
 |
 |
 |
 |
 |
Artigo
|
|
| Corrosão localizada do alumínio em meios aerados e em meios com baixo teor oxigênio: estudo e comparação por meio de curvas de polarização Localized corrosion of aluminum in aerated environments and in environments with low oxygen content: study and comparison through polarization curves |
|
João Victor de S. Araujo* Centro de Ciência e Tecnologia de Materiais, Instituto de Pesquisas Energéticas e Nucleares, Universidade de São Paulo, 05508-000 São Paulo - SP, Brasil Recebido: 12/07/2024 *e-mail: jvaraujo@usp.br In this study, the influence of deaeration on the electrochemical behavior of aluminum was studied by polarization techniques. The advantages of deaeration for the evaluation of localized corrosion on aluminum are discussed. The influence of the corrosive environment with low oxygen contents on the electrochemical response of aluminum was evaluated by anodic and cathodic potentiodynamic polarization curves. The polarization tests were carried out in chloride solutions followed by optical and scanning electron microscopy characterization. It was observed that deaeration of the solution significantly influences the electrochemical behavior of aluminum, as predicted by corrosion kinetics theories. For the cathodic curves, a decrease in the limiting current density for the oxygen reduction reaction (ORR) was observed for low oxygen solutions, while the "apparent" initiation of the hydrogen evolution reaction (HER) was shifted to higher potentials. In the anodic curves, a common passive behavior was not observed in aerated conditions, however, at the corrosion potential, localized pitting was observed indicating that the material was already above the breakdown potential (Ebr). In the low oxygen content solution, however, a passive region with a breakdown potential, was observed on the anodic curves. INTRODUÇÃO Há várias técnicas para avaliar a cinética da corrosão de metais, incluindo técnicas eletroquímicas, como as curvas de polarização. Técnicas eletroquímicas são, em geral, mais rápidas que ensaios de imersão, além de fornecerem parâmetros para avaliar o mecanismo de corrosão. Os ensaios eletroquímicos são bastante utilizados para avaliações comparativas de diferentes metais, ligas ou tratamentos de superfície - ou para comparar a corrosão de um metal em diferentes meios. No entanto, a correta interpretação das curvas de polarização para estudo da corrosão requer um conhecimento prévio do comportamento do metal no meio em estudo e, em geral, caracterizações metalográficas da superfície exposta ao meio corrosivo após os ensaios. A interpretação das curvas de polarização para metais em meios em que o material é ativo, ou em meios que se encontra passivo, é intrinsicamente diferente.1 O alumínio, por exemplo, por ser passivo em alguns meios, e apresentar corrosão localizada em outros, como por exemplo em meios neutros com cloretos, torna a interpretação das curvas de polarização complexa, necessitando caracterizações adicionais após os ensaios.2 Desta forma, um estudo detalhado da termodinâmica e cinética de corrosão do alumínio se faz necessário para a correta interpretação das curvas de polarização desse material. A revisão das teorias da termodinâmica e cinética de corrosão nos próximos tópicos tem como objetivo fornecer uma base teórica para embasar o trabalho, assim como justificar o estudo e a análise dos resultados nas seções subsequentes. Para uma compreensão abrangente e detalhada das teorias fundamentais de termodinâmica e cinética de corrosão, recomenda-se a consulta a textos e livros especializados.1,3-6 Termodinâmica da corrosão do alumínio Do ponto de vista termodinâmico, os diagramas de Pourbaix são ferramentas essenciais para se compreender o comportamento eletroquímico dos metais, conforme apresentado na Figura 1.
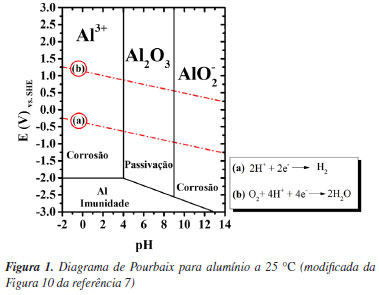
Por meio de diagramas de equilíbrio de Pourbaix pode-se avaliar as regiões de pH e potencial onde um metal, imerso em uma solução de seus próprios íons, se encontra em estado de corrosão ativa (íons), passivo (óxidos ou hidróxidos) ou imune (estado metálico). Sendo assim, a partir do diagrama de Pourbaix, pode ser indicado se o alumínio irá: (i) corroer formando Al3+ e AlO2- em faixas de pH baixo e alto; (ii) formar um filme passivo de Al2O3 (ou hidróxidos) em uma faixa de pH aproximadamente neutra; ou (iii) ser imune à dissolução em valores de potenciais menores que -2.0 VSHE (potencial reversível ou de equilíbrio do alumínio; SHE: do inglês standard hydrogen electrode). O diagrama de Pourbaix, no entanto, não fornece informações sobre a cinética da corrosão, ou