
 |
 |
 |
 |
 |
Artigo
| Multiresidue and multiclass determination of Current-use Pesticides (CUPs) in water samples BY SPE and UHPLC-MS/MS |
|
Állisson A. da S. Avellar; Gabriel A. B. Prates; Larissa da S. Alves; Osmar D. Prestes; Martha B. Adaime; Renato Zanella* Departamento de Química, Universidade Federal de Santa Maria, 97105-900 Santa Maria- RS, Brasil Received: 02/21/2024 *e-mail: renato.zanella@ufsm.br Given the extensive use of pesticides in modern times, residues are frequently detected in water samples. Therefore, this study developed and validated a method utilizing solid-phase extraction (SPE) with a polymeric sorbent for sample preparation in combination with ultra-high performance liquid chromatography coupled to tandem mass spectrometry (UHPLC-MS/MS). The aim was to determine a variety of current-use pesticides (CUPs) in different types of water. The preconcentration step, employing the polymeric sorbent Oasis HLB (60 mg), combined with the high selectivity and sensitivity of the UHPLC-MS/MS analysis, enabled the multiclass determination of pesticide residues in water samples at very low concentrations. The proposed method underwent validation, demonstrating satisfactory linearity, precision, and accuracy. The accuracy results, determined through recovery assays at varying spike levels (0.04-0.4 µg L-1), ranged from 71 to 117%, with precision expressed as relative standard deviations consistently below 19%. The practical limits of detection and quantification for most compounds were 0.01 and 0.04 µg L-1, respectively. This method was effectively employed to analyze drinking water, artesian well water, dam water, and river water, leading to the detection of various pesticides at concentrations of up to 2.24 µg L-1. INTRODUCTION Brazil is recognized as one of the largest agricultural producers in the world, playing a crucial role in the global economy through the export of agricultural products. To sustain such high levels of production, Brazil extensively relies on transgenic seeds and chemical inputs, including fertilizers and pesticides.1 Consequently, Brazil ranks among the top consumers of pesticides globally. Regrettably, the widespread and indiscriminate use of pesticides poses significant risks, including soil, surface water, and food contamination. These hazards can negatively impact terrestrial and aquatic organisms, as well as lead to human poisoning through the ingestion of contaminated water and food.2 In addition to the direct threats to human health, the presence of pesticides in the environment can trigger unintended consequences such as disruptions to natural biochemical processes and alterations in the functionality of affected ecosystems.3,4 Water contamination manifests as changes in its intrinsic properties due to anthropogenic pollutants, rendering it unfit for human consumption or incapable of supporting natural ecosystems.5 Research has shown that soils, sediments, surfaces, and groundwater in agroecosystems harbor various levels of pesticide residues. Multiple mechanisms facilitate the contamination of the environment by pesticides, resulting in the dispersion of these substances across different environmental compartments.6 Predominantly, leaching and runoff serve as primary pathways for pesticides entering aquatic systems. Factors such as soil porosity, hydrogeology, biodegradation, and aquifer characteristics influence the scale of groundwater contamination. Simultaneously, the physicochemical properties of pesticides are pivotal in determining their leaching potential,7 in addition to other parameters such as water solubility, octanol-water partition coefficient (Kow), acidic compound dissociation constant (Ka), and half-life (t1/2), which are crucial for comprehending how these compounds interact with the environment.8 Information regarding the structure and physicochemical properties of pesticides has proven to be invaluable in developing effective methods for pesticide residue sample preparation and chromatographic detection methods. Efforts have been undertaken to identify pesticide residues in various water samples, including drinking water,9-13 river water,14,15 surface water,13,16 subsurface water,16 spring water,14 artesian wells,15 groundwater,13,17,18 and wastewaters.13 The analysis of these samples is challenging due to the diverse physicochemical properties of pesticides and their presence in extremely low concentrations in water. Consequently, analytical detection techniques with high sensitivity and selectivity are necessary, along with suitable sample preparation methods, in order to conduct these analyses.19 Solid-phase extraction (SPE) is a commonly employed technique for preparing aqueous samples and enables one to obtain suitable concentration factors for analyzing contaminants in water samples at low concentration levels. This method involves utilizing a solid sorbent to retain analytes, followed by elution with a small volume of an appropriate organic solvent.20 Among its advantages are convenience, cost-effectiveness, simplicity, reduced organic solvent consumption, potential for multiresidue analysis, and compatibility with various detection techniques. Notably, a key advantage of solid-phase extraction lies in the availability of a broad range of sorbents with different chemical structures, resulting in diverse extraction mechanisms that can accommodate pesticides with varied physicochemical properties.21 Multiresidue methods allow for the analysis of numerous compounds with high recovery rates and address potential sample interferences.22 These methods provide good precision, robustness, cost-effectiveness, efficiency, and safety, as they involve minimal volumes of low-toxicity solvents. Chromatographic techniques paired with a mass spectrometer detector are particularly valuable for multiresidue analyses.23 Specifically, high-performance liquid chromatography coupled with a triple quadrupole mass spectrometer detector has proven effective for analyzing pesticide residues in water samples.24-26 Liquid chromatography is a well-established tool for current-use pesticide (CUP) analysis. The utilization of ultra-high performance liquid chromatography (UHPLC) with tandem mass spectrometry (MS/MS) has revolutionized multiresidue sample preparation.27 The coupling of UHPLC-MS/MS is widely utilized as it combines high selectivity, separation efficiency, and the acquisition of structural information, molar mass, and additional selectivity.28,29 Given this context, the main objective of this study was to develop, validate, and apply a multiclass and multiresidue method to determine 94 CUPs in different types of water samples. Pesticides were selected based on their frequency of application and the occurrence of residues in water samples. The preconcentration process was evaluated by optimizing sorbent type and quantity, sample pH, and elution conditions. The sample preparation involved SPE with a polymeric sorbent followed by UHPLC-MS/MS utilizing selected reaction monitoring (SRM) as the acquisition mode. The developed method was successfully applied to different types of water samples.
