
 |
 |
 |
 |
 |
Artigo
| Novel dihydropyrimidine-thione derivatives as cholinesterase inhibitors targeting alzheimer's disease: advanced structural characterization, computational modelling and biological evaluation |
|
Aisha A. AlsfoukI; Pervaiz Ali ChannarII; Aftab AhmedIII; Syeda Abida EjazIII,* I Department of Pharmaceutical Sciences, College of Pharmacy, Princess Nourah bint Abdulrahman University, P.O Box 84428, 11671 Riyadh, Saudi Arabia Received: 04/24/2025; *e-mail: abida.ejaz@iub.edu.pk, abidaejaz2010@gmail.com;
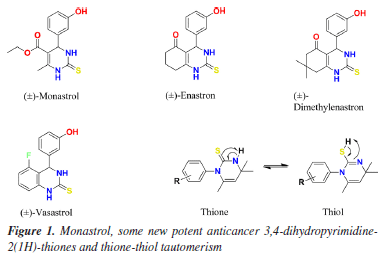
asaeed@qau.edu.pk, aamersaeed@yahoo.com Editor handled this article: Nelson H. Morgon Green synthesis of two dihydropyrimidine-thione derivatives, i.e., 1-(4-bromo-2-fluorophenyl)-4,4,6-trimethyl-3,4-dihydropyrimidine-2(1H)-thione (SA1) and 1-(3-chloro-4-fluorophenyl)-4,4,6-trimethyl-3,4-dihydropyrimidine-2(1H)-thione (SA2), was accomplished through a convenient and simple protocol by the reaction of acetone, potassium thiocyanate, and corresponding anilines (4-bromo-2-fluoroaniline and 3-chloro-4-fluoroaniline). The single crystal X-ray diffraction (SCXRD) studies along with Hirshfeld surface analysis were conducted to gain insight of the structural details and intermolecular interactions within the crystal lattice. An extensive network of intermolecular interactions was observed in the SA2 crystal packing implying its higher reactive potential. The density functional theory (DFT) calculations conducted to estimate the global reactivity parameters underscored the high reactivity of both derivatives. Our in vitro experiments demonstrated that compound SA2 showed greater inhibition than SA1 against the targeted enzymes, acetylcholinesterase and butyrylcholinesterase, which were inhibited with IC50 (half-maximal inhibitory concentration) values of 27.2 ± 2.4 µM and 51.8 ± 2.7 µM, respectively. Also, the correlation between the descriptors of DFT reactivity and the biological activity was empirically observed. The favorable binding and interaction potential for SA2 can be explained by the optimized electronic structure, which increased the binding to the enzyme targets. In addition, the potential drug-like properties of both compounds, including their toxicity profiles, were evaluated through computational predictive modeling. INTRODUCTION Alzheimer's disease is a progressive neurodegenerative disorder marked by neurodegeneration, cognitive impairment, and deposition of amyloid-β plaques and neurofibrillary in brain tissues.1 Published statistical data of WHO 20232 reported that over 55 million people are living with dementia across the world. A sharp rise to 139 million cases were expected by 2050 affecting the low-income countries in disproportionate manner. Among 100 different kinds of dementia, Alzheimer's disease (AD) is the well-known type making up 50 to 75% of all cases.3-5 AD causes the disruption of neurotransmission by affecting the neurons and cellular structures especially neural pathways involved in the storage of cognitive information. The gradual memory loss is the first sign of AD yet other symptoms include trouble with problem-solving, choosing the proper term, and correctly identifying others.6 Alzheimer disease associated co-morbidities, i.e., diabetes mellitus, neurodegenerative disorders, cardiovascular diseases, and renal diseases may make the pathophysiology of AD more complex.7 In spite the high prevalence of AD in some diabetic groups, co-existence of both pathological disorders is not imperative.8 Large number of hypotheses have been proposed to explain the neuropathology of Alzheimer's disease which include cholinergic, amyloid, tau propagation, mitochondrial cascade, calcium homeostasis, neurovascular, inflammatory, metal ion and lymphatic system. The cholinergic hypothesis was emerged from the comparative study of neurotransmitter-related enzyme activities in Alzheimer's disease (AD) and healthy brains. There was a significant decline in choline acetyltransferase (ChAT) activity, a crucial enzyme for ACh synthesis in the amygdala, hippocampus, and cortex of AD brains, correlating with reduced synaptic acetylcholine levels. In contrast, other enzymes, such as glutamic acid decarboxylase, tyrosine hydroxylase, and monoamine oxidase, remained within normal ranges. Since ChAT depends on choline, acetyl-CoA, and adenosine triphosphate (ATP) for its catalytic function, these findings supported the novel concept of AD as a cholinergic system dysfunction. Later studies confirmed similar cholinergic deficits in other neurological and psychiatric conditions, including Parkinson's disease (PD) and depression. To counteract cognitive decline in AD, acetylcholinesterase inhibitors (AChEIs) were developed to prevent acetylcholine breakdown. Following the Food and Drug Administration (FDA) approval of tacrine, the first reversible AChEI, in 1995,9 these drugs became a cornerstone of AD treatment for over two decades. However, due to hepatotoxic potential of tacrine, its use declined with the introduction of safer alternatives like donepezil, rivastigmine, and galantamine. Rivastigmine and galantamine show superior effects on daily functioning compared to donepezil. Despite their clinical use, current AD medications primarily improve the patient's quality of life without halting disease progression. There is a need to develop newer medicinal agents having activity against the targets involved in the progression of AD.10-13 Neuropathology of Alzheimer's disease is characterized by coexistence of intracellular neurofibrillary tangles (NFTs), which consist of abnormally hyperphosphorylated tau proteins that aggregate into paired helical filaments, while extracellular Aβ deposits result in parenchymal senile plaques and cerebrovascular accumulations known as cerebral amyloid angiopathy (CAA) or congophilic angiopathy.14 The revised amyloid cascade hypothesis proposed by Volloch and Rits-Volloch15 highlights the soluble Aβ oligomers as the primary molecular instigators in the Alzheimer's disease progression diverting the focus on amyloid plaques. These small soluble aggregates are responsible for neuronal impairment and initiate the tau related damage. Despite the progress in therapeutics development against AD, challenges persist, including inefficiencies in trial design, incomplete biomarker validation, and insufficient demographic diversity, with only 23% of trials actively addressing population representativeness.16,17 Among the other strategies in drug design for AD, concept of targeting the specific enzymes contributing to pathogenesis of AD has been proven effective. A decreased amount of neurotransmitter acetylcholine (ACh), a neurotransmitter hydrolyzed by the enzymes acetylcholinesterase (AChE) and butyrylcholinesterase (BChE), has been associated to Alzheimer's disease. Hence, AChE and BChE are the promising targets in this regard. Acetylcholinesterase inhibitors (AChEI) such as tacrine, donepezil, and galantamine are being used for the symptomatic treatments of patients suffering from AD.18 Among the different heterocyclic compounds, thiones have been shown to have some pharmacological properties, including antimicrobial, anti-cancer and anti-inflammatory activity.19-21 Thiones heterocyclic ring has been investigated as a potential therapeutic agent for neurodegenerative diseases such as Alzheimer's and Parkinson's disease.22 The pyrimidine ring system, which is found in many biomolecules such as vitamins and nucleic acids, plays an important role in various biological processes.23,24 Dihydropyrimdin-2(1H)-thiones, are the most important class of compounds that contain pyrimidine ring in their structure.25 Physicochemical properties of dihydropyrimidine and pyridine derivatives are influenced by thione-thiol tautomerism (Figure 1) which is a solvent dependent equilibrium. Polar solvents favor the thione tautomer whereas thiol form is preferentially stabilized by non-polar solvents.26 These organosulfur compounds demonstrate wide pharmacological active profile including the antimicrobial, antifungal and platelet aggregation inhibitory activity. Advanced structural elucidation through X-ray diffraction, spectroscopic analysis, and quantum chemical computations has provided comprehensive understanding of their molecular architecture and electronic configurations.27,28 Therefore, the synthesis of dihydropyrimidine-2-thiones has become a major focus of organic and medical chemistry. Unfortunately, the potential of this class of compounds remained unexplored for some time until the 1980s. However, the synthesis of many other heterocyclic compounds has been traced back to these fully flexible precursors.29,30 Monastrol was the prototype anticancer drug based upon the dihydropyrimidinones scaffold. Several other analogues of monastrol including the enastron, dimethylenastron and vasastrol (Figure 1) were synthesized to explore the highly potent anticancer agent.31-35
Given the significance of 3,4-dihydropyrimidine-2(1H)-thiones, this study investigates the structural properties and biological potential of two derivatives: 1-(4-bromo-2-fluorophenyl)-4,4,6-trimethyl-3,4-dihydropyrimidine-2(1H)-thione (SA1) and 1-(3-chloro-4-fluorophenyl)-4,4,6-trimethyl-3,4-dihydropyrimidine-2(1H)-thione (SA2), using well-established computational methods. Furthermore, the study explores their inhibitory effects against key enzymes implicated in Alzheimer's disease. Computational and experimental analyses suggest that both compounds exhibit promising chemical reactivity, indicating their potential as lead molecules for the development of novel anti-cholinesterase agents.