seja, não define o quão rápido a reação irá ocorrer, por se tratar de construções para equilíbrio termodinâmico.7 Revisão de conceitos de curvas de polarização utilizadas em estudos de corrosão Quando há corrosão na superfície de um metal, ocorrem pelo menos dois processos eletroquímicos: um anódico e outro catódico, cada um caracterizado por um potencial de equilíbrio diferente. O potencial de corrosão, Ecorr, que pode ser medido por meio de um voltímetro utilizando um eletrodo de referência, é necessariamente intermediário entre os dois potenciais de equilíbrio.5 No potencial de corrosão, os processos anódicos e catódicos ocorrem com a mesma taxa, chamada corrente de corrosão, ou densidade de corrente de corrosão, icorr, se a área onde ocorrem as reações catódicas e anódicas forem iguais.8 Entende-se, portanto, que na corrosão, a densidade de corrente anódica é aquela devido à reação de dissolução do metal:  Enquanto a densidade de corrente catódica será a soma das densidades de corrente devido às possíveis reações catódicas observadas em corrosão em meios aquosos aerados ácidos: 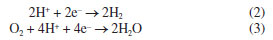 Ou em meios aquosos aerados neutros ou alcalinos: 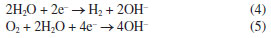 É importante destacar que tanto as reações anódicas quanto as catódicas podem apresentar controle por ativação, ou por concentração. Para os processos catódicos observados em corrosão em meios aquosos aerados, o efeito mais importante para o controle da taxa da reação por concentração é a difusão do gás oxigênio (O2) dissolvido no meio para a superfície do metal, ocorrendo a subsequente redução do oxigênio pela reação de eletrodo 3 ou 5 descritas acima.8 Para que a reação de redução do oxigênio ocorra, as moléculas de O2 primeiro difundem até a superfície do eletrodo para, em seguida, serem reduzidas em sítios catódicos na superfície do metal.3 Quando a taxa de difusão das moléculas de O2 é menor que a taxa de redução dessas moléculas na superfície do metal, cria-se uma diferença de concentração entre o gás dissolvido próximo da superfície e nas regiões mais distantes. Essa diferença, conduz a uma densidade de corrente limite (iL) para redução de O2, ou seja, em determinado momento, independentemente do potencial aplicado, a corrente permanece praticamente constante por estar limitada por difusão das espécies reagentes até o local da reação.8 O fenômeno descrito acima pode ser observado na corrosão de alguns metais em alguns meios, como, por exemplo, para o alumínio em meios aquosos aerados, e pode ser interpretado por meio das curvas de polarização catódica. A Figura 2 apresenta uma curva de polarização catódica esquemática mostrando as regiões dominadas pela reação de redução do oxigênio com controle por difusão, e pela reação de evolução de hidrogênio sobre um metal.3,4
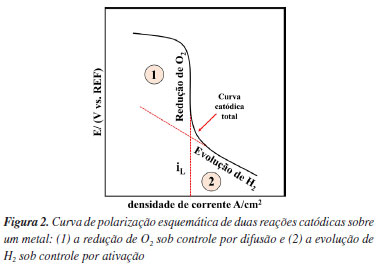
A Figura 2 mostra que a curva catódica é caracterizada por dois estágios: (1) o primeiro relacionado à reação de redução de oxigênio, e o (2) segundo dominado pela evolução de gás hidrogênio (H2). Entende-se, portanto, que a curva de polarização catódica total é a soma dos processos catódicos individuais.5 Nesse caso, a reação de redução de O2 está sob controle por difusão com densidade de corrente limite (iL). Já a reação de redução de H+ está sob controle por ativação.9 Isso é observado em potenciais onde ocorre mudança no coeficiente angular da curva catódica total (coeficiente angular de Tafel, conforme previsto para um processo com controle por ativação), como indicado na Figura 2. Abaixo desse potencial tem-se predominantemente a reação de redução dos cátions H+. Cabe ressaltar que, assim como na reação de redução de O2, em sobretensões catódicas muito elevadas, a redução de H+ também pode sofrer controle por difusão.10 Evans11,12 analisou as curvas de polarização, desenvolvendo conceitos que posteriormente se tornaram conhecidos como diagramas de Evans. Estes diagramas auxiliam na interpretação das curvas de polarização experimentais obtidas em diferentes sistemas.10 Sendo assim, a seguir é descrita a interpretação por meio dos diagramas de Evans para um sistema com controle por ativação e com controle por difusão e, por fim, diagramas esquemáticos para metais passivos, neste caso do alumínio e suas ligas, serão discutidos. Interpretação do diagrama de Evans para um sistema sob controle por difusão e por ativação A Figura 3 apresenta diagramas esquemáticos de Evans para a curva de polarização experimental para um metal (M) com dissolução sob controle por ativação e suas principais características em meios aerado, semi-desaerado e desaerado.