EXPERIMENTAL Chemicals and reagents All standards were purchased from LGC Standards (Germany), Sigma-Aldrich (USA) and Chem Service (USA), with the highest purity available. Solvents as acetonitrile (MeCN) and methanol (MeOH) (HPLC grade), were purchased by J. T. Baker (USA), ethyl acetate and dichloromethane (analytical grade) were purchased by Scharlab (Spain) and Honeywell (USA), respectively, and the ultrapure water was obtained with a Milli-Q Direct 3UV® system (resistivity of 18.2 Ω) from Millipore (France). Vortex mixer model VX-38 was acquired by Ionlab (Brazil). The SPE cartridges Strata-X (60 and 200 mg; 3 mL) were purchased by Phenomenex (USA), Oasis HLB (60 and 200 mg; 3 mL) were from Waters (USA), Bond Elut C18 (500 mg; 3 mL) and Bond Elut Plexa (200 and 500 mg; 3 mL) were purchased from Agilent (USA) and Supel-Select HLB (200 mg; 6 mL) was from Sigma-Aldrich (USA). A stock standard solution (1000 mg L-1) was prepared for each pesticide from solid standards. These solutions were prepared in MeOH or MeCN according to their solubility, considering the purity of each standard. Carbendazim was prepared in MeCN with 8% v/v 0.1 mol L-1 HCl. A multicomponent working standard solution (5 mg L-1) containing all compounds was prepared. In addition, two individual standard solutions of 10 mg L-1 each were prepared for atrazine-d5 and triphenylphosphate, used as surrogate standard (SS) and internal standard (IS), respectively. The SS used is an analyte chemically similar to those being extracted and has been added to the samples at a known concentration prior to sample preparation to control extraction efficiency. The IS was added at a constant concentration to the final extract and standards to monitor the response of the equipment. In our case it was not used to correct analyte concentrations during analysis. All analytical solutions were stored in amber flasks at –10 ºC. Instrumentation and conditions UHPLC-MS/MS analyses were performed in a Waters Acquity UPLCTM system with Xevo TQ mass spectrometer (USA) equipped with an electrospray source, an Acquity binary pump with autosampler and column temperature controller. The chromatographic column was an Acquity BEH C18 column (50 mm × 2.1 mm, 1.7 µm) from Waters (USA). The mobile phase (A) was aqueous solution containing 2% v/v of MeOH, and the organic mobile phase (B) was MeOH, both containing 0.1% v/v of formic acid and ammonium formate 5 mmol L-1. Gradient program was applied with eluent B as follows: 0 min, 5% B; 7.75 min, 100% B; and 8.51 min, 5% B. The flow-rate was 0.225 mL min-1, the injection volume was 10 µL, the total chromatographic run time was 10 min and the column temperature was set at 40 ºC. The mass spectrometer was operated in the electrospray ionization positive mode (ESI+) using selected reaction monitoring (SRM) acquisition mode. The MS source conditions were: capillary voltage 0.5 kV; source temperature 150 ºC; desolvation temperature 500 ºC; and desolvation gas (N2) flow 600 mL min-1. The transition with the highest intensity was selected for quantification and the second highest intensity was used as a qualitative ion. MassLynx 4.1 software was used for data processing and instrument control. Evaluation of sample preparation In order to stablish the sample preparation conditions, the SPE method was evaluated for different parameters aiming to extract multiclass pesticides in a quick SPE procedure. The evaluated variables for sample preparation were, sample pH, type of extraction sorbent, elution solvent and elution solvent volume. Water samples, after being filtered with a cellulose acetate membrane (47 mm diameter and 0.45 µm porosity, from Agilent Technologies, USA) were placed in volumetric flasks and then transferred to conditioned SPE cartridges through polytetrafluoroethylene (PTFE) tubes. The percolation flow was maintained at around 10 mL min-1. An aliquot of 3 mL ultrapure water was passed through the cartridge after the sample percolating to clean the sorbent. The blank samples (uncontaminated tap water from the laboratory) were spiked at 0.32 µg L-1 for the evaluation of the SPE procedure. Initially, an adaptation of the Fortuny et al.30 method was evaluated using the Bond Elut Plexa 500 mg cartridge and ethyl acetate and dichloromethane as the elution solvent. The elution solvent was used for sorbent conditioning to remove possible interferences present in the cartridges that could be eluted together with the analytes.31 The method was effective for the extraction of most of the compounds. Even so, because the sample pH can be critical for the SPE method, influencing the retention of pesticides in the sorbent, in general it is necessary to adjust the pH of the sample to stabilize pesticides and increase their retention in the solid phase.32 Therefore, it was