METHODOLOGY General procedure for the preparation of 1-(aryl)-4,4,6-trimethyl-3,4-dihydropyrimidine-2(1H)-thione derivatives Initially, potassium thiocyanate (1.0 mmol) was added to 4-methylpent-3-en-2-one (1.20 mmol) in a reaction flask, and the mixture was stirred continuously at room temperature until a suspension formed. Subsequently, substituted aniline (1.0 mmol) was added portion-wise under continuous stirring. The reaction mixture was then heated to 50-60 °C for 3-5 h, with progress monitored by thin-layer chromatography (TLC). Upon completion, the mixture was cooled to room temperature and poured into ice-cold water to induce precipitation. The resulting solid was collected by filtration and washed with ice-cold water. The crude product was then recrystallized from ethanol to obtain the purified dihydropyrimidine-2-thiones (1-2) in good yield. For further characterization, the dried products were dissolved in ethyl acetate (after confirming solubility), and colorless crystals suitable for X-ray analysis were grown via slow evaporation. The synthesis procedure is given in Scheme 1. Detailed synthesis protocol can be studied by our previous papers.36
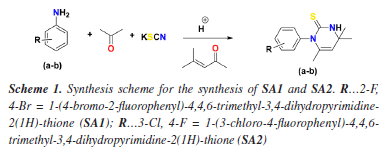
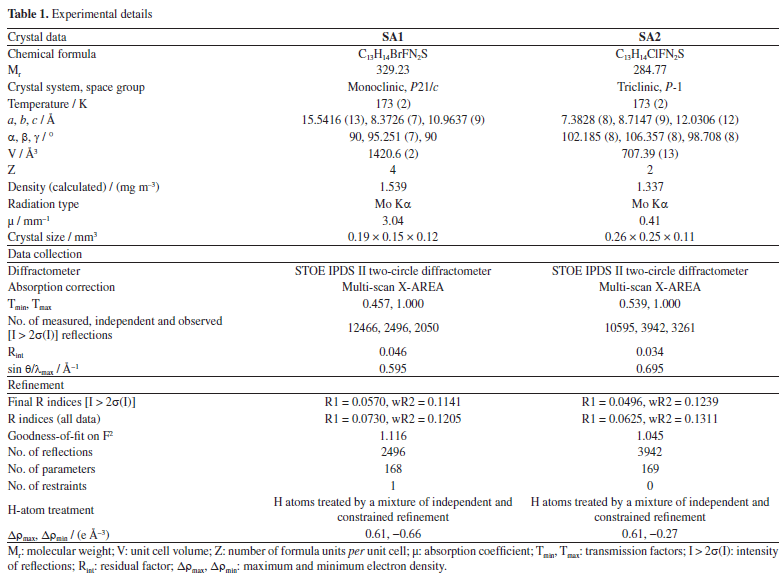
1-(4-Bromo-2-fluorophenyl)-4,4,6-trimethyl-3,4-dihydropyrimidine-2(1H)-thione (SA1) Yield 85%; M.P. (melting point) 151 °C; Rf 0.43 (EtOAc:petroleum ether 1:9), IR (neat) ν / cm-1 3217 (N-H), 3052 (=C-H stretch), 1697 (S-NH), 1597 (C=C), 1262 (C-N); 1H NMR (300 MHz, DMSO-d6) d 11.21 (s, 1H, NH), 7.99-7.66 (m, 3H, Ar-H), 5.36 (s, 1H, Csp2-H), 2.80 (s, 3H, Csp2-CH3), 1.78 (s, 6H, 2 × CH3); 13C NMR (75 MHz, DMSO-d6) d 168.6 (C=S), 160.2, 138.3, 136.5, 135.4, 131.9, 131.3, 130.5, 128.5 (Ar-C), 112.8, 56.2, 31.5, 20.2; anal. calcd. for C13H14BrFN2S: C, 47.43; H, 4.29; N, 8.51; S, 9.74; found: C, 47.40; H, 4.24; N, 8.55; S, 9.71; HRMS calcd. C13H14BrFN2S [M+]: 328.0045, found 328.0042. 1-(3-Chloro-4-fluorophenyl)-4,4,6-trimethyl-3,4-dihydropyrimidine-2(1H)-thione (SA2) Yield 85%, M.P. 158 °C; Rf 0.48 (EtOAc:petroleum ether 1:9), IR (neat) ν / cm-1 3215 (N-H), 3058 (=C-H stretch), 1696 (S-NH), 1578 (C=C), 1268 (C-N); 1H NMR (300 MHz, CDCl3) d 9.76 (s, 1H, NH), 7.82-7.53 (m, 3H, Ar-H), 5.23 (s, 1H, Csp2-H), 2.09 (s, 3H, Csp2-CH3), 1.79 (s, 6H, 2 × CH3); 13C NMR (75 MHz, CDCl3) d 179.0 (C=S), 154.3, 137.5, 134.5, 133.7, 131.6, 127.4, 125.0 (Ar-C), 100.0, 55.7, 29.5, 22.0; anal. calcd. for C13H14ClFN2S: C, 54.83; H, 4.96; N, 9.84; S, 11.26; found: C, 54.78; H, 4.98; N, 9.80; S, 11.26; HRMS calcd. C13H14ClFN2S [M+]: 284.0550, found 284.0547. X-ray crystallography X-ray diffraction data were collected on a STOE IPDS II two-circle diffractometer using Mo Kα radiation (λ = 0.71073 Å). The structures were solved and refined using the SHELX program package SHELXL-2014/6 (Sheldrick, University of Göttingen, Germany, 2014) and analyzed with XP in SHELXTL-Plus (Bruker AXS Inc., Madison, WI, USA). A multi-scan absorption correction was applied using X-AREA (Stoe & Cie, Germany, 2001). Molecular graphics and structural analysis were performed using ORTEP-3 (Johnson, Oak Ridge National Laboratory, Oak Ridge, TN, USA, 1994) and PLATON (Spek, Utrecht University, The Netherlands, 2020). The NH hydrogen atoms were located from difference Fourier maps and refined isotropically. All other hydrogen atoms were placed in idealized positions and refined using a riding model, with Uiso(H) = 1.2 Ueq(C) for CH groups and 1.5 Ueq(C) for methyl groups. The crystallographic data for the structure reported in the document has been deposited with the Cambridge Crystallographic Data Centre (CCDC) as Supplementary Material.37 The experimental details of both compounds SA1 and SA2 are given in Table 1.
Hirshfeld surface analysis Hirshfeld surface analysis was performed to quantify and visualize the intermolecular interactions in the crystal structures of SA1 and SA2 using CrystalExplorer 21.5 (University of Western Australia, Australia, 2021). The normalized contact distance (dnorm) surfaces were generated and mapped with a red-white-blue color scheme, where red regions correspond to contacts shorter than the sum of van der Waals radii, white areas represent distances near van der Waals separation, and blue zones indicate longer interactions.38 Density functional theory (DFTs) studies Density functional theory (DFT) has emerged as a pivotal tool in theoretical medicinal chemistry, with its foundation rooted in the Hohenberg-Kohn theorem which establishes that electron density exclusively determines the ground state energy and molecular properties.39 For this study, initial molecular structures were constructed using Gaussian 09 software (Revision D.01, Gaussian, Inc., Wallingford, CT, USA, 2013), followed by geometry optimization employing the hybrid B3LYP functional (Becke's three-parameter exchange with Lee-Yang-Parr correlation).40,41 The B3LYP functional was selected due to its well-established accuracy for organic systems, offering an optimal balance between computational cost and reliability for predicting molecular geometries and electronic properties.42 Comparative analyses were performed using two basis sets: the polarized triple-zeta 6-311G(d,p) basis set, which includes d-type polarization functions on heavy atoms and p-type on hydrogens,43 and the correlation-consistent aug-cc-pVDZ basis set that incorporates diffuse functions for improved description of electron density distribution and non-covalent interactions.44 Final DFT calculations were executed at the B3LYP/6-311G(d,p) level of theory, chosen for its proven accuracy in similar medicinal chemistry applications.45 All quantum mechanical computations were conducted on multi-core processing systems, with molecular visualization and orbital analysis performed using GaussView 6 software (Gaussian, Inc., USA, 2017). Enzyme inhibition assay The cholinesterase inhibitory activity was evaluated using a modified Ellman's method.46-48 Briefly, 96-well microplates were prepared by sequentially adding 0.2 U of enzyme (AChE or BChE), 25 μL of 0.5 mM 5,5'-dithiobis(2-nitrobenzoic acid) (DTNB) as substrate, 50 μL of 0.1 M sodium phosphate buffer (pH 8.0), and 25 μL of test compounds (a and b) at concentrations of 200, 100, and 50 ppm. After incubation at 37 °C for 30 min, the absorbance was measured at 405 nm using an ELx-800 microplate reader (BioTek Instruments, USA). Galantamine hydrobromide (at equivalent concentrations) served as the positive control. The percentage inhibition was calculated using the formula:  Galantamine hydrobromide were used as positive controls for cholinesterase assays. The experiments were performed in triplicates to ensure the reproducibility of the results. Docking studies Docking studies were performed to study the binding interactions between ligand and protein molecule. AutoDock Vina 1.2.3 (The Scripps Research Institute, USA, 2022) was employed for docking procedure due to its highly accurate algorithms. Proteins structures were retrieved from Protein Data Bank under PDB IDs: 7E3H49 and 5DYW49 for acetylcholinesterase and butyrylcholinesterase, respectively. The co-crystal ligand of each protein was docked along with proposed ligands to validate the docking procedure employing the concept of self-docking. Active sites for the docking purpose were located based upon literature50-52 review and considering the coordinates of co-crystal ligand. Grid box center coordinates were x = -44.849679 Å, y = +9.506214 Å, z = -81.321214 Å for AChE (PDB: 7E3H) and x = 16.283906 Å, y = -24.849469 Å, z = 41.551094 Å for BChE (PDB: 5DYW), with size (dimension) value of 40 Å (cubic box). Exhaustiveness value of 20 with default energy range of 3.0 kcal mol-1 was employed. Best docking pose (solution) was used for analysis. Discovery Studio Visualizer (Version 2021, BIOVIA, Dassault Systèmes, USA, 2020) was used for protein-ligand interactions analysis of the best docking poses. Normal mode analysis (NMA) The iMODs server53 was used to conduct normal mode analysis (NMA) on April 15, 2025. For our analysis, we took the rigid protein structures resulting from the docking of SA1 and SA2. These structures allowed the modelling of transition pathways of the enzyme-ligand complexes at 300 K and 1 atm. iMODs offers online enhanced NMA with a friendly graphical interface that employs internal coordinates like dihedral angles, which allows the holistic functional description of movements of large biological molecules.54-56 As a caveat, although the proteins were kept rigid during docking, NMA had no regard to the conformational and flexibility changes due to ligand attachment, as the analysis was conducted considering the proteins' conformational flexibility unbound to ligands. ADMET (absorption, distribution, metabolism, excretion, and toxicity) evaluation The drug-likeness of the synthesized compounds was assessed using the ADMET analysis. An ADMETlab 3.0 online server57 was employed to evaluate physicochemical properties, pharmacokinetics parameters as well as toxicological and medicinal properties. These investigations were conducted to learn more about the synthetic compounds drug-like properties, which may facilitate the future drug development efforts. The effectiveness and safety of possible medication candidates need to be further validated through experiments.