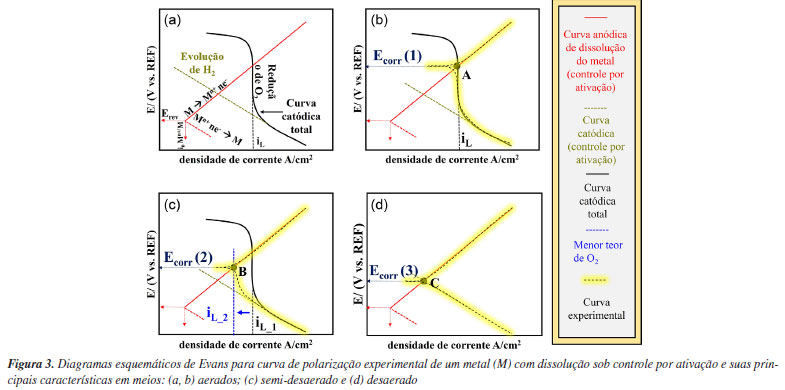
Nos diagramas de Evans apresentados na Figura 3a estão representadas a curva de polarização catódica, conforme descrito na Figura 3, e uma reação de dissolução para um metal (M) sob controle por ativação. No potencial reversível (Erev), a reação de eletrodo está em equilíbrio, sendo a taxa da reação igual à densidade de corrente de troca (i0) nos dois sentidos (redução e oxidação), conforme indicado na Figura 3 para a reação anódica.3 Quando há um sobrepotencial positivo e uma corrente anódica, a reação ocorrerá principalmente na direção da oxidação, resultando na dissolução do metal. Em contrapartida, com um sobrepotencial negativo e uma corrente catódica, a reação predominante será na direção da redução. A soma de todas as reações anódicas e catódicas resultará na curva experimental obtida. Conforme a teoria do potencial misto, no ponto de intersecção entre a soma das curvas anódicas e a das catódicas, onde a soma das correntes anódicas é igual à soma das correntes catódicas, o potencial de corrosão (Ecorr) é estabelecido, conforme apresentado na Figura 3b. Cabe ressaltar que a interpretação para a densidade de corrente só é válida se a área para as reações anódicas e catódicas for igual, ou seja, nesse caso assume-se que esta é a área total da superfície do metal exposta ao meio. Com a diminuição do teor de oxigênio na solução uma nova curva experimental é estabelecida, como apresentado na Figura 3c para um meio "semi-desaerado". Um novo potencial de corrosão (Ecorr (2)) é definido, por se tratar de um sistema semi-desaerado, e a corrente limite (iL) associada à redução de O2 (polarização por concentração) é deslocada para menores valores. Na ausência total de oxigênio, a reação de evolução do hidrogênio (ou, similarmente, a redução da água) é o único processo catódico possível; consequentemente, a condição de corrosão é representada pelo ponto C com a interseção da curva anódica e catódica, conforme apresentado na Figura 3d, sendo o valor de potencial de corrosão (Ecorr (3)). Logo, a diminuição do potencial de corrosão do ponto A (Figura 3b) para o B (Figura 3c) pode ser explicada pela diminuição da concentração de O2 no meio. À medida que o O2 diminui na solução, tanto o potencial de corrosão (Ecorr), como a taxa de corrosão, diminuem. A interpretação acima é válida quando a reação de dissolução do metal é controlada por ativação, o que, em geral, ocorre em processos de corrosão uniforme. Contudo, para o alumínio e suas ligas em meios aquosos neutros e aerados, a relação estabelecida acima é diferente, pois nestes meios o alumínio é espontaneamente passivo, conforme previsto pelo diagrama de Pourbaix. A região associada à passividade do alumínio e suas ligas também pode ser avaliada por meio de curvas de polarização, bem como a ruptura do filme passivo, responsável pela iniciação da corrosão localizada.13 Sendo assim, a seção a seguir apresenta o comportamento eletroquímico esperado para o alumínio e suas ligas em meios neutros contendo cloretos, por meio do diagrama de Evans. Interpretação do diagrama de Evans para o alumínio em meios neutros aerados e desaerados A Figura 4a apresenta uma curva de polarização anódica esquemática para o alumínio e suas ligas em um meio neutro com cloretos. Tanto a densidade de corrente de troca (i0) quanto o potencial reversível (Erev) são apresentados com base nos mesmos conceitos explicados na Figura 3. Contudo, são observadas características típicas de uma região passiva associada à formação do óxido protetor. A região passiva se caracteriza pela estabilização da corrente independente do potencial aplicado, ou seja, nesta condição tem-se a densidade de corrente passiva (ipass).3 Em potenciais mais elevados observa-se um aumento abrupto da corrente indicando a ruptura do filme passivo, devido à iniciação da corrosão localizada. Cabe ressaltar que outros fenômenos podem causar esse aumento da corrente, como a reação de evolução de oxigênio, ou corrosão transpassiva. Entretanto, em geral, esse tipo de comportamento não é observado para o alumínio por se tratar de um metal com potenciais de ruptura do filme passivo menores em relação ao aço inoxidável.