evaluated whether the acidification of the sample to pH 2.5 influences the recovery of the compounds. For a more comprehensive assessment, eight types of cartridges available on the market were evaluated: Strata-X 60 mg, Strata-X 200 mg, Oasis HLB 60 mg, Oasis HLB 200 mg, Bond Elut C18 500 mg, Bond Elut Plexa 200 mg, Bond Elut Plexa 500 mg and Supel-Select HLB 200 mg. The cartridges were evaluated according to the type and amount of stationary phase and the percentage of recovery of the analytes. The elution solvent must allow efficient elution of the analytes from the cartridge and keep the interferents present in the matrix retained in the cartridge as much as possible. In the attempt to find an appropriate elution solvent were evaluated solvents like MeCN, MeOH, ethyl acetate and dichloromethane, as well as, the organic solvent acidified with acetic acid and mixtures of these organic solvents. For this evaluation, 100 mL of blank sample was used. The elution with MeCN and MeOH were done with 2 mL of the respective solvent. In the case of ethyl acetate and dichloromethane, 3 aliquots of 2 mL dichloromethane/ethyl acetate 1:1 v/v were used, followed by evaporation using N2 and redissolution with 2 mL MeCN. In all cases, the eluent was added with the stopcock of the cartridge holder closed and the eluent was kept in contact with the sorbent for 1 min before elution. The best elution solvent was chosen considering the analytical signal of the compounds, the percentage of recovery and the compatibility of the solvent with the chromatographic system. The final extract was diluted twice in ultrapure water for all tests in order to obtain better chromatographic peaks. The SPE method was validated using uncontaminated tap water from the laboratory as blank sample. Throughout the method development and validation, analyses of blank samples were performed to verify the occurrence of contamination. Method validation In order to evaluate the selected procedure for the pesticide residues analysis in water samples, the method was validated according to the guideline SANTE33 for the parameters: selectivity, analytical curve and linearity, limit of detection (LOD), limit of quantification (LOQ), accuracy (recovery) and precision (repeatability and intermediate precision). Selectivity was evaluated by comparing the chromatograms obtained from the UHPLC-MS/MS system by injections of the blank and blank spiked extract. The calibration curves were prepared in MeCN with 1% v/v acetic acid at the concentrations 1, 2, 5, 10, 12, 15 and 20 µg L-1. The accuracy and precision of the method were evaluated in terms of recovery and relative standard deviation (RSD), with the blank spiked at 0.04, 0.1, 0.2 and 0.4 µg L-1 for the repeatability assay, and at 0.1 and 0.4 µg L-1 for the intermediate precision assay. LOQ was established as the lowest spiked level, which presented a signal/noise ratio greater than 10, recoveries between 70 and 120% with RSD ≤ 20%. The LOD values was established by dividing the LOQ value by 3.33. Application of the proposed method in water samples The proposed method was applied in different types of water samples (rivers, artesian wells, dam sample and tap water) collected during 2023 in the Central Region of the Rio Grande do Sul State, Brazil. Samples were collected in amber bottles of 500 mL, maintained between 4 and 10 ºC during the transport to prevent the possible change in the sample characteristics and degradation of the analytes. Samples were prepared within 24 h.
RESULTS AND DISCUSSION UHPLC-MS/MS analysis The chromatographic conditions used in this work are frequently used in our laboratory for pesticide residues in routine analyses. Some analytes were included in the method after the establishment of the MS conditions by infusion of the standard in the MS system. The ionization mode and the transitions for the quantification and qualification of the analytes were chosen using selection reaction monitoring (SRM). The two most intense transitions in the SRM acquisition mode were selected for each compound. The most intense transition was used for quantification and the second highest intensity was selected for qualifying the analyte. The selected conditions for the pesticides analyzed by UHPLC-MS/MS are available in the Supplementary Material (Table 1S). The Acquity BEH C18 chromatographic column provided high resolution and good peak shape for all evaluated compounds. The mobile phase provided high analytical signals for the studied compounds. An adequate separation was achieved with the gradient elution selected. The UHPLC-MS/MS system allowed the determination of 94 pesticides with a quick analysis of a total of 10 min. The chromatogram obtained by UHPLC-MS/MS of the ions selected in the SRM acquisition mode is shown in Figure 1.