RESULTS AND DISCUSSION Chemistry The synthesis of compounds under study has been already reported.36 For the current study, the green synthesis procedure was carried out according to the previously reported method with slight modifications.58 The procedure for the preparation of 1-(R-phenyl)-4,4,6-trimethyl-3,4-dihydropyrimidine-2(1H)-thione derivatives involved the reaction of substituted anilines with potassium thiocyanate in the presence of 4-methylpent-3-en-2-one as a solvent. X-ray structure In the molecule of the SA1 (Figure 2), atoms Br1, F1 and N1 are -0.0331(7), 0.0375(33) and -0.0417(39) Å away from the best least-squares plane of ring A (C11-C16), respectively, while ring B (N1/N2/C1-C4) is in envelope conformation where atom C2 is at the flap position and -0.2286(49) Å away from the best least-squares plane of the remaining atoms of the ring. In the molecule of SA2 (Figure 2), atoms Cl1, F1 and N1 are 0.0413(6), 0.0229(16) and 0.1033(16) Å away from the best least-squares plane of ring A (C11-C16), respectively, while ring B (N1/N2/C1-C4) is in envelope conformation where atom C2 is at the flap position and -0.2268(23) Å away from the best least-squares plane of the remaining atoms of the ring.
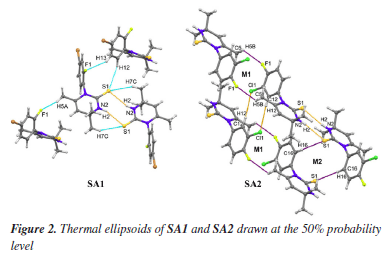
Supramolecular chemistry techniques play a vital role in drug discovery and the development, as biological functions are heavily influenced by non-covalent interactions. These interactions involve both strong and weak hydrogen bonds. Supramolecular synthons are the specific patterns of interactions that describe the manner in which complementary molecular components align and interact with each other. Identification of synthons are important for crystal structure analysis. Screening appropriate synthons can facilitate the design of ligand molecules that exhibit optimal binding affinity for specific protein pockets.59,60 Crystal lattice of SA1 was stabilized by large number of hydrogen bonds. Co-planar supramolecular synthon was formed due to N2-H2···S1 hydrogen bond. H7C···S1 and its reciprocal contact formed a dimer enclosing NH···S synthon with identical bond lengths of 2.940 Å. This directional interaction is complemented by additional hydrogen bonds (H13, H12, H7C, H5A) that collectively form a three-dimensional framework, while the halogen atoms (F1) participate in conventional hydrogen bonds C-X···H (H5A, H13) as shown in Figure 3. These strong directional interactions (hydrogen and halogen bonds) create a stable supramolecular architecture. Hydrogen bond geometry of selected atoms is given in Table 2.
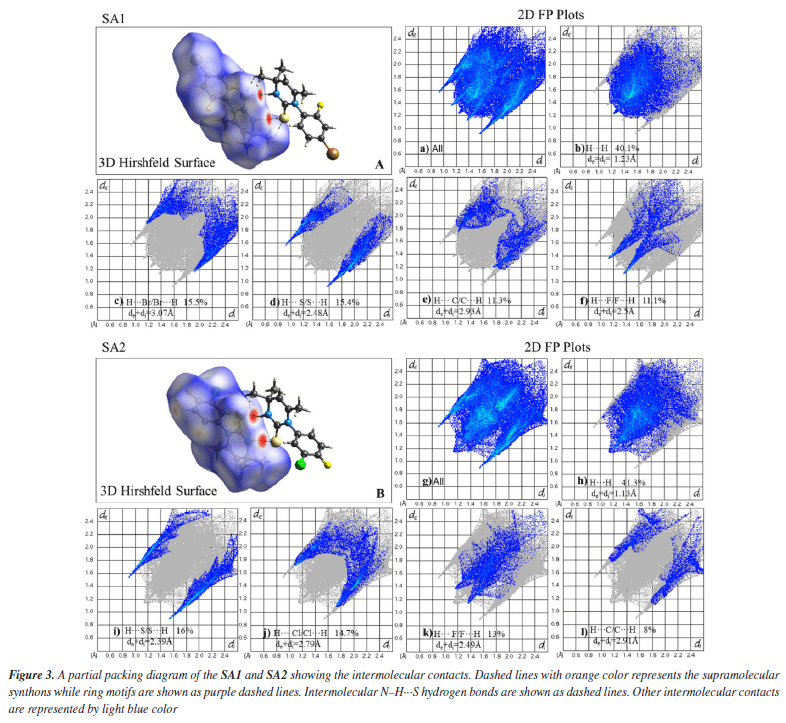

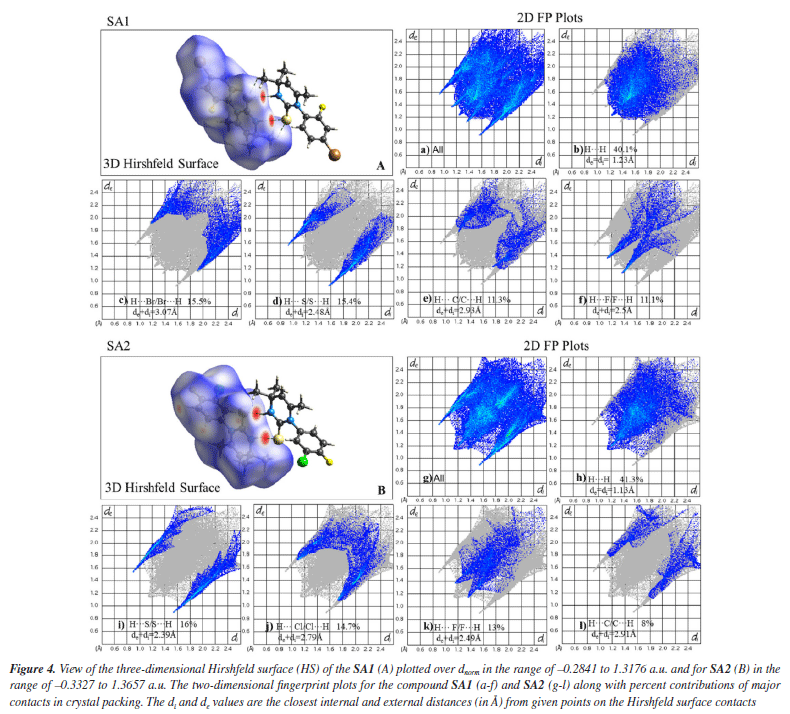
In the crystal lattice of SA2, ring motifs and supramolecular synthons were formed as a result of hydrogen bonding and short contacts. Ring motif (M1) formation resulted due to C5-H5B···F1 and the reciprocal contacts with graph-set notation R12(18) while C16-H16···S1 and the reciprocal contacts lead to the formation of ring motif (M2) with graph-set notation R12(12). Two supramolecular synthons were formed due to N2-H2···S1 hydrogen bonds and C12-H12···C11 contacts as shown in Figure 3. Upon careful analysis of the interaction pattern exists among both crystals, it can be observed that presence of an extensive network of strong intermolecular interactions imparts higher stability to SA2 crystal lattice as compared to SA1. Hirshfeld surface analysis The 3D Hirshfeld surface analysis enables the quantification of various molecular properties, such as shape, volume, surface area, globularity, and asymmetry. The outcomes of this analysis provide a deeper understanding of the molecular characteristics, facilitating the design of new compounds with specific and targeted properties.61 Hirshfeld surface analysis provides a detailed understanding of intermolecular interactions in the crystal structure based on the electron density at each contact point.62 In order to visualize the intermolecular interactions in the crystal of the synthesized compounds, a Hirshfeld surface (HS) analysis was carried out by using Crystal Explorer 21.5. Based on the Hirshfeld surface, the 2D fingerprint plots offer a visual representation of the distribution and frequency of de and di values across the molecular surface. These plots highlight the intermolecular interactions as well as relative areas of the surface that contribute to each interaction type. The most significant interaction to the total crystal packing in both SA1 and SA2 is H···H interaction. This is depicted in Figure 4 as widely scattered spots of high density due to the considerable hydrogen content of the molecule. There is a symmetrical distribution of points between the pairs of spikes in the fingerprint plots that are defined as H···Br/Br···H and H···S/S···H in SA1 while characterized as H···S/S···H interactions H···Cl/Cl···H contacts and H···F/F···H contacts in SA2. The pair of typical wings in the fingerprint plot are representing H···C/C···H contacts in both crystals. The Hirshfeld surface study provides more evidence that forming connections between H-atoms is critical to the packing process. Because of the high number of interactions of the types H···H, H···S/S···H, H···Cl/Cl···H, and H···F/F···H, it is likely that van der Waals interactions and hydrogen bonding play the most important roles in the crystal packing.63
DFT studies Density functional theory calculations For the calculation of optimized geometries, B3LYP level of theory and two basis set 6-311G d,p (Gaussian basis set with added diffuse function) and aug cc-pVDZ (larger Dunning-type basis set, double zeta) were employed. B3LYP was used due to its cost effectiveness, accuracy and empirical calibration. DFT calculated theoretical bond lengths and bond angles of selected atoms of SA1 and SA2 were compared with the experimentally calculation as shown in Tables 3 and 4, respectively. Gaussian default labelling scheme was applied for both structures.