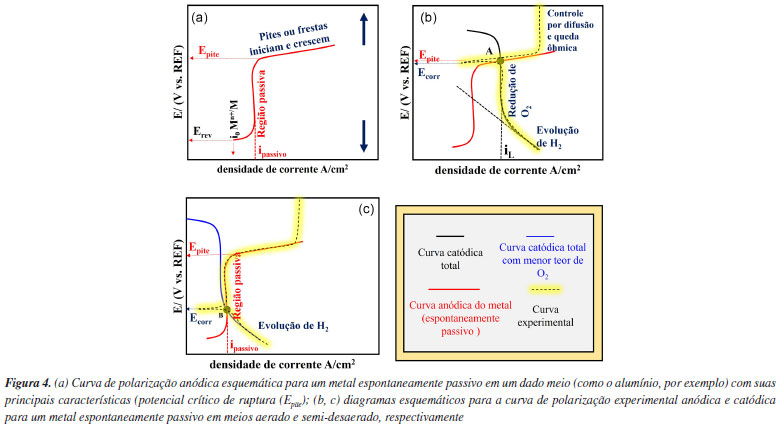
De fato, a corrosão localizada é comumente observada no alumínio e suas ligas devido à presença de heterogeneidades que tendem a causar descontinuidade no filme passivo. Essa quebra é observada nas curvas de polarização do alumínio pelo aumento abrupto da corrente.13 O potencial associado à quebra é denominado potencial de pite (Epite) ou de fresta, e, em geral, tende a ser maior que o potencial de corrosão (Ecorr). Tanto o pite como a fresta são formas de corrosão localizada. A primeira se caracteriza por acontecer de forma puntiforme na superfície do material, enquanto a segunda acontece em regiões oclusas, como as formadas por uma fresta, nas quais o meio corrosivo pode entrar e permanecer em condições estagnadas e com difusão limitada de espécies.14 Em meios aerados e, portanto, com O2 dissolvido, a visualização da região passiva e do potencial de ruptura do filme passivo pode ser dificultada e, como consequência, a interpretação das curvas eletroquímicas experimentais obtidas para o alumínio13 pode ser complexa devido ao fato de que o potencial de ruptura do filme passivo do alumínio puro coincide com a região de redução do oxigênio sobre a superfície. Por exemplo, levando em consideração a obtenção de curvas de polarização anódicas e catódicas para o alumínio puro e algumas ligas em meios neutros aerados, pode ser observada a curva apresentada na Figura 4b em amarelo. Na presença de oxigênio (O2) o potencial de corrosão (Ecorr) é estabelecido no ponto de intersecção das curvas anódicas e catódicas no ponto A, e encontra-se na mesma faixa de valores que o potencial de pite (Epite). Isso sugere que no potencial de corrosão já ocorre a ruptura do filme passivo. Esse comportamento é comumente observado em curvas de polarização do alumínio na presença de íons Cl- em meios aerados.13 Contudo, quando se remove o O2 do meio corrosivo podem ser observadas a região passiva e o potencial de pite, como mostra a Figura 4c. Neste caso, o processo de desaeração permite a visualização da região passiva e do potencial de ruptura do filme passivo devido à diminuição do potencial de corrosão (ponto B). Explorando a resistência à corrosão do alumínio e suas ligas através das curvas de polarização: aplicações e análise crítica A utilização de ligas de alumínio na indústria vem sendo impulsionada nas últimas décadas principalmente devido à redução de peso de estruturas e componentes metálicos (em comparação com estruturas tradicionais de aço) possibilitando ganhos econômicos e ambientais. O desenvolvimento de técnicas robustas e confiáveis para avaliação da corrosão localizada (por pite e fresta) em ligas de alumínio é, portanto, essencial para a prevenção de falhas estruturais com impacto econômico e garantia da segurança e confiabilidade dessas estruturas metálicas. Além disso, estudos visando elucidar o mecanismo de corrosão localizada no alumínio contribuem para o desenvolvimento de ligas com melhores propriedades de resistência à corrosão. Nos últimos anos, houve um aumento significativo no número de estudos focados na avaliação da corrosão localizada em ligas de alumínio15-24 e nas estratégias para sua prevenção.25-29 Conforme discutido nas seções anteriores, a análise da cinética de corrosão do alumínio por meio das curvas de polarização requer atenção especial, uma vez que o alumínio corrói de forma localizada e não uniforme em meios neutro com cloreto e sendo a principal reação catódica de redução do oxigênio limitada por difusão em meios aerados. Logo, análises que pressupõe corrosão uniforme e reações com controle por ativação, como, por exemplo, aplicação da lei de Faraday para cálculo de perda de massa ou espessura e extrapolação de retas de Tafel, não podem ser aplicadas neste caso. Ao investigar a corrosão por pites em ligas de alumínio, as curvas de polarização não devem ser utilizadas isoladamente como o único recurso para essa avaliação. Os dados extraídos dessas curvas devem ser cuidadosamente examinados. Além disso, os detalhes experimentais de como as curvas de polarização são obtidas podem ter um impacto significativo nos resultados, levando a interpretações equivocadas ou até mesmo a conclusões imprecisas. Nesse contexto, o presente estudo tem como propósito apresentar, através de resultados experimentais claros e contundentes, uma detalhada interpretação das curvas de polarização do alumínio em meios neutro contento cloreto, tanto aerados quanto com baixo teor de oxigênio, com base nas teorias de termodinâmica e cinética eletroquímica. Além disso, busca-se investigar e elucidar o processo de corrosão localizada no alumínio, com foco na corrosão por pites. Para enriquecer os resultados e discussões, uma análise comparativa é apresentada, contrastando a caracterização da corrosão localizada com técnicas metalográfica, como microscopia óptica e microscopia eletrônica de varredura.