 Figure 1. Total ion chromatogram obtained by UHPLC-MS/MS from a blank sample spiked with the analytes at 0.1 μg L-1
Establishment of the sample preparation conditions The evaluation of the Bond Elut Plexa 500 mg cartridge using ethyl acetate and dichloromethane as the elution solvent indicated that this condition is effective for most of the analytes evaluated. The evaluation of pH adjustment to 2.5 and without adjustment (pH 5.5 to 6.5) resulted in adequate recovery results for most of the compounds. Dichloromethane and ethyl acetate are not compatible with the UHPLC-MS/MS system and there is a need to evaporate the eluate and redissolve the analytes in an appropriate solvent. The evaporation stage makes the procedure more expensive, with risks of contamination of the final extract. Thus, it was evaluated the use of MeCN as elution solvent, which is compatible with the UHPLC-MS/MS system. MeCN improved the recovery of analytes due to its intermediate polarity.34 It allows the elution of analytes with a wide range of polarities and does not require an evaporation step, simplifying the procedure. In order to evaluate different SPE sorbents, eight types of cartridges with different amounts of sorbent were evaluated: Bond Elut Plexa 200 and 500 mg, Strata-X 60 and 200 mg, Supel-Select HLB 200 mg, Bond Elut C18 500 mg and Oasis HLB 60 and 200 mg. The number of compounds recovered for each cartridge is shown in Figure 2. MeCN was used as the elution solvent in all cases.
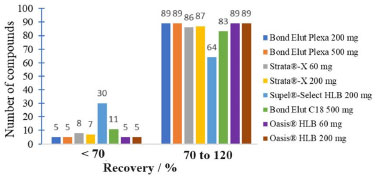 Figure 2. Evaluation of different SPE sorbents considering the obtained recoveries
Bond Elut C18 sorbents and Supel®-Select HLB showed the least satisfactory analytes recovery results in acceptable range (70-120%). These cartridges are often used for nonpolar compounds with log Kow greater than 1.35,36 The polarity of the compounds under study is intermediate to high. So, these sorbents are not the most suitable for the retention of the analytes in this study. Bond Elut Plexa is a polymeric sorbent with a hydroxylated exterior and hydrophobic interior. It is universally applicable, and is efficient for extracting a wide range of acidic, neutral, and basic analytes. Due to its advanced polymer structure of uniform particles with a limited size distribution this cartridge allows favorable working conditions such as larger flow rate and higher versatility.30 Strata-X and Oasis HLB cartridges are polymers accompanied by a modified surface with divinylbenzene (non-polar) and N-vinylpyrrolidone (polar). These sorbents are recommended for the extraction of acidic, basic and neutral compounds of medium to high polarity, and present broader retention capacity than silica-based sorbents.9 When comparing the different cartridges evaluated it was observed that the Strata-X, Oasis HLB and Bond Elut Plexa sorbents showed better recovery results (Figure 2), which was expected due to the versatility of these cartridges. The best sorbent was chosen based on the recovery results, laboratory availability and the cartridge price. Therefore, it was decided to use the Oasis HLB 60 mg cartridge for subsequent tests. The elution solvents evaluated were MeCN, MeOH, MeCN/MeOH 1:1 v/v, MeCN with 1% v/v acetic acid, MeOH with 1% v/v acetic acid and MeCN/MeOH 1:1, v/v with 1% v/v acetic acid. This choice was based on the literature9,37 and on the compatibility of the solvents with the UHPLC-MS/MS system. Figure 3 shows the difference in recovery between the evaluated elution solvents.