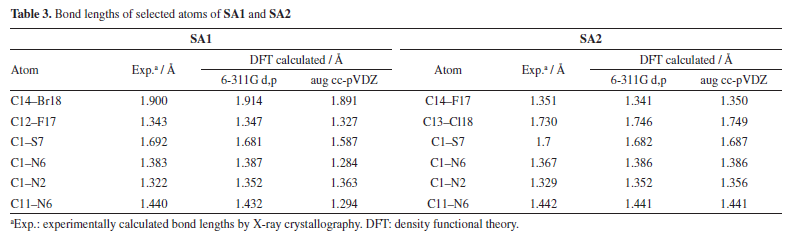
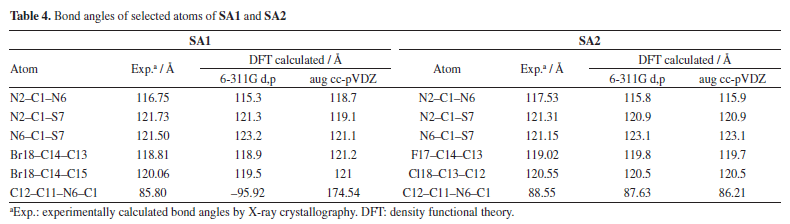
Tables 3 and 4 indicated that values of bond lengths and bond angles obtained from B3LYP/6-311G d,p basis set were found in a good agreement with the experimentally calculated results. Keeping in view the results of geometry optimization results B3LYP/6-311G d,p basis set were used for further DFT studies. Optimized structures of SA1 and SA2 calculated from B3LYP/6-311G d,p basis set are shown in Figure 5.
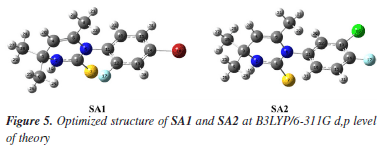
To investigate potential atropisomerism and conformational preferences, we performed a relaxed dihedral scan around the phenyl-pyrimidine torsion (C12-C11-N6-C1) using the B3LYP/6‑311G(d,p) level of theory. The potential energy surface (PES) revealed two local minima for both SA1 and SA2, suggesting the presence of stable (P)- and (M)-like conformers. However, the torsional energy barriers were found to be much lower than 20 kcal mol-1 (EB ~4 kcal mol-1 for SA1 and EB ~3.5 kcal mol-1 for SA2), indicating that these compounds are not typical atropisomers that can be isolated at room temperature. This observation contradicts the definition of atropisomerism as outlined in the literature,64,65 where the torsional energy barriers for isolable atropisomers are usually greater than 20 kcal mol-1. Additionally, we analyzed the torsion angles from the highest-ranked docking poses with the X-ray diffraction (XRD) and density functional theory (DFT) calculations. As illustrated in Table 5, SA1 showed more pronounced variability in the torsion angle through the methods applied, while SA2 conserved geometry across XRD, DFT, and docking. Importantly, SA1 showed greater torsional flexibility which correlates with the lower torsional energy barriers observed, while SA2 adopted a more rigid conformation.
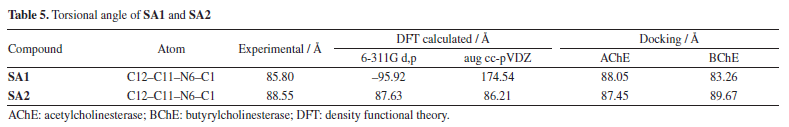
The torsional angle of SA1 using the aug cc-pVDZ basis set computed torsional angle showed almost co-planar rings which is in contrast to the experimental results, which is why the 6-311G d,p basis set was then used in further DFT calculations. The PES scan plot of SA1 and SA2 which incorporates torsional rotation of an aryl moiety and a pyrimidine ring, is available in the Supplementary Material (Figure 6S). Even though the angles were calculated differently than the experimental results, the discrepancy was likely a result of the basis set used or the force field used to parameterize docking calculations. Overall, SA1 exhibited larger torsional flexibility across methods while SA2 showed consistent geometry throughout XRD, DFT, and docking. This is suggestive that the biologically active conformation of SA2 is likely approximating the geometry of the crystal, reinforcing the notion that SA2 displays characteristics suggesting atropisomeric behavior. The low barriers to torsional rotation mean that neither SA1 nor SA2 could be classified as true isolable atropisomers.
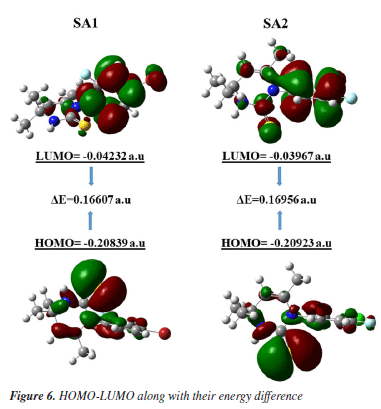
Frontier molecular orbital analysis Reactivity patterns of a molecule can be studied by frontier molecular orbital, i.e., HOMO-LUMO (highest occupied molecular orbital-lowest unoccupied molecular orbital) analysis. Global reactivity descriptors were assessed by using the DFT calculated values of HOMO and LUMO. Physical and quantum mechanical descriptors of SA1 and SA2 calculated by employing the B3LYP/6-311G d,p are given in Table 6.
Figure 6 shows that, for both SA1 and SA2, the electron density is localized in almost the same regions. Specifically, in the LUMO, the electron density is concentrated on the phenyl ring where the halogen atom is attached, while in the HOMO, the maximum electron density is observed around the areas containing sulfur and nitrogen heteroatoms. This trend can be correlated with experimental results of XRD where NH···S interactions among SA1 and SA2 crystal lattice played the major role in the stability of crystal lattice by forming the supramolecular synthons. DFT derived HOMO, LUMO, and their energy difference values for SA1 and SA2 indicated that SA1 exhibits a slightly higher HOMO energy (-0.20839 vs. -0.20923 a.u.) and lower LUMO energy (-0.04232 vs. -0.03967 a.u.) compared to SA2, indicating marginally greater electron-donating ability and electron affinity, which could enhance its reactivity in redox processes. Conversely, higher HOMO-LUMO gap in SA2 justifies its superior kinetic stability. SA1 has a significantly lower electronic energy compared to SA2, indicating greater thermodynamic stability. Polarizability is a fundamental property of a system that measures the extent to which the distribution of valence electrons can be distorted in response to an external electric field. It offers valuable information about the bonding properties and geometrical parameters of a molecule.66 Higher polarizability of SA1 indicates that the compound might exhibit stronger dispersion forces. Dipole moments provide a fundamental measure of the electron density distribution in polar molecules and play a crucial role in determining intermolecular interactions.67 SA2 exhibits a higher dipole moment than SA1 that encompasses the better solubility of SA2 in polar solvents (e.g., water) and stronger interactions with biological targets. Employing the energy values of HOMO and LUMO global reactivity descriptors were calculated by using the reported methods. Global hardness refers to the resistance to distortion of the electron cloud in a chemical species, such as atoms, ions, or molecules. Softness describes a molecule's capacity to accept electrons, which is closely associated with the characteristics of its functional groups. It plays a significant role in understanding various chemical properties, including molecular solubility, chemical reactivity, and the stability.68,69 Hardness and softness parameter of studied compound were calculated using the Equations 2 and 3, respectively. 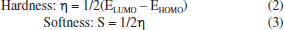 Table 6 indicated the higher softness and lower hardness value for SA1 while reverse trend was observed in case of SA2 suggesting the higher reactivity and propensity of SA1 to participate in covalent interactions. The quantitative measurement of electronegativity based on the thermodynamic properties of compounds describe the degree of ionic character in chemical bonds. Density functional theory calculated electronegativity values utilizes the concept of electronegativity as the function of electron density.70 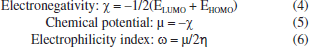 Both compounds showed nearly identical values of electronegativity suggesting similar electron-attracting tendencies. The chemical potential reflects the tendency of an electron to escape from equilibrium and is mathematically defined as the partial derivative of the system's energy with respect to the number of electrons.69 The electrophilicity index (ω) quantifies the ability of a species to accept electrons, providing a measure of its electron accepting capacity.71 Electronegativity, hardness and electrophilicity index are the global descriptors that offer a way to understand the molecular reactivity based on initial electronic distribution.72 No significant difference was found between electrophilicity index, electron donating and electron accepting ability of both compounds. Overall, both compounds showed excellent profile for the global reactivity parameter implying their appropriateness in drug design. Molecular electrostatic potential (MEP) The charge distributions of molecules can be depicted three dimensionally on the molecular electrostatic potential surface. MEP surface enables the visualization of charged areas within a molecule.73 Probable attack sites of nucleophile and electrophilic areas are conventionally represented as blue and red, respectively. Sulfur and nitrogen atoms are presumably more prone toward the electrophilic attack where as insignificant blue region extended over terminal methyl group makes it a preferred site for nucleophilic attack. MEP surfaces calculated at B3LYP/6-311G d,p basis set are given in Figure 7.