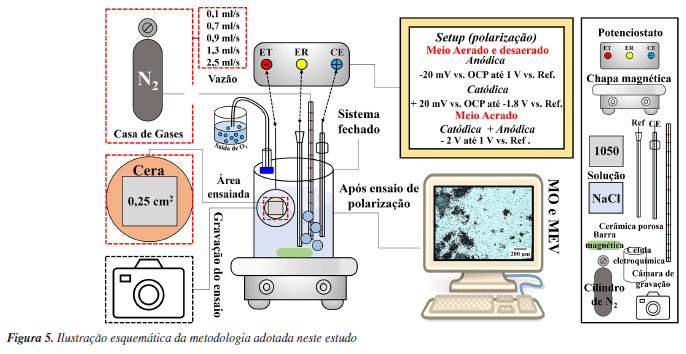
PARTE EXPERIMENTAL Material Neste estudo foi utilizada a liga de alumínio 1050 que é classificada como alumínio comercialmente puro.30 A composição química nominal, em % massa, é da ordem de 99,50 Al; 0,03 Cu; e 0,01 Fe. Preparação metalográfica das amotras Antes dos ensaios eletroquímicos, a superficie do alumínio para exposição ao meio corrosivo foi lixada sequencialmente usando lixas de carbeto de sílicio (SiC) #P2000, #P2500 e #P4000. Em seguida, a superfície foi polida sequencialmente com suspensão de diamante de 3 e 1 µm. A superfície exposta ao ensaio foi de 0,25 cm2, sendo esta delimitada por cera de abelha, com intuito de evitar a corrosão por fresta. Ensaios eletroquímicos A resistência à corrosão do alumínio foi avaliada por curvas de polarização em uma célula fechada com entrada e saída para a desaeração do meio com gás nitrogênio, utilizando o potenciostato SP-200 Biologic®. Todas as medições foram conduzidas em 250 mL de solução de NaCl 0,1 mol L-1 a uma temperatura de aproximadamente 25 ºC, utilizando uma célula eletroquímica com configuração de três eletrodos. Nesta configuração, a amostra de alumínio desempenhou o papel de eletrodo de trabalho, enquanto o eletrodo de referência (REF) foi o Ag/AgCl/KCl 3 mol L-1. Um fio de platina, com área aproximada de 3 cm2, foi empregado como contra eletrodo. Diferentes procedimentos foram utilizados para investigar o comportamento eletroquímico do alumínio por meio de curvas de polarização:
O potencial de circuito aberto foi medido por aproximadamente 3 min antes da varredura das curvas de polarização em todos os testes. As condições experimentais descritas acima (a-c) foram realizadas em condição aerada. Ensaios desaerados foram realizados apenas para as condições descritas em (a, b). O processo de desaeração foi feito com o borbulhamento de gás nitrogênio na solução de NaCl, em diferentes vazões 0,1; 0,7; 0,9; 1,3; 2,5 mL s-1 durante 24 h antes do ensaio para investigação da influência da concentração de oxigênio dissolvido sobre o PCA e nas curvas de polarização anódica. A vazão de 0,9 mL s-1 foi utilizada para comparação da influência da desaeração no comportamento das curvas anódicas e catódicas descritas em (a) e (b). Para garantir a reprodutibilidade, foram realizadas três repetições para todas as condições (a-c) dos ensaios eletroquímicos descritos anteriormente com vazão de nitrogênio de 0,9 mL s-1, conforme apresentado no Material Suplementar. A vazão de gás N2 foi estimada com base no tamanho e no número de bolhas por tempo observadas na saída do gás durante o processo de borbulhamento em um béquer contendo água. Cabe ressaltar que durante o processo de desaeração da solução por 24 h, a amostra de alumínio foi mantida dentro de célula eletroquímica acima da solução. Apenas após as 24 h de desaeração, a amostra de alumínio foi inserida na solução e o ensaio eletroquímico foi iniciado. A vazão de N2 foi reduzida ao mínimo durante o ensaio eletroquímico para evitar interferências devido a agitação intensa da solução. Observação da corrosão durante e após os ensaios eletroquímicos Os ensaios de polarização descritos foram filmados com o auxílio de um microscópio portátil com resolução máxima de 50×. Apenas os ensaios na condição aerada foram gravados (vídeos 1-3 do Material Suplementar). A superfície da liga após os ensaios eletroquímicos descritos em (a), (b) e (c) foi observada por microscopia óptica usando um equipamento Leica Modelo DMLM e, por microscopia eletrônica de varredura, após remoção dos produtos de corrosão. Para remoção dos produtos de corrosão, as amostras foram imersas em solução de HNO3 30% v/v durante 3 min em um banho ultrassônico a temperatura de ~25 ºC.31 As micrografias obtidas por microscopia eletrônica de varredura (MEV) foram feitas utilizando o microscópio Hitachi Tabletop Microscope TM3000. As imagens foram coletadas usando o detector de elétrons retroespalhados (ERP) e uma tensão de aceleração de 20 kV. A profundidade dos locais de corrosão localizada no alumínio, após polarização anódica, seja na condição aerada ou desaerada, foi calculada utilizando a norma ASTM G46-21,32 na qual a diferença de profundidade é determinada pelo foco na superfície e no interior dos pites, sendo estimada por microscopia óptica. Para cada condição de ensaio, foram analisados 25 pites. Também foi realizado corte de precisão com disco de diamante na seção transversal dos locais associados à corrosão por pites, seguido por desbaste até lixa P#4000, e polimento com pasta de diamante de 1 μm, para em seguida a amostra ser analisada por microscopia eletrônica de varredura. A profundidade da corrosão (perda de espessura) foi calculada utilizando: (Equação 6) os valores de densidade de carga total consumida durante a polarização anódica e (Equação 7) os valores de icorr obtidos por extrapolação anódica das retas de Tafel. Esses cálculos foram aplicados para simular uma comparação dos resultados com casos de corrosão uniforme e corrosão controlada por ativação, respectivamente, considerando ambas as condições, aerada e desaerada. A densidade de carga total, q (C cm-2), para as curvas de polarização anódica, foram estimadas diretamente pelo software EC-Lab®33 calculando a área sob a curva de i (A cm-2) versus tempo (s). Valores de perda de espessura foram calculados utilizando a equação de Faraday (Equação 6 ou 7), onde qa na Equação 6 é a densidade de carga total consumida durante polarização anódica e icorr-a na Equação 7 é a densidade de corrente de corrosão obtida por extrapolação anódica de retas de Tafel até o PCA e ta é o tempo médio de duração da varredura de polarização anódica. As outras variáveis têm significado usual, sendo M a massa molar do alumínio (26,98 g mol-1), Z o número de elétrons envolvidos na reação de dissolução do alumínio (3 e-), F a constante de Faraday (96.485 C mol-1), e ρ a densidade do alumínio (2,7 g cm-3). A análise foi realizada em triplicata para cada curva de polarização obtida e apresentadas no Material Suplementar. 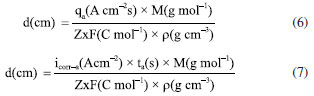 Cabe ressaltar que a avaliação por extrapolação anódica de retas de Tafel não é válida para metais em meios onde são passivos, como será discutido nos resultados. A Figura 5 apresenta um resumo da metodologia adotada nesse estudo.