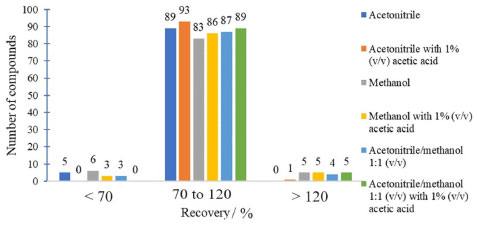 Figure 3. Comparison of elution solvents based on pesticide recoveries obtained by UHPLC-MS/MS
Figure 3 shows that the use of MeCN as an elution solvent had a better result in the recovery of analytes than MeOH and MeCN/MeOH 1:1 v/v. MeCN has intermediate polarity that resembles the polarity of most of the compounds in study. In addition, the use of MeCN acidified with 1% v/v acetic acid shows an increase in the number of compounds recovered. The elution solvent having an acid character favored the elution of some compounds. Therefore, it was decided to use MeCN 1% v/v acetic acid as the elution solvent. For the evaluation of the elution solvent, 2 mL of solvent was used. After choosing the solvent, the elution volumes of 1 and 2 mL were evaluated. The number of analytes adequately recovered using 1 and 2 mL of MeCN containing 1% v/v acetic acid were the same. Elution with 1 mL of solvent provides a concentration factor twice as high as using 2 mL. Therefore, it was decided to use 1 mL of eluent, leaving 1 min of contact with the sorbent before elution. The polymeric sorbent Oasis HLB was reported as effective for the preconcentration of pesticide residues in water samples. Oasis HLB 200 mg was used to determine acetamiprid, sulfoxaflor and flupyradifurone, and the common metabolite 6-chloronicotinoic acid in surface waters by LC-MS/MS. The sample (200 mL) was adjusted to pH 4.6, the cartridge was eluted with 5 mL MeCN with 0.03% v/v formic acid followed by drying with nitrogen and redissolution with 1 mL 15:85 v/v ACN:water containing 0.1% v/v formic acid.38 The same sorbent and amount was used in a multiresidue method for the determination of 42 pesticides by HPLC-MS/MS and 64 by GC-MS/MS using 500 mL of sample, elution with 3 aliquots of 2 mL dichloromethane/ethyl acetate 1:1 v/v, evaporation using N2, redissolution with 1 mL MeOH for HPLC-MS/MS and 1 mL n-hexane for GC-MS/MS.39 Chen et al.40 used Oasis HLB 500 mg for the determination of 22 pesticides by LC-MS/MS. A 200 mL water sample was preconcentrated, eluted with 4 mL ACN containing 5% v/v formic acid, concentrated to 1 mL using N2 before analysis. Selected sample preparation method Oasis HLB 60 mg cartridges were conditioned with 3 mL MeCN acidified with 1% v/v acetic acid and 3 mL ultrapure water. Later the sample was percolated (100 mL at 10 mL min-1; pH 5.5 to 6.5), then the cartridges were washed with 3 mL ultrapure water and left to dry for 20 min under vacuum in the manifold. Finally, the elution step was performed with 1 mL MeCN acidified with 1% v/v acetic acid. The final extract was diluted twice in ultrapure water in order to improve the performance of the UHPLC-MS/MS system. Method validation The selectivity of the method was ensured due to the blank samples did not show the presence of the evaluated compounds and interferents in the same quantification and qualification ions of all the analytes. The solvents and reagents