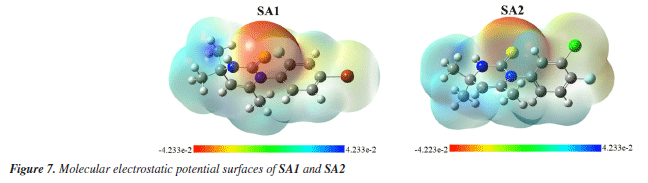
In our analysis, DFT calculated global reactivity chemical descriptors such as hardness (η), electronegativity (χ), electrophilicity index (ω) along with dipole moment were applied not only to assess the intrinsic chemical reactivity and stability of SA1 and SA2 but also to explain potential interactions with biological targets. Kinetic stability may be assessed through the chemical hardness and the HOMO-LUMO energy gap in which a larger value is preferred. The marginally higher HOMO-LUMO gap for SA2 indicates it is slightly more stable than SA1, supporting the greater binding stability seen during molecular docking, as well as lower IC50 (half-maximal inhibitory concentration) values recorded during most in vitro assays. Here, stability means that the compound retains its intended structure (conformation) under experimental conditions without undergoing degradation. This facilitates the efficient interaction of compound with the enzyme's active site. Moreover, the electrophilicity index (ω) and softness (S) are one of the many parameters that the charge-transfer dynamics, especially with regard to the active site nucleophiles, are keen to focus on. For example, the active dihydropyrimidine-2-thione scaffold bears sulfur and nitrogen heteroatoms which are capable of forming specific hydrogen bonding and pi type interactions that were evidenced in our docking studies. The softness values for both compounds, though modestly high, are not so labile that they would become deactivated before being able to participate in such specific interactions. The further elucidation of the potential physiological activities of compounds SA1 and SA2 under study is facilitated by considering molecular electrostatic potential (MEP) surfaces, which reflect the distribution of electric charge and show potential nucleophilic and electrophilic regions aligned with the binding pockets of important enzymes, most noteworthy of which is AChE. This indicates that complementarity of electronic properties is essential for inhibition of the enzyme, linking reactiveness and descriptors of activity with a biological mechanism. As much as DFT descriptors do not self-evidently predict biological activity, their offering of an electron donor-acceptor framework allows for the prediction of the positioning of the molecule as well as rationalization of why SA2 with less bulky groups shows stronger inhibition of most enzymes relative to SA1. Enzyme inhibition assays The compounds SA1 and SA2 were evaluated for anticholinesterase activities using microplate method and IC50 values are given in Table 7. The compound SA2 showed highest activity against acetylcholinesterase with lowest IC50 value of 27.2 ± 2.4 µM while compound SA1 exhibited less potential as compared to SA2. Against butyrylcholinesterase, SA2 showed comparatively moderate activity with an IC50 = 51.8 ± 2.7 µM. Against both targeted enzymes, the compound SA2 showed maximum potential as is depicted in Table 7 by their respective IC50 values.

Conclusively, the favorable potentials of binding and interaction of SA2 are attributed to an optimized electronic structure which improves binding to its target enzymes. A correlation between some DFT-based reactivity characterization such as HOMO-LUMO energy gap, electronegativity, and hardness which were derived from DFT calculations and the observed biology was noted, which greatly enhanced understanding into the reactivity and inhibition of the enzymes by the compound. The energy gap of the HOMO and LUMO levels of a compound is one of the primary descriptors of its reactivity. A smaller energy gap means a compound is more reactive as it can easily donate or accept electrons (B3LYP/6-311G(d,p)). For SA1, the HOMO-LUMO gap was 0.166 eV, while SA2 exhibited a slightly smaller gap of 0.1695 eV. This support the observation made that SA2 is more reactive and more potent in enzyme inhibition.74 This reactivity is in agreement with the in vitro IC50 values for the inhibition of acetylcholinesterase and butyrylcholinesterase, where SA2 showed a lower IC50 of 27.2 ± 2.4 µM for AChE and 51.8 ± 2.7 µM for BChE, while SA1 was 53.7 ± 2.4 µM and 65.9 ± 0.2 µM, respectively. Moreover, the parameter impacts how readily a molecule would interact with the electrophilic sites on the enzyme. The differences in the data are minimal, with SA1 at 0.125 and SA2 at 0.124. However, the dipole moment of SA2 at 6.44 Debye surpassing SA1's 5.22 Debye suggests greater electrostatic interactions with the enzyme's active site. Thus, these findings explain the stronger binding affinity and biological activity of SA2 compared to SA1. In addition to differences in reactivity, the hardness descriptor indicates the degree of resistance to electron transfer of a molecule, where SA1 and SA2 have values of 0.083 and 0.085, respectively. The dipole moment and HOMO-LUMO gap is lower in SA2, increasing its reactivity and subsequently enzyme inhibition. Coupled with the strong relationship observed between descriptors such as DFT-derived reactivity, and IC50 values, the predicted reactivity and binding potency of SA2 by DFT models indicates its biological activity.75 The integration of these DFT calculations with experimental enzyme inhibition assays exemplifies the efficacy of in silico techniques in biochemistry with respects to predicting and explaining the biological actions of new compounds. Global reactivity descriptors provide the overall view of the electronic characteristics of the compound that may justify its inhibition of the relevant cholinergic enzymes involved in the Alzheimer's disease process, acetylcholinesterase and butyrylcholinesterase. Molecular docking Compounds SA1 and SA2 were docked as ligands against both protein targets, acetylcholinesterase (AChE) and butyrylcholinesterase (BChE). Torsional angles between phenyl ring and aromatic ring in synthesized compounds were monitored. The best pose for each protein-ligand complex was selected for the interaction analysis. Binding interaction analysis Acetylcholinesterase Docking study of SA1, SA2 and reference ligand with AChE showed the binding energy values of -9.3, -9.5 and -11.3 kcal mol-1, respectively, for the best pose. Torsional angles for SA1 and SA2 were 88.05° and 87.45°. SA1 has relatively more deviation from experimentally calculated torsions suggesting the structural flexibility upon binding with AChE. Interaction analysis of SA1 with AChE (Figure 8) revealed that hydrogen atom of -NH group of pyrimidine ring made hydrogen bonding with amino acid residue TRP286. Another hydrogen bond was found between Br atom and amino acid residue PHE295. π-Alkyl interactions were observed between Br atom and peripheral anionic site (PAS) amino acid TYR341.76 Amino acid residue TRP 286 is located at the gorge entrance of AChE, so it can be hypothesized that our studied ligand possibly block the entry of incoming substrate toward the catalytic region of AChE.77
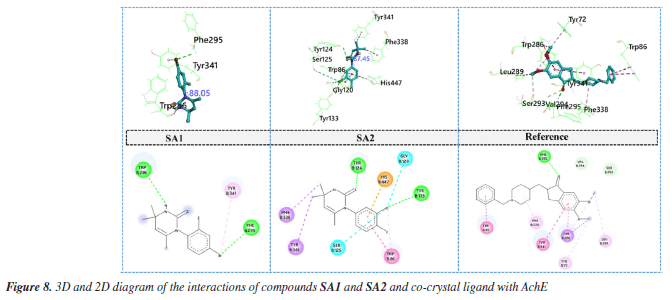
In case of SA2, sulfur atom of pyrimidine-2(1H)-thione made hydrogen bond with amino acid residue TYR124 located at the peripheral anionic site of AChE. One of critical amino acid residue that is the part of catalytic triad of the enzyme HIS44778 is involved in the formation of π-cation contact. π-Sigma and π-π stacked interactions was observed between SA2 and amino acids residues PHE338, TYR341 and TRP86.79 Halogen interactions mediated by chlorine atom involving the amino acid residues GLY120 and SER125 contributed to the enhanced stability of protein ligand complex. Binding pattern of SA2 involved the key residues of peripheral anionic site as well as catalytic triad of enzyme may suggest the efficient inhibition of AChE. In case of co-crystal ligand, oxygen atom attached with cyclopentane ring was involved in the formation of hydrogen bond with PHE295 while carbon hydrogen bond with amino acid residue VAL294. π-Sigma interactions and π- π stacking involved the amino acid residues TRP286 and TYR341, respectively. Protein ligand complex is further stabilized by alkyl and π-alkyl interactions with amino acids residues LEU289, TYR72 and PHE338. Butyrylcholinesterase Docking study of SA1, SA2 and reference ligand with BChE showed the binding energy values of -6.6, -6.8 and -8.4 kcal mol-1, respectively, for the best conformation. Torsional angels for SA1 and SA2 were 83.26° and 89.67°, respectively. Measured torsions were very close to the experimentally calculated XRD values suggesting the structural compactness upon interaction with BChE. Interaction analysis of SA1 with BChE revealed that bromine atom is responsible for variety of interactions with amino acid residues of BChE. Bromine atom made conventional hydrogen bond, carbon hydrogen bond, alkyl interaction and the halogen contact with amino acid residues TYR128, GLY121, LEU125 and THR122, respectively. Protein ligand complex was further stabilized by sulfur mediated π-sulfur contacts with amino acid residues TRP82 and HIS438. π-Sigma interaction was observed between methyl group and active site wall amino acid residue TYR332.80 Hydrogen bonding involving important amino acid residue of BChE TYR124, TYR133 and sulfur and chlorine atoms of SA2, respectively, was found during the interactions analysis. Terminal methyl groups at pyrimidine ring made π-sigma contacts with amino acid residues PHE338 and TYR341. Aromatic ring of SA2 facilitated the formation of π-cation contact and π-π stacked interaction with amino acid residues HIS447 and TRP86, respectively. Further stability to protein ligand complex was imparted by halogen interactions by chlorine atoms with amino acid residues GLY120 and SER125. Oxygen atom of sulfonamide moiety of co-crystal ligand took part in the formation of hydrogen bond with amino acid residue THR120 (Figure 9). Complex is further stabilized by π-π T‑shaped and amide-π stacked interactions with amino acid residues TRP231, GLY116, PHE329 and TRP231 as well as π-alkyl interactions with LEU286, ALA328 and TYR332.