RESULTADOS E DISCUSSÃO A Figura 6 apresenta as curvas de polarização anódicas e catódicas experimentais para o alumínio obtidas nesse estudo em meios aerado e semi-desaerado.
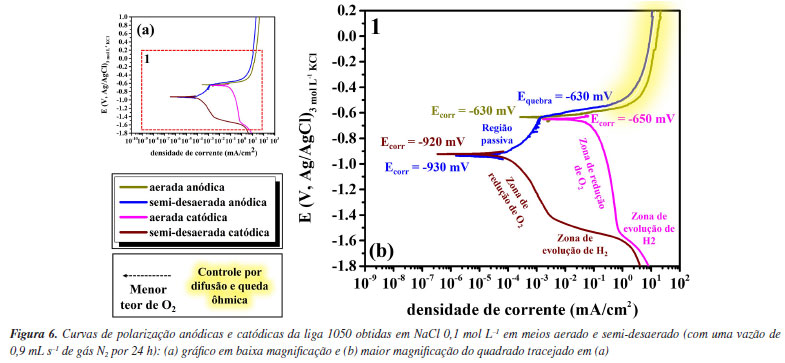
Comparando a curva catódica em meio aerado (rosa) e em meio semi-desaerado (vinho), observa-se que o potencial de corrosão da curva catódica aerada é aproximadamente 270 mV maior que o potencial de corrosão da curva obtida em meio semi-desaerado. Além disso, observa-se diminuição da densidade de corrente limite da reação de redução de O2 para a condição do meio semi-desaerado. Entretanto, ainda se observa uma região associada à reação de redução de O2 no meio semi-desaerado. Esses resultados mostram que o meio não foi completamente desaerado, mas sim semi-desaerado com uma diminuição da densidade de corrente limite associada ao controle por difusão de O2 de aproximadamente 1 mA cm-2 para 1 μA cm-2. Na curva catódica em meio aerado observa-se que a região dominada pela reação de evolução de H2 inicia em potenciais (em torno de -1550 mV vs. Ag/AgCl/ KCl 3 mol L-1) inferiores aos obtidos para as curvas catódicas em meio semi-desaerado (cerca de -1400 mV vs. Ag/AgCl/ KCl 3 mol L-1). Em ambos os casos, o potencial reversível para a reação de evolução de hidrogênio é igual, logo a reação inicia no mesmo potencial. Contudo, como a curva experimental catódica é a soma das duas reações catódicas, redução do oxigênio e evolução de hidrogênio, a região associada à evolução de hidrogênio só é observada quando a densidade de corrente dessa reação é maior que a densidade de corrente limite para a reação de redução do oxigênio. Além disso, é possível observar uma região bem definida com controle por ativação para a reação de evolução de hidrogênio no meio semi-desaerado com um coeficiente de Tafel de aproximadamente -65 mV dec-1. Essa região não é observada no meio aerado porque para ambas as curvas catódicas experimentais, ocorre desvio de linearidade para a reação de evolução de hidrogênio em densidades de corrente maiores que aproximadamente 1 mA cm-2, decorrente de efeitos de difusão e queda ôhmica devido ao acúmulo de bolhas de H2 na superfície. No vídeo 1, apresentado no Material Suplementar, é possível observar a intensa evolução de bolhas de H2 em correntes elevadas. Nas curvas experimentais anódicas também são observados diferentes comportamentos para a curva em meio aerado (marrom) e em meio semi-desaerado (azul). Assim como nas curvas catódicas, houve diminuição do Ecorr para a condição em meio semi-desaerado, devido à cinética das reações conforme previsto pela teoria do potencial misto e detalhada na Introdução (Figura 3). É importante destacar que para a condição semi-desaerada observa-se uma região passiva clara. Além disso, observa-se a iniciação e propagação da corrosão por pites, ou seja, a ruptura do filme passivo, que ocorre em aproximadamente -630 mV (Ag/AgCl/KCl 3 mol L-1). Cabe ressaltar que o potencial de ruptura do filme passivo no meio semi-desaerado é aproximadamente igual ao Ecorr para a liga na condição aerada, ou seja, a corrosão localizada já se inicia no PCA ou potencial de corrosão na condição aerada. No entanto, na condição aerada, esse fato só pode ser confirmado após análises metalográficas após ensaios, visto que não se observa a região passiva na curva anódica, e um comportamento eletroquímico semelhante ao observado poderia ser devido à corrosão uniforme. Para altas densidades de corrente anódica, maiores que aproximadamente 10 mA cm-2, observa-se uma corrente limite para a reação anódica em ambas as condições, aerada e semi-desaerada. Isso ocorre porque em altas correntes ocorre limitação da movimentação das espécies Al3+ e efeitos de queda ôhmica.34-37 