were also evaluated, and no interferents were observed. Linearity was evaluated by the linear regression of the calibration curves and the results of determination coefficients (R2) obtained. The response was linear in the evaluated range (1 to 20 µg L-1), the linearity of all compounds was adequate showing R2 ≥ 0.99. The significance of linearity regression was evaluated by the analysis of variance (F test) and the lack of adjustment test. ANOVA showed that the analyte calibration curves presented linearity with well-adjusted models without lack-of-fit. The results of accuracy and precision were evaluated through the recovery and RSD values. Table 1 shows the recovery (n = 7) and RSD values for the analytes at the concentration levels 0.04, 0.1, 0.2 and 0.4 µg L-1, as well as the results of the intermediate precision test. Recovery was calculated using the calibration curves prepared in MeCN 1% v/v acetic acid. Only recovery values between 70 and 120% and RSD ≤ 20% were accepted.33
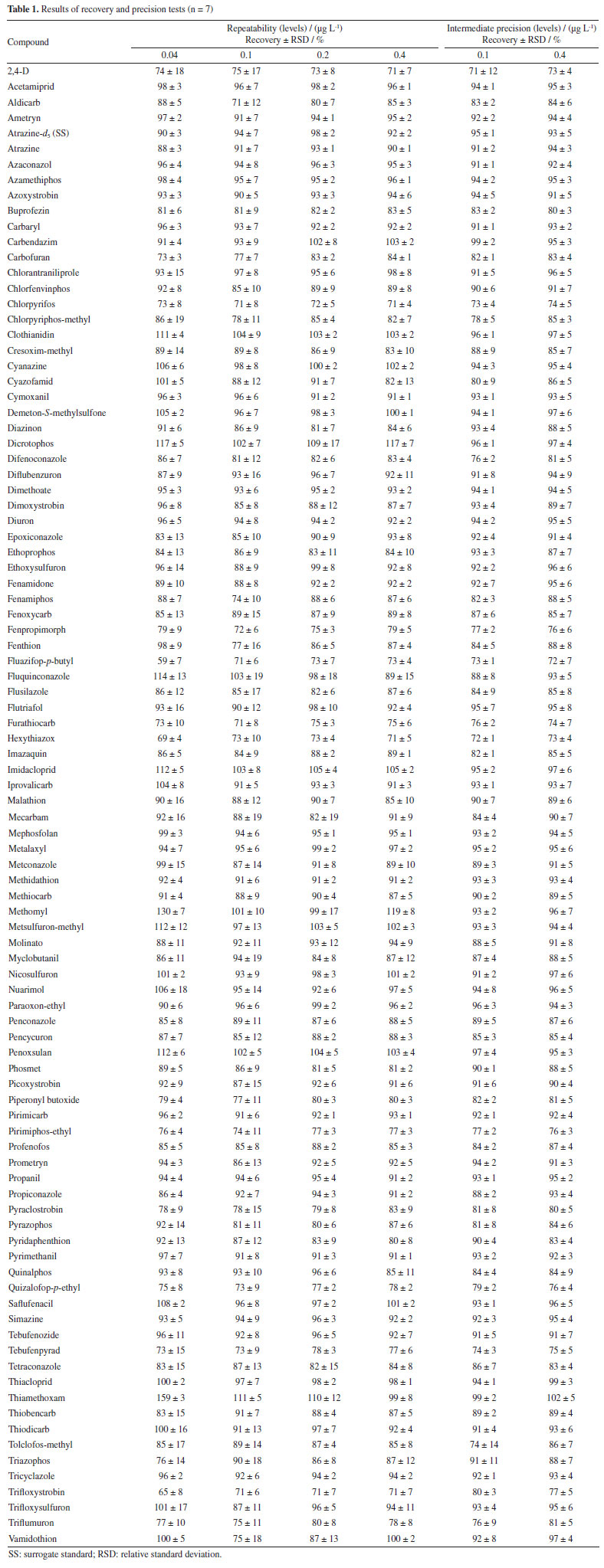
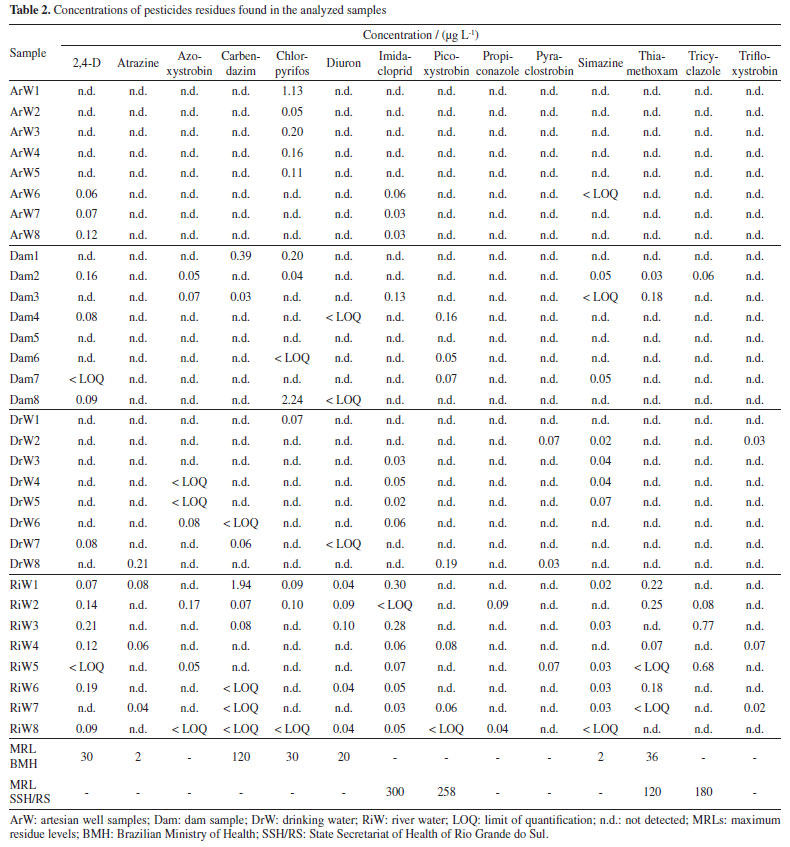
The accuracy and precision, in terms of repeatability and intermediate precision, presented acceptable results for most of the tests with recoveries between 71 and 117%, with RSD ≤ 19%. Except fluazafop-p-butyl, hexithiazoxy, methomyl, thiamethoxam and trifloxystrobin which showed recoveries outside the acceptable range at the level of 0.04 µg L-1. The LOD and LOQ of the method were 0.01 and 0.04 µg L-1, respectively, for most of the compounds analyzed. For fluazafop-p-butyl, hexithiazoxy, metomyl, thiamethoxam and trifloxystrobin the LOD and LOQ values, obtained considering the recovery and RSD values, were 0.03 and 0.10 µg L-1, respectively. These method limits were satisfactory regarding the limits establish in Brazilian and European standards. Thus, the developed method has great potential for laboratory routines and it can be used to perform determinations at trace levels analyses. Application of the method in water samples The validated method was applied for the determination of pesticide residues in 32 water samples from the Central Region of the Rio Grande do Sul State, Brazil. Eight samples of each different types of water samples (artesian well (ArW), dam (Dam), drinking water (DrW), and river water (RiW)) were analyzed. Table 2 shows the concentration values of pesticide residues for each sample. All the samples analyzed showed pesticide residues. The concentration values in the water samples are lower than the limits established in Ordinance GM/MS No. 88841 of the Brazilian Ministry of Health and Ordinance 32042 of the State Health Secretariat of Rio Grande do Sul for the 14 pesticides found. The concentrations of most of the pesticides were higher than the limits established by the European standards43 for drinking water. The results also showed that even with the treatment of the water distributed to the population, it still has pesticide residues, even though the concentrations found are below the limits of Brazilian legislation. SRM chromatogram of chlorpyrifos in drinking water samples and an artesian well sample overlaid on its LOQ (0.04 µg L-1) and the blank sample is available in Supplementary Material (Figure 1S).