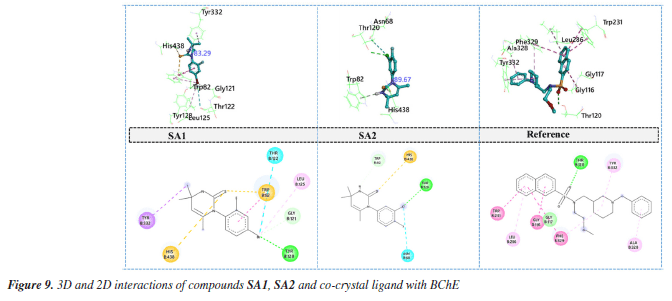
Both co-crystals ligands showed similar interactions (excluding the interactions due to water molecules) as was found in experimental crystallographic structures suggesting the correctness and reliability of docking procedure. Superimposed structures of both co-crystal ligands are given in Supplementary Material (Figure 7S). Amino acid residues involved in the binding of SA1 and SA2 were found in the reported inhibitors of acetylcholinesterase and butyrylcholinesterase.81,82 The results of molecular docking studies suggested the favorable contacts between proposed ligands, i.e., SA1 and SA2 with the critical amino acids of enzyme targets warrants the inhibition potential of compounds under study. Normal mode analysis (NMA) Assessing the deformability alongside the B-factors provides insight into the mobility of the proteins in question, namely AChE and BChE, derived from the original PDB files (7E3H and 5DYW), excluding the docked complexes. The deformability and B-factor information reveals peaks in the data corresponding to the flexible regions of the proteins. The most significant peaks correlate with regions of maximum deformability, meaning they correspond to greater flexibility. Such plots aid in evaluating the results from NMA, which, alongside the 3D structures, were performed on the PDB files and not on the docked complexes (AChE-SA1 and SA2, BChE-SA1 and SA2). For this study, the iMODs server was employed to determine the ease of motion of the proteins, where the proteins were held rigid in the docking phase, and the results represent the docking-structure-averaged flexibility of the enzyme structures. The B-factor graphs were analyzed alongside NMA and PDB data, as shown in Figures 10 and 11. These results emphasize the mobility of the proteins, as derived from their original structures, 7E3H for AChE and 5DYW for BChE.
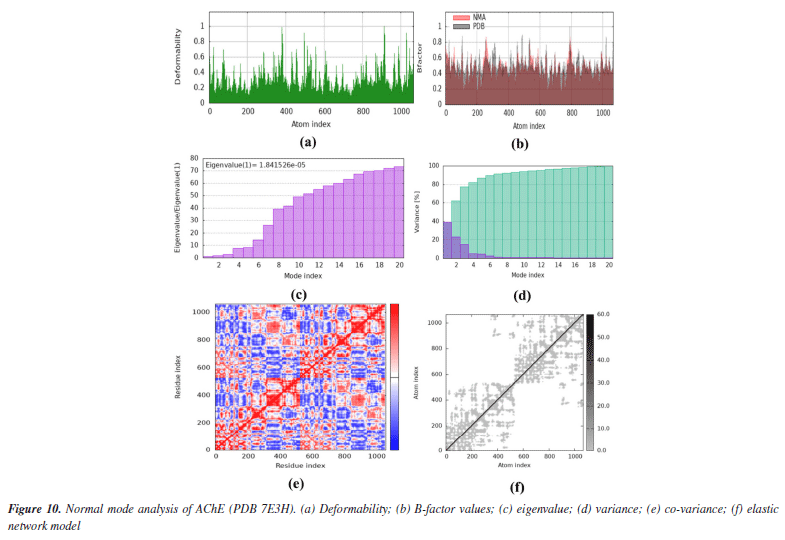
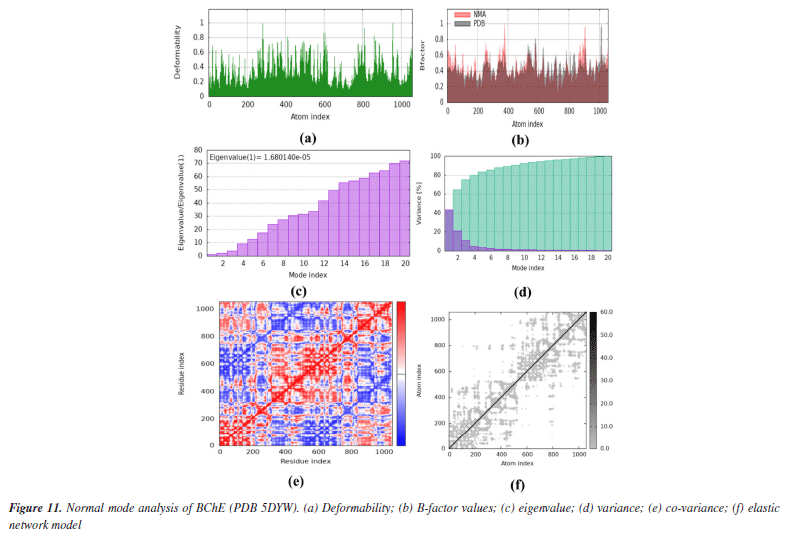
In addition, the eigenvalue and variance for each protein normal mode are proportional inverse relationships. For AChE and BChE, the eigenvalues for SA1 were 1.84 × 10-5 and 1.68 × 10-5, respectively. Figures 10 and 11 illustrate the variance graph, where individual variances (purple bars) and cumulative variance (green-shaded bars) are depicted. The covariance matrix for the complex portrays the correlation among residues of the complex. Strong correlations are shown in red, no correlation in white, and blue indicates anti-correlations. The greater the correlation, the greater the benefit to the interaction within the complex. Lastly, the elastic maps showing the interatomic connections highlight rigid regions in darker gray, indicating low mobility and greater stiffness. Finally, the elastic maps from the docked proteins highlight the connections between atoms, with darker gray regions symbolizing greater rigidity and therefore decreased movement. ADMET studies The hydrophilicity of a compound can be determined by looking at its LogP value; if the value is negative, the compound is considered to be hydrophilic.83 Both of the compounds listed in Table 8 have lipophilic character. The LogS value is a measure of solubility; a higher solubility corresponds to a lower LogS value, which in turn increases absorption.84 LogD+2 represent the ideal lipophilicity for medicines having central nervous system (CNS) activity in terms of their ability to penetrate the blood-brain barrier. When it comes to medications for the CNS, a LogD greater than 4 is considered unsuitable. In the process of anticipating the oral absorption of drug-like substances, the topological polar surface area, abbreviated TPSA, is utilized. A higher TPSA score shows that the membrane's permeability has been reduced. As a result, a lower TPSA level is considered to be appropriate for drug-likeness. In order to get the best possible CNS diffusion, the TPSA value should be low.85 If a derivative has a TPSA score of 70, then that derivative is deemed to have adequate bioavailability. Our investigation revealed that TPSA values were slightly higher than expected; however, this result can be altered in the future by eliminating polar atoms from the synthesized molecules. The number of atoms that may form hydrogen bonds, denoted by the symbol nHD, is equal to the total of all OH and NH groups, whereas the number of atoms that can form hydrogen bonds, denoted by the symbol nHA, is equal to the sum of all nitrogen and oxygen atoms that do not have a positive charge. The ideal ranges for nHA are 0-12, and 0-7 for nHD.86
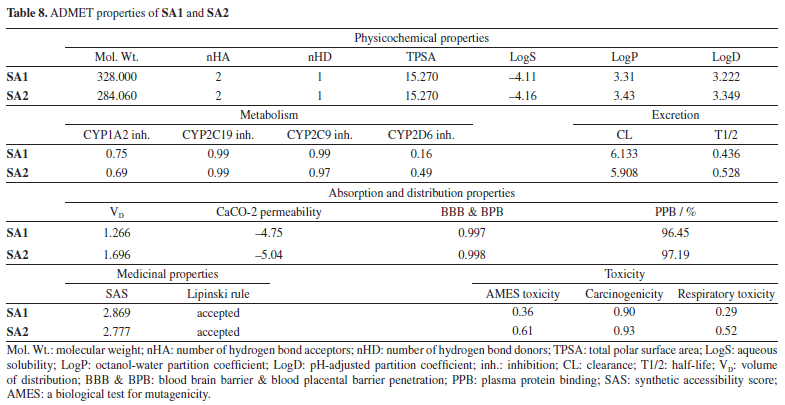
According to the absorption and distribution properties, the ideal range for the volume of distribution (VD) is between 0.04 and 20 L kg-1. The CaCO-2 permeability represents the amount of medication that is absorbed in vivo by the intestinal tract. The ideal values should be more than -5.15 on the logarithmic scale.87 The ideal value for the binding of plasma proteins should be less than 90%. If the PPB is high, this indicates that the therapeutic index of the chemical is low. Compounds under study exhibited ideal VD and CaCO-2 permeability values as is shown in Table 8. Both compounds are able to get cross the BBB (blood brain barrier) because of their lipophilic character. According to our observations, the substances SA1 and SA2 both demonstrated PPB (plasma protein binding) values that were higher than 90%. Both proposed drugs acted as inhibitors of the CYP1A2, CYP2C19, CYP2C9, and CYP3A4 and CYP2D6 enzymes during the metabolic and excretory processes. It is possible to classify the rate at which a drug is eliminated from the body as high (more than 15), moderate (5-15), or low (less than 5). The clearance rate for all of our compounds was quite low. The results of the tests that were done on the compounds toxicity profiles are presented in Table 8. The level of difficulty associated with the synthesis of drug-like molecules can be determined by using the synthetic accessibility score. The chemicals that have a synthetic accessibility score (SAS) that is lower than six are simple to produce. Compounds under evaluation have an SAS that places them within the desirable range; this indicates that they were simple to manufacture. The AMES (a biological test for mutagenicity) toxicity test is carried out to determine whether or not the substance in question is a mutagen. Both of the compounds exhibited low to high levels of carcinogenicity, with low respiratory toxicity profile. It is possible to infer that both compounds have a good ADMET profile. Compound SA1 displayed a profile that was significantly less hazardous than that of the SA2 compound. The findings were shown to have a correlation with the other data obtained in vitro. It is necessary to conduct additional research in order to improve the toxicity profile of substances.