Vale ressaltar que na região passiva da curva anódica, em meio semi-desaerado, é possível observar a formação de pites metaestáveis abaixo do potencial de ruptura do filme passivo. Estes são caracterizados por pequenos transientes (aumento e diminuição) da corrente anódica. Pites metaestáveis são pequenos pites, de alguns micrômetros, que se iniciam, mas logo repassivam durante o aumento do potencial no sentido positivo na curva de polarização anódica.13 No potencial de ruptura do filme passivo, ou no potencial de pite (Epite), os pites crescem, se estabilizam e, como consequência, se propagam sendo observado um aumento abrupto, de algumas ordens de grandeza, da densidade de corrente anódica. É importante destacar também que durante o desenvolvimento da corrosão por pites em ligas de alumínio é observada intensa evolução de bolhas de hidrogênio, ou seja, uma reação catódica ocorre dentro de um sítio anódico. Algumas teorias e explicações sobre o fenômeno, conhecido como efeito da diferença negativa ou evolução de hidrogênio anômalo, podem ser encontrados na literatura.21,38-42 No vídeo 2, apresentado no Material Suplementar, é possível observar a evolução de pequenas bolhas na superfície do alumínio durante o ensaio de polarização anódica. Essas bolhas são observadas em potenciais bem menores que o previsto para a reação anódica de evolução de oxigênio (que é aproximadamente +729 mV vs. Ag/AgCl/KCl 3,5 mol L-1 em pH 5) e, além disso, levam a um aumento do pH da solução portanto estão associadas a reação catódica de evolução de hidrogênio. A Figura 4S, disponível no Material Suplementar, confirma o aumento do pH da solução após o ensaio de polarização (pH inicial da solução na faixa de 5 e pH após ensaio de polarização em torno de 7). Além disso, observa-se a precipitação de produtos de corrosão em solução devido ao aumento de pH resultando em uma coloração turva. Após os ensaios de polarização anódica e catódica, a superfície do alumínio, em ambas as condições aerada e semi-desaerada, foi analisada por microscopia óptica, conforme apresentado na Figura 7.
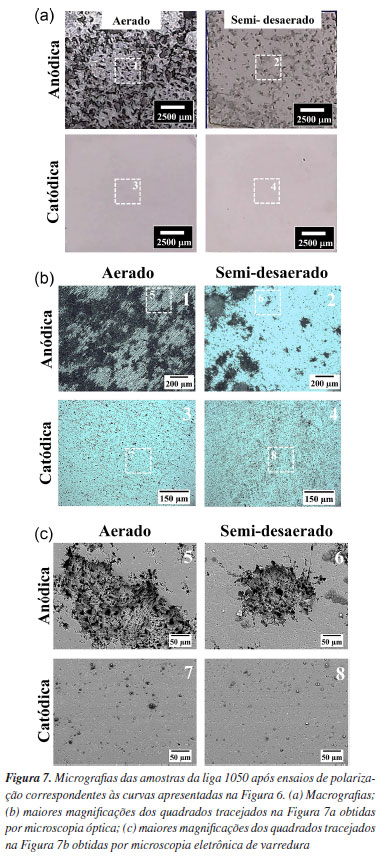
Observa-se corrosão localizada nas macrografias (Figura 7a) após polarizações anódicas, enquanto que após polarizações catódicas nenhum tipo de ataque é observado nas macrografias. Também é observado ataque mais intenso na liga polarizada anodicamente em solução aerada. Isso se deve ao fato de a liga polarizada anodicamente em meio semi-desaerado permanecer algum tempo na região passiva (Figura 6), antes da ruptura do filme com a iniciação da corrosão por pites. Dessa forma, a liga passa por espessamento do filme passivo anódico, o que dificulta a iniciação de pites. Além disso, durante o ensaio, a liga polarizada anodicamente em meio semi-desaerado recebe menor carga total, conforme será discutido a seguir. Nas micrografias por MO e MEV apresentadas nas Figuras 7b e 7c, com maiores magnificações, é possível observar na superfície das amostras polarizadas catodicamente, em ambas as condições, aerada e semi-desaerada, ataque uniforme e ataque associado às intermetálicos micrométricas presentes no alumínio e suas ligas. O ataque uniforme do alumínio durante polarização catódica se deve ao aumento local do pH na superfície (devido às reações de redução do O2 e evolução de H2) e, por ser um metal anfótero, ocorre dissolução do filme de óxido em pHs elevados. Além disso, as partículas micrométricas no alumínio tendem a ter em sua composição Al, Cu, Fe, Mn ou Mg, dependendo da composição química e das impurezas na liga, e correspondendo a sítios anódicos ou catódicos em relação à matriz de alumínio.19 Durante a polarização catódica, o ataque da matriz ao redor das partículas causa a remoção destas, ou a precipitação de óxidos/hidróxidos sobre elas, revelando assim pequenos buracos (da ordem de poucos mícrons) e pontos pretos, conforme observado na Figura 7c.20 Já as superfícies polarizadas anodicamente, apresentaram ataque localizado em ambas as condições dos meios, aerado e semi-desaerado (Figuras 7b e 7c). Esse tipo de ataque, como já mencionado, está relacionado à corrosão por pites que tende a se iniciar nos defeitos/descontinuidades do filme passivo e se propagar localmente.43 Na imagem de microscopia óptica para a liga polarizada anodicamente na condição aerada (Figura 7b), a morfologia característica de pites não pôde ser observada devido ao ataque excessivo da matriz decorrente da alta carga aplicada durante o ensaio anódico. No entanto, nas imagens de MEV (Figura 7c) com maiores magnificações, a morfologia de corrosão por pite é observada com maior resolução, o que foi confirmado nas imagens da secção transversal que serão apresentadas em seguida. Vale ressaltar que os pites formados em ambas as condições, aerada e semi-desaerada, apresentam morfologia de ataque cristalográfico em oposição à clássica morfologia semiesférica com superfície polida. Pites cristalográficos também são encontrados em alguns outros materiais e meios e a literatura44 apresenta várias explicações sobre o mecanismo de propagação desse tipo de morfologia de pite. Em maiores magnificações, a morfologia deste ataque fica evidente, como mostrado na Figura 8.

As Figuras 8a e 8b mostram os pites associados às amostras submetidas aos ensaios de polarização em meio aerado e semi-desaerado, respectivamente. Em maiores magnificações nas regiões associadas a esses ataques (Figuras 8c e 8d) verifica-se a morfologia do ataque associado à corrosão cristalográfica. É amplamente aceito que o início da corrosão cristalográfica no Al puro é consequência da ruptura do filme passivo na superfície.45 Os pites cristalográficos em Al puro são delimitados por planos cristalográficos {001} e são caracterizados por uma forma de meio cubo bem definida com arestas vivas,46,47 conforme indicado pelas retas azuis tracejadas nas Figuras 8c e 8d.48 As Figuras 9a e 9b mostram as superfícies das amostras submetidas aos ensaios em meios aerado e semi-desaerado, respectivamente. Em maiores ampliações, a microscopia ótica revela (Figuras 9c e 9d) a morfologia da corrosão por pites. Imagens dos pites em 3D, obtidas por microscopia confocal (Figuras 9e e 9f), indicam uma maior profundidade de corrosão para os pites nas amostras submetidas ao ensaio na condição semi-desaerada. Isso foi confirmado pelo corte da seção transversal dos pites destacados nas Figuras 9g e 9h, conforme indicado pelas linhas vermelhas tracejadas. As imagens da seção transversal desses pites estão apresentadas nas Figuras 9i e 9j que mostraram uma diferença na profundidade do ataque. O pite formado na condição aerada (30 μm) apresentou uma profundidade aproximadamente duas vezes menor que o pite na condição semi-desaerada (70 μm).
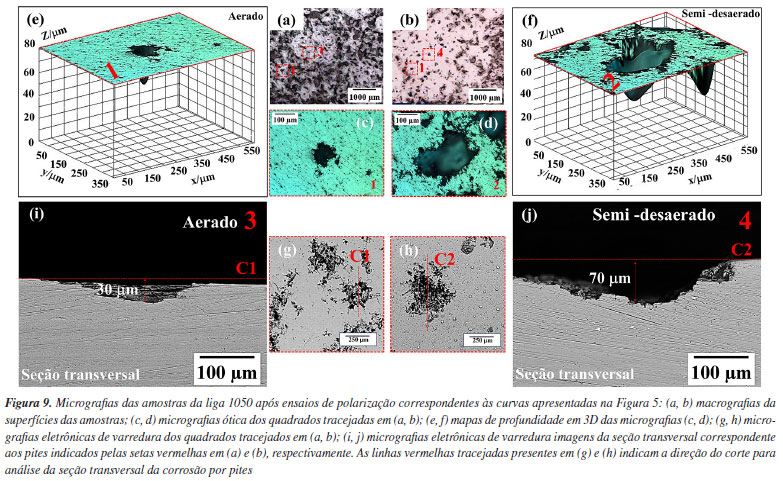
A profundidade de penetração da corrosão, para cada condição de ensaio, foi também calculada por meio de técnicas eletroquímicas e medida por microscopia ótica (método de ajuste de foco segundo descrito na norma ASTM G46-21),32 conforme apresentado na Tabela 1.

Observa-se na Tabela 1 que a perda de espessura média devido ao ataque corrosivo estimada pela equação de Faraday utilizando a densidade de carga total consumida durante o ensaio de polarização anódica, para as condições aerada e semi-desaerada, foi cerca de 7,3 ± 0,3 e 5,3 ± 0,2 μm, respectivamente, enquanto, que a avaliação realizada por microscopia óptica aponta valores de profundidade de pite da ordem de aproximadamente 34 ± 6