CONCLUSIONS The method proposed proved effective in determination of 94 current-use pesticides in various water samples, including drinking water, artesian well water, dam water, and river water. Through numerous tests conducted, the best sample preparation results were achieved using the SPE cartridge Oasis HLB (60 mg/3 mL) and MeCN with 1% v/v acetic acid as the elution solvent. Moreover, utilizing UHPLC-MS/MS with an electrospray ionization source in positive mode, operating in selected reaction monitoring mode, yielded satisfactory results in terms of detectability, selectivity, accuracy, and precision during the method validation phase. The linearity of the method was assessed within the range of 1-20 µg L-1, along with testing its precision and accuracy, which yielded satisfactory outcomes. Additionally, the limits of detection and quantification values obtained for the 94 CUPs were deemed acceptable compared to the maximum residue limits established by the European Union and Brazilian guidelines. The method was then applied to 12 different water samples, revealing that nine of the samples contained residues of one to four pesticides per sample, with concentrations of up to 2.24 µg L-1, thereby underscoring the significance of investigating pesticide residue levels in different types of water samples. In conclusion, our findings affirm that the proposed method exhibits high reliability in determining different classes of pesticides in water samples, showcasing its suitability for routine analysis and efficiency in conducting pesticide residue analyses at trace levels. The results that can be obtained with the application of the method will enable the generation of results that can guide measures to protect the environment and the population.
SUPPLEMENTARY MATERIAL Figure 1S and Table 1S related to the results obtained in this work are available at http://quimicanova.sbq.org.br, as PDF file, with free access.
ACKNOWLEDGMENTS The authors are grateful to the financial support and fellowship grants from the Brazilian Funding Agencies CNPq and CAPES (Finance Code 001).
REFERENCES 1. Pignati, W. A.; Lima, F. A.; Lara, S. S.; Correa, M. L.; Barbosa, J. R.; Leão, L. H.; Pignatti, M. G.; Ciência & Saúde Coletiva 2017, 22, 3281. [Crossref] 2. Spadotto, C. A.; Gomes, M. A.; Luchini, L. C.; Andréa, M. M.; Monitoramento do Risco Ambiental de Agrotóxicos: Princípios e Recomendações, 1ª ed.; Embrapa Meio Ambiente: Jaguariúna, 2004. 3. Salimov, Y.; Journal of Veterinary Medicine and Animal Sciences 2021, 4, 1070. [Link] accessed in June 2024 4. Ganaie, M. I.; Jan, I.; Mayer, A. N.; Dar, A. A.; Mayer, I. A.; Ahmed, P.; Sofi, J. A.; Int. J. Anal. Chem. 2023, 2023, 1. [Crossref] 5. Mojiri, A.; Zhou, J. L.; Robinson, B.; Ohashi, A.; Ozaki, N.; Kindaichi, T.; Farraji, H.; Vakili, M.; Chemosphere 2020, 253, 126646. [Crossref] 6. Vryzas, Z.; Current Opinion in Environmental Science & Health 2018, 4, 5. [Crossref] 7. Dugan, S. T.; Muhammetoglu, A.; Uslu, A.; Sci. Total Environ. 2023, 901, 165892. [Crossref] 8. Ghosh, R. K.; Ray, D. P.; Internacional Journal of Bioresource Science 2016, 3, 73. [Crossref] 9. Syafrudin, M.; Kristanti, R. A.; Yuniarto, A.; Hadibarata, T.; Rhee, J.; Al-Onazi, W. A.; Algarni, T. S.; Almarri, A. H.; Al-Mohaimeed, A. M.; Int. J. Environ. Res. Public Health 2021, 18, 468. [Crossref] 10. Panis, C.; Candiotto, L. Z. P.; Gaboardi, S. C.; Gurzenda, S.; Cruz, J.; Castro, M.; Lemos, B.; Environ. Int. 2022, 165, 107321. [Crossref] 11. El-Nahhal, I.; El-Nahhal, Y.; J. Environ. Manage. 2021, 299, 113611. [Crossref] 12. Voutchkova, D. D.; Schullehner, J.; Skaarup, C.; Wodschow, K.; Ersbøll, A. K.; Hansen, B.; GEUS Bulletin 2021, 47, 6090. [Crossref] 13. Kruć-Fijałkowska, R.; Dragon, K.; Drożdżyński, D.; Górski, J.; Sci. Rep. 2022, 12, 3317. [Crossref] 14. Estevão, P. L.; Zamora, P. P.; Nagata, N.; J. Braz. Chem. Soc. 2018, 29, 2104. [Crossref] 15. Mas, L. I.; Aparicio, V. C.; de Gerónimo, E.; Costa, J. L.; SN Appl. Sci. 2020, 2, 691. [Crossref] 16. Rocha, A. A.; Monteiro, S. H.; Andrade, G. C.; Vilcab, F. Z.; Tornisielo, V. L.; J. Braz. Chem. Soc. 2015, 26, 2269. [Crossref] 17. Caldas, S. S.; Demoliner, A.; Primel, E. G.; J. Braz. Chem. Soc. 2009, 20, 125. [Crossref] 18. Caldas, S. S.; Demoliner, A.; Costa, F. P.; D'Oca, M. G.; Primel, E. G.; J. Braz. Chem. Soc. 2010, 21, 642. [Crossref] 19. Wille, K.; Brabander, H. F.; Wulf, E.; Caeter, P. V.; Janssen, C. R.; Vanhaecke, L.; TrAC, Trends Anal. Chem. 2012, 35, 87. [Crossref] 20. Queiroz, S. C.; Collins, C. H.; Jardim, C. S.; Quim. Nova 2001, 24, 68. [Crossref] 21. Faraji, M.; Yamini, Y.; Gholami, M.; Chromatographia 2019, 82, 1207. [Crossref] 22. Mandal, S.; Poi, R.; Hazra, D. K.; Ansary, I.; Bhattacharyya, S.; Karmakar, R.; J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2023, 1215, 123587. [Crossref] 23. Prestes, O. D.; Adaime, M. B.; Zanella, R.; Scientia Chromatographica 2011, 3, 51. [Crossref] 24. Maldaner, L.; Jardim, I. C.; Talanta 2012, 100, 38. [Crossref] 25. Kharbouche, L.; García, M. D.; Lozano, A.; Hamaizi, H.; Galera, M. M.; Talanta 2019, 199, 612. [Crossref] 26. Elfikrie, N.; Ho, Y. B.; Zaidon, S. Z.; Juahir, H.; Tan, E. S.; Sci. Total Environ. 2020, 712, 136540. [Crossref] 27. Kemmerich, M.; Rizzetti, T. M.; Martins, M. L.; Prestes, O. D.; Adaime, M. B.; Zanella, R.; Food Analytical Methods 2015, 8, 728. [Crossref] 28. Tsipi, D.; Botitsi, H.; Economou, A.; Mass Spectrometry for the Analysis of Pesticide Residues and Their Metabolites, 1st ed.; John Wiley & Sons: Hoboken, 2015. 29. Donato, F. F.; Bandeira, N. M. G.; dos Santos, G. C.; Prestes, O. D.; Adaime, M. B.; Zanella, R.; J. Chromatogr. A 2017, 1516, 54. [Crossref] 30. Fortuny, G.; Pineda, L.; Rúbies, A.; Centrich, F.; Companyó, R.; Int. J. Environ. Anal. Chem. 2013, 93, 707. [Crossref] 31. Caldas, S. S.; Gonçalves, F. F.; Primel, E. G.; Prestes, O. D.; Martins, M. L.; Zanella, R.; Quim. Nova 2011, 34, 1604. [Crossref] 32. Jardim, I. C. In Preparo de Amostras para Análise de Compostos Orgânicos, 1st ed.; Figueiredo, E.; Borges, K. B.; Queiroz, M. E., eds.; LTC: Barueri, 2015, ch. 8. 33. European Commission Analytical Quality Control and Method Validation Procedures for Pesticide Residues Analysis in Food and Feed; Guidance SANTE 11312/2021-Analytical Quality Control and Method Validation Procedures for Pesticide Residues Analysis In Food and Feed, 2021. [Link] accessed in June 2024 34. Komsta, L.; Stepkowska, B.; Skibinski, R.; J. Chromatogr. A 2017, 1483, 138. [Crossref] 35. Cherta, L.; Beltran, J.; Portolés, T.; Hernández, F.; Anal. Bioanal. Chem. 2012, 402, 2301. [Crossref] 36. Santasania, C. T.; Trinh, A.; Sarker, M.; Reporter (Sigma-Aldrich) 2009, 27.3, 8. [Link] accessed in June 2024 37. Casado, J.; Santillo, D.; Johnston, P.; Anal. Chim. Acta 2018, 1024, 1. [Crossref] 38. Łukaszewicz, P.; Stepnowski, P.; Haliński, Ł. P.; Chemosphere 2023, 310, 136868. [Crossref] 39. Li, W.; Xin, S.; Deng, W.; Wang, B.; Liu, X.; Yuan, Y.; Wang, S.; Water Res. 2023, 245, 120570. [Crossref] 40. Chen, F.; Zheng, Y.; Mao, L.; Zhang, L.; Zhu, L.; Zhang, Y.; Jiang, H.; Liu, X.; Int. J. Environ. Anal. Chem. 2022 [Crossref] 41. Ministério da Saúde; Portaria GM/MS No. 888, de 4 de maio de 2021, Altera o Anexo XX da Portaria de Consolidação GM/MS No. 5, de 28 de setembro de 2017, para Dispor sobre os Procedimentos de Controle e de Vigilância da Qualidade da Água para Consumo Humano e seu Padrão de Potabilidade; Diário Oficial da União (DOU), Brasília, No. 85, de 07/05/2021, p. 127. [Link] accessed in June 2024 42. Secretaria da Saúde do Rio Grande do Sul; Portaria No. 320/2014, de 24 de abril de 2014, Estabelece Parâmetros Adicionais de Agrotóxicos ao Padrão de Potabilidade para Substâncias Químicas, no Controle e Vigilância da Qualidade da Água para Consumo Humano no RS; Diário Oficial do Estado do Rio Grande do Sul, Porto Alegre, 2014. [Link] accessed in June 2024 43. Official Journal of the European Union; Directive (EU) 2020/2184 of the European Parliament and of the Council of 16 December 2020, on the Quality of Water Intended for Human Consumption, 2020. [Link] accessed in June 2024
Guest Editor handled this article: Marco T. Grassi |

On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access