CONCLUSIONS Structural data and intermolecular interactions in 1-(aryl)-4,4,6-trimethyl-3,4-dihydropyrimidine-2(1H)-thiones derivatives was studied by single crystal X-ray diffraction (SCXRD) studies and Hirshfeld surface analysis. Results of DFT studies helps in understanding the quantum mechanical descriptors of compounds SA1 and SA2. In vitro enzyme assays proved the synthesized compounds (SA1 and SA2) as inhibitors of both cholinesterases, AChE and BChE. Despite the varying degree of IC50 values against AChE and BChE, SA2 having electron withdrawing group fluoro and chloro substitution at meta and para position, respectively, was found more potent than SA1. Global reactivity indices calculated by DFT studies suggested the higher reactivity of SA2 as compared to SA1. Molecular docking highlighted the favorable interactions between synthesized compounds and the amino acid residues of their respective enzyme targets. The results of the NMA, which utilized the rigid protein structures from the PDB files 7E3H for AChE and 5DYW for BChE, indicate both these enzymes possess considerable intrinsic flexibility. These results may further explain the manner in which the enzymes may associate with ligands. However, due to the rigid nature of the docking approach, the examined pose only reflects the enzymes' natural flexibility without considering changes from ligand-induced conformational changes. During ADMET evaluation, both compounds behaved as drug-like. So, it can be concluded that the both compounds SA1 and SA2 may inhibit the cholinesterases for the treatment of Alzheimer's disease.
SUPPLEMENTARY MATERIAL Supplementary material for this work is available at http://quimicanova.sbq.org.br/, as a PDF file, with free access. The CCDC number of compounds SA1 and SA2 are 1966994 and 1504417, respectively. The copies can be requested to the CCDC at 12 Union Road, Cambridge CB2 1EZ, UK via fax at +44 1223 336033 or e-mail at deposit@ccdc.cam.ac.uk, or can be accessed online.
DATA AVAILABILITY STATEMENT All data generated or analyzed during this study are included in this published article and its supplementary material.
ACKNOWLEDGMENTS The work was supported by Princess Nourah bint Abdulrahman, Univeristy Researchers supporting project number (PNURSP2025R116), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. The funding body played potential role in the design of the study, purchasing, sample characterization, analysis, interpretation of data, and in writing the manuscript.
AUTHOR CONTRIBUTIONS Conceptualization: Syeda Abida Ejaz, Aamer Saeed; data curation: Aisha A. Alsfouk, Ulrich florke; formal analysis: Tahira Shamim; funding acquisition: Aisha A. Alsfouk; investigation: Pervaiz Ali Channar, Michael Bolte; project administration: Syeda Abida Ejaz, Aamer Saeed, Tuncer Hökelek; resources: Hammad Ismail; software: Aftab Ahmed; validation: Syeda Abida Ejaz; visualization: Aftab Ahmed; writing original draft: Hammad Ismail, Rabail Ujan; writing-review and editing: Syeda Abida Ejaz.
REFERENCES 1. Hussain, M. S.; Mishra, A. K.; Int. J. Res. Med. Sci. 2024, 12, 1789. [Crossref] 2. World Health Organization; Global Status Report on the Public Health Response to Dementia; WHO: Geneva, 2021. [Link] accessed in October 2025 3. Strachan, M. W.; Reynolds, R. M.; Frier, B. M.; Mitchell, R. J.; Price, J. F.; Br. Med. Bull. 2008, 88, 131. [Crossref] 4. Baumgart, M.; Snyder, H. M.; Carrillo, M. C.; Fazio, S.; Kim, H.; Johns, H.; Alzheimer's Dementia 2015, 11, 718. [Crossref] 5. Crous-Bou, M.; Minguillón, C.; Gramunt, N.; Molinuevo, J. L.; Alzheimer's Res. Ther. 2017, 9, 71. [Crossref] 6. Zhang, X.-X.; Tian, Y.; Wang, Z.-T.; Ma, Y.-H.; Tan, L.; Yu, J.-T.; The Journal of Prevention of Alzheimer's Disease 2021, 8, 313. [Crossref] 7. Cai, H.; Cong, W.-n.; Ji, S.; Rothman, S.; Maudsley, S.; Martin, B.; Curr. Alzheimer Res. 2012, 9, 5. [Crossref] 8. Mittal, K.; Katare, D. P.; Diabetes Metab. Syndr.: Clin. Res. Rev. 2016, 10, S144. [Crossref] 9. Clarke, R. S. J.; Dundee, J. W.; Br. J. Anaesth. 1965, 37, 779. [Crossref] 10. Liu, P.-P.; Xie, Y.; Meng, X.-Y.; Kang, J.-S.; Signal Transduction Targeted Ther. 2019, 4, 29. [Crossref] 11. Dierckx, R. I. R.; Vandewoude, M. F. J.; Acta Clin. Belg. 2008, 63, 339. [Crossref] 12. Hansen, R. A.; Gartlehner, G.; Webb, A. P.; Morgan, L. C.; Moore, C. G.; Jonas, D. E.; Clin. Interventions Aging 2008, 3, 211. [Link] accessed in October 2025 13. AlQot, H. E.; Rylett, R. J.; Sci. Rep. 2024, 14, 27614. [Crossref] 14. Yarns, B. C.; Holiday, K. A.; Carlson, D. M.; Cosgrove, C. K.; Melrose, R. J.; Psychiatric Clinics of North America 2022, 45, 663. [Crossref] 15. Volloch, V.; Rits-Volloch, S.; Journal of Alzheimer's Disease Reports 2022, 7, 21. [Crossref] 16. Cummings, J.; Zhou, Y.; Lee, G.; Zhong, K.; Fonseca, J.; Cheng, F.; Alzheimer's & Dementia: Translational Research & Clinical Interventions 2023, 9, e12385. [Crossref] 17. van Dyck, C. H.; Swanson, C. J.; Aisen, P.; Bateman, R. J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; Froelich, L.; Katayama, S.; Sabbagh, M.; Vellas, B.; Watson, D.; Dhadda, S.; Irizarry, M.; Kramer, L. D.; Iwatsubo, T.; N. Engl. J. Med. 2023, 388, 9. [Crossref] 18. Shaikh, S.; Verma, A.; Siddiqui, S.; Ahmad, S.; Rizvi, S. M. D.; Shakil, S.; Biswas, D.; Singh, D.; Siddiqui, M. H.; Shakil, S.; Tabrez, S.; Kamal, M. A.; CNS Neurol. Disord.: Drug Targets 2014, 13, 391. [Crossref] 19. Jasiewicz, B.; Babijczuk, K.; Warżajtis, B.; Rychlewska, U.; Starzyk, J.; Cofta, G.; Mrówczyńska, L.; Molecules 2023, 28, 708. [Crossref] 20. Faihan, A. S.; Hatshan, M. R.; Alqahtani, A. S.; Nasr, F. A.; Al‑Jibori, S. A.; Al-Janabi, A. S.; J. Mol. Struct. 2022, 1247, 131291.[Crossref] 21. Arshad, N.; Jawaid, S.; Hashim, J.; Ullah, I.; Gul, S.; Aziz, A.; Wadood, A.; Khan, A.; Bioorg. Med. Chem. Lett. 2023, 79, 129068. [Crossref] 22. Arble, D. M.; Ramsey, K. M.; Bass, J.; Turek, F. W.; Best Pract. Res., Clin. Endocrinol. Metab. 2010, 24, 785. [Crossref] 23. Di Mascio, P.; Martinez, G. R.; Miyamoto, S.; Ronsein, G. E.; Medeiros, M. H. G.; Cadet, J.; Chem. Rev. 2019, 119, 2043. [Crossref] 24. Dhanjai; Sinha, A.; Lu, X.; Wu, L.; Tan, D.; Li, Y.; Chen, J.; Jain, R.; TrAC, Trends Anal. Chem. 2018, 98, 174. [Crossref] 25. Andleeb, S.; Imtiaz-ud-Din; Rauf, M. K.; Azam, S. S.; Badshah, A.; Sadaf, H.; Raheel, A.; Tahir, M. N.; Raza, S.; RSC Adv. 2016, 6, 79651. [Crossref] 26. Álvarez, L.; Brovelli, F.; Baggio, R.; Peña, O.; Muñoz, J.; Soto-Delgado, J.; Moreno, Y.; J. Chil. Chem. Soc. 2022, 67, 5702. [Crossref] 27. Sąsiadek, W.; Bryndal, I.; Ptak, M.; Lisiecki, R.; Lis, T.; Hanuza, J.; J. Mol. Struct. 2023, 1286, 135531. [Crossref] 28. Marinescu, M.; Popa, C.-V.; Int. J. Mol. Sci. 2022, 23, 5659. [Crossref] 29. de Fátima, Â.; Braga, T. C.; Neto, L. S.; Terra, B. S.; Oliveira, B. G. F.; da Silva, D. L.; Modolo, L. V.; J. Adv. Res. 2015, 6, 363. [Crossref] 30. Dallinger, D.; Stadler, A.; Kappe, C. O.; Pure Appl. Chem. 2004, 76, 1017. [Crossref] 31. Matache, M.; Dobrota, C.; Bogdan, N. D.; Funeriu, D. P.; Curr. Org. Synth. 