organic Chemistry, 1st ed.; The MacMillan Company: New York, 1968.
18. Stock, L. M.; Reações de Substituição Aromáticas; Edgard Blücher: São Paulo, 1969.
19. McMurry, J.; Organic Chemistry, 9th ed.; Thomson Learning: Stanford, 2015.
20. Solomons, T. W. G.; Fryhle, C. B.; Química Orgânica, vol. 1, 10ª ed.; LTC: Rio de Janeiro, 2012.
21. Vollhardt, K. P. C.; Schore, N. E.; Química Orgânica: Estrutura e Função, 6ª ed.; Bookman: Porto Alegre, 2013.
22. Anslyn, E. V.; Dougherty, D. A.; Modern Physical Organic Chemistry; University Science Books: New York, 2005.
23. Clayden, J.; Greeves, N.; Warren, S.; Organic Chemistry, 2nd ed.; Oxford University Press: Oxford, 2012.
24. Cox, B. G.; Acids and Bases: Solvent Effects on Acid-Base Strength; Oxford University Press: Oxford, 2013.
25. Hendrickson, J. B.; Cram, D. J.; Hammond, G. S.; Organic Chemistry, 3rd ed.; McGraw-Hill: New York, 1970.
26. Lowry, T. H.; Richardson, K. S.; Mechanism and Theory in Organic Chemistry, 3rd ed.; Harper & Row: New York, 1987.
27. Smith, M. B.; March's Advanced Organic Chemistry, 7th ed.; Wiley-Interscience: New Jersey, 2013.
28. Morrison, R. T.; Boyd, R. N.; Organic Chemistry, 6th ed.; Prentice Hall International, Inc.: New Jersey, 1992.
29. Streitwieser, A.; Heathcock, C. H.; Kosower, E. M.; Introduction to Organic Chemistry, 4th ed; Macmillan Publishing Company: New York, 1992.
30. Smith, M. B.; Organic Chemistry: An Acid-Base Approach; CRC Press: Boca Raton, 2011.
31. Robert, J. D.; Caserio, M. C.; Basic Principles of Organic Chemistry, 2nd ed.; W. A. Benjamin, Inc.: Menlo Park, 1977.
32. Bruckner, R.; Organic Mechanisms Reactions, Stereochemistry and Synthesis, Tradução: Karin Beifuss, 3rd ed.; Springer-Verlag: Berlin, 2010.
33. Schreck, J. O.; Organic Chemistry: Concepts and Applications; C. V. Mosby Company: St. Louis, 1975.
34. Geissman, T. A.; Principles of Organic Chemistry, 3rd ed.; W. H. Freeman and Company: San Francisco, 1968.
35. Terra, B.; Química Orgânica e Noções Elementares de Alguns Assuntos de Bioquímica, vols. 1 e 2, 4ª ed.; Editora Científica: Rio de Janeiro, 1952.
36. Finar, I. L.; Organic Chemistry; Longmans, Green and CO: London, New York, Toronto, 1951.
37. Noller, C. R.; Chemistry of Organic Compounds, 3rd ed.; W. B. Saunders Company: Philadelphia, 1965.
38. Fessenden, R. J.; Fessenden, J. S.; Organic Chemistry, 3rd ed.; Brooks Cole Pub Co: California. 1998.
39. Hine, J.; Physical Organic Chemistry, International Student Edition, 2nd ed.; McGraw-Hill: New York, 1962.
40. Gould, E. S.; Mechanism and Structure in Organic Chemistry; Holt Rinehart and Winston, Inc.: New York, 1964.
41. Webster, M. S.; J. Chem. Soc. 1956, 2841. [Crossref]
42. Gowenlock, B. G.; Lüttke, W.; Q. Rev. Chem. Soc. 1958, 12, 321. [Crossref]
43. Beaudoin, D.; Wuest, J. D.; Chem. Rev. 2016, 116, 258. [Crossref]
44. Gowenlock, B. G.; Richter-Addo, G. B.; J. Chem. Educ. 2008, 85, 1243. [Crossref]
45. Ingold, C. K.; J. Chem. Soc., Trans. 1925, 127, 513. [Crossref]
46. Hammick, D. L.; Illingworth, W. S.; J. Chem. Soc. 1930, 2358. [Crossref]
47. Robertson, P. W.; Hitchings, T. R.; Will, G. M.; J. Chem. Soc. 1950, 808. [Crossref]
48. Yamamoto, H.; Momiyama, N.; Chem. Commun. 2005, 3514. [Crossref]
49. Schnatter, W. F. K.; Rogers, D. W.; Zavitsas, A. A.; J. Phys. Chem. A 2013, 117, 13079. [Crossref]
50. Hansch, C.; Leo, A.; Taft, R. W.; Chem. Rev. 1991, 91, 165. [Crossref]
51. Ingold, C. K.; Vass, C. C. N.; J. Chem. Soc. 1928, 417. [Crossref]
52. De la Mare, P. B. D.; Robertson, P. W.; J. Chem. Soc. 1948, 100. [Crossref]
53. Eaborn, C.; Taylor, R.; J. Chem. Soc. 1961, 2388. [Crossref]
54. Ault, A.; J. Chem. Educ. 1966, 43, 329. [Crossref]
55. Robertson, P. W.; de la Mare, P. B. D.; Swedlund, B. E.; J. Chem. Soc. 1953, 782. [Crossref]
56. Rosenthal, J.; Schuster, D. I.; J. Chem. Educ. 2003, 80, 679. [Crossref]
57. Ingold, C. K.; Shaw, F. R.; J. Chem. Soc. 1927, 2918. [Crossref]
58. Holmes, E. L.; Ingold, C. K.; J. Chem. Soc. 1926, 1328. [Crossref]
59. Holleman, A. F.; Recl. Trav. Chim. Pays-Bas Belg. 1900, 19, 188. [Crossref]
60. Holleman, A. F.; Recl. Trav. Chim. Pays-Bas Belg. 1900, 19, 364. [Crossref]
61. Holleman, A. F.; Recl. Trav. Chim. Pays-Bas Belg. 1903, 22, 263. [Crossref]
62. Holleman, A. F.; Recl. Trav. Chim. Pays-Bas Belg. 1905, 24, 140. [Crossref]
63. Holleman, A. F.; Recl. Trav. Chim. Pays-Bas Belg. 1913, 32, 134. [Crossref]
64. Avery, H. E.; Cinética Química Básica y Mecanismos de Reacción; Reverté: Barcelona, 1982.
65. Atkins, P. W.; Paula, J.; Físico-Química, vol. 2, 7ª ed.; LTC: Rio de Janeiro, 2004.
66. Levenspiel, O.; Engenharia das Reações Químicas; Blucher: São Paulo, 2000.
67. Stewart, R.; A Investigação de Reações Orgânicas; Edgard Blücher: São Paulo, 1969.
68. Neese, F.; WIREs Comput. Mol. Sci. 2012, 2, 73. [Crossref]
69. Costa, P.; Pilli, R.; Pinheiro, S.; Vasconcellos, M.; Substâncias Carboniladas e Derivados, 1ª ed.; Bookman: São Paulo, 2003. Ver em especial os Apêndices desta edição.
70. Nigst, T. A.; Antipova, A.; Mayr, H.; J. Org. Chem. 2012, 77, 8142. [Crossref]
Editor responsável pelo artigo: Jorge M. David