2011, 8, 356. [Crossref] 32. Heravi, M. M.; Zadsirjan, V.; Curr. Org. Chem. 2020, 24, 1331. [Crossref] 33. Kappe, C. O.; Acc. Chem. Res. 2000, 33, 879. [Crossref] 34. Müller, C.; Gross, D.; Sarli, V.; Gartner, M.; Giannis, A.; Bernhardt, G.; Buschauer, A.; Cancer Chemother. Pharmacol. 2007, 59, 157. [Crossref] 35. Dowarah, J.; Patel, D.; Marak, B. N.; Yadav, U. C. S.; Shah, P. K.; Shukla, P. K.; Singh, V. P.; RSC Adv.2021, 11, 35737. [Crossref] 36. Khurshid, A.; Saeed, A.; J. Chin. Chem. Soc. 2017, 64, 224. [Crossref] 37. CCDC, Structural Chemistry Data, Software, and Insights, https://www.ccdc.cam.ac.uk/, accessed in October 2025. 38. Spackman, M. A.; Jayatilaka, D.; CrystEngComm 2009, 11, 19. [Crossref] 39. Hohenberg, P.; Kohn, W.; Phys. Rev. 1964, 136, B864. [Crossref] 40. Becke, A. D.; J. Chem. Phys. 1993, 98, 5648. [Crossref] 41. Lee, C.; Yang, W.; Parr, R. G.; Phys. Rev. B 1988, 37, 785. [Crossref] 42. Sousa, S. F.; Fernandes, P. A.; Ramos, M. J.; J. Phys. Chem. A 2007, 111, 10439. [Crossref] 43. Krishnan, R.; Binkley, J. S.; Seeger, R.; Pople, J. A.; J. Chem. Phys. 1980, 72, 650. [Crossref] 44. Dunning Jr., T. H.; J. Chem. Phys. 1989, 90, 1007. [Crossref] 45. Karelson, M.; Lobanov, V. S.; Katritzky, A. R.; Chem. Rev. 1996, 96, 1027. [Crossref] 46. Ellman, G. L.; Courtney, K. D.; Andres Jr., V.; Featherstone, R. M.; Biochem. Pharmacol. 1961, 7, 88. [Crossref] 47. Siddiqui, S. Z.; Abbasi, M. A.; Ashraf, M.; Mirza, B.; Ismail, H.; Pak. J. Pharm. Sci. 2017, 30, 1735. 48. Mehfooz, H.; Saeed, A.; Faisal, M.; Larik, F. A.; Muqadar, U.; Khatoon, S.; Channar, P. A.; Ismail, H.; Bilquees, S.; Rashid, S.; Shafique, S.; Mirza, B.; Dilshad, E.; Ahmad, F.; Res. Chem. Intermed. 2020, 46, 2437. [Crossref] 49. PDB ID: 7E3H, https://www.rcsb.org/structure/7E3H, accessed in October 2025; PDB ID: 5DYW, https://www.rcsb.org/structure/5DYW, accessed in October 2025. 50. Cheung, J.; Rudolph, M. J.; Burshteyn, F.; Cassidy, M. S.; Gary, E. N.; Love, J.; Franklin, M. C.; Height, J. J.; J. Med. Chem. 2012, 55, 10282. [Crossref] 51. Maurus, R.; Begum, A.; Williams, L. K.; Fredriksen, J. R.; Zhang, R.; Withers, S. G.; Brayer, G. D.; Biochemistry 2008, 47, 3332. [Crossref] 52. Sim, L.; Willemsma, C.; Mohan, S.; Naim, H. Y.; Pinto, B. M.; Rose, D. R.; J. Biol. Chem. 2010, 285, 17763. [Crossref] 53. iMODS, https://imods.iqf.csic.es/, accessed in October 2025. 54. López-Blanco, J. R.; Aliaga, J. I.; Quintana-Ortí, E. S.; Chacón, P.; Nucleic Acids Res. 2014, 42, W271. [Crossref] 55. Lopéz-Blanco, J. R.; Garzón, J. I.; Chacón, P.; Bioinformatics 2011, 27, 2843. [Crossref] 56. Kovacs, J. A.; Chacón, P.; Abagyan, R.; Proteins: Struct., Funct., Bioinf. 2004, 56, 661. [Crossref] 57. Fu, L.; Shi, S.; Yi, J.; Wang, N.; He, Y.; Wu, Z.; Peng, J.; Deng, Y.; Wang, W.; Wu, C.; Lyu, A.; Zeng, X.; Zhao, W.; Hou, T.; Cao, D.; Nucleic Acids Res. 2024, 52, W422. [Crossref] 58. Venkatesan, P.; Thamotharan, S.; Ilangovan, A.; Liang, H.; Sundius, T.; Spectrochim. Acta, Part A 2016, 153, 625. [Crossref] 59. Nangia, A.; Desiraju, G. R. In Design of Organic Solids; Weber, E.; Aoyama, Y.; Caira, M. R.; Desiraju, G. R.; Glusker, J. P.; Hamilton, A. D.; Meléndez, R. E.; Nangia, A., eds.; Springer: Berlin, Heidelberg, 1998, p. 57. [Crossref] 60. Bojarska, J.; Remko, M.; Breza, M.; Madura, I. D.; Kaczmarek, K.; Zabrocki, J.; Wolf, W. M.; Molecules 2020, 25, 1135. [Crossref] 61. Al-Dies, A.-A. M.; Alblewi, F. F.; Okasha, R. M.; Alsehli, M. H.; Borik, R. M. A.; Ihmaid, S.; Amr, A. E.-G. E.; Ghabbour, H. A.; Elhenawy, A. A.; El-Agrody, A. M.; Discover Appl. Sci. 2025, 7, 71. [Crossref] 62. Bakheit, A. H.; Abuelizz, H. A.; Al-Salahi, R.; Crystals 2023, 13, 1410. [Crossref] 63. Hathwar, V. R.; Sist, M.; Jørgensen, M. R. V.; Mamakhel, A. H.; Wang, X.; Hoffmann, C. M.; Sugimoto, K.; Overgaard, J.; Iversen, B. B.; IUCrJ 2015, 2, 563. [Crossref] 64. Toenjes, S. T.; Gustafson, J. L.; Future Med. Chem. 2018, 10, 409. [Crossref] 65. LaPlante, S. R.; Edwards, P. J.; Fader, L. D.; Jakalian, A.; Hucke, O.; ChemMedChem 2011, 6, 505. [Crossref] 66. Suvitha, A.; Venkataramanan, N. S.; J. Theor. Comput. Chem. 2015, 14, 1550049. [Crossref] 67. Hait, D.; Head-Gordon, M.; J. Chem. Theory Comput. 2018, 14, 1969. [Crossref] 68. Yadav, P.; Tandon, H.; Malik, B.; Chakraborty, T.; Theor. Chem. Acc. 2021, 140, 60. [Crossref] 69. Rouhani, M.; Comput. Theor. Chem. 2021, 1195, 113096. [Crossref] 70. Sproul, G. D.; ACS Omega 2020, 5, 11585. [Crossref] 71. Kannan, K. S.; Thamaraikannan, P.; Kannan, S.; Kalaipoonguzhali, V.; Kumar, R. A.; Venkatachalam, K.; AIP Conf. Proc. 2020, 2270, 040008. [Crossref] 72. Glossman-Mitnik, D.; Procedia Computer Science 2013, 18, 816. [Crossref] 73. Lakshminarayanan, S.; Jeyasingh, V.; Murugesan, K.; Selvapalam, N.; Dass, G.; J. Photochem. Photobiol. 2021, 6, 100022. [Crossref] 74. Radhi, A. H.; Du, E. A.; Khazaal, F. A.; Abbas, Z. M.; Aljelawi, O. H.; Hamadan, S. D.; Kadhim, M. M.; NeuroQuantology 2020, 18, 37. [Crossref] 75. Bourouai, M. A.; Larbi, K. S.; Bouchoucha, A.; Terrachet-Bouaziz, S.; Djebbar, S.; BioMetals 2023, 36, 153. [Crossref] 76. Kherachi, R.; Daoud, I.; Melkemi, N.; Kenouche, S.; Mettai, M.; Mesli, F.; Biologia 2023, 78, 3691. [Crossref] 77. Codony, S.; Pont, C.; Griñán-Ferré, C.; Di Pede-Mattatelli, A.; Calvó‑Tusell, C.; Feixas, F.; Osuna, S.; Jarné-Ferrer, J.; Naldi, M.; Bartolini, M.; Loza, M. I.; Brea, J.; Pérez, B.; Bartra, C.; Sanfeliu, C.; Juárez-Jiménez, J.; Morisseau, C.; Hammock, B. D.; Pallàs, M.; Vázquez, S.; Muñoz-Torrero, D.; J. Med. Chem. 2022, 65, 4909. [Crossref] 78. Zhang, Y.; Kua, J.; McCammon, J. A.; J. Am. Chem. Soc. 2002, 124, 10572. [Crossref] 79. Barak, D.; Kronman, C.; Ordentlich, A.; Ariel, N.; Bromberg, A.; Marcus, D.; Lazar, A.; Velan, B.; Shafferman, A.; J. Biol. Chem. 1994, 264, 6296. [Crossref] 80. Macdonald, I. R.; Martin, E.; Rosenberry, T. L.; Darvesh, S.; Biochemistry 2012, 51, 7046. [Crossref] 81. Khan, Y.; Rehman, W.; Hussain, R.; Khan, S.; Malik, A.; Khan, M.; Liaqat, A.; Rasheed, L.; Begum, F.; Fazil, S.; Khan, I.; Abdellatif, M. H.; J. Heterocycl. Chem. 2022, 59, 2225. [Crossref] 82. Jamal, Q. M. S.; Alharbi, A. H.; Environ. Sci. Pollut. Res. 2022, 29, 61972. [Crossref] 83. Heiden, W.; Moeckel, G.; Brickmann, J.; J. Comput.-Aided Mol. Des. 1993, 7, 503. [Crossref] 84. Hughes, L. D.; Palmer, D. S.; Nigsch, F.; Mitchell, J. B. O.; J. Chem. Inf. Model. 2008, 48, 220. [Crossref] 85. Thai, N. Q.; Theodorakis, P. E.; Li, M. S.; J. Chem. Inf. Model. 2020, 60, 3057. [Crossref] 86. Tantawy, M. A.; Shalby, A. B.; Barnawi, I. O.; Kattan, S. W.; Abd-Rabou, A. A.; Elmegeed, G. A.; Steroids 2023, 193, 109187. [Crossref] 87. Wood, M.; Anesth. Analg. 1986, 65, 786. [Link] accessed in October 2025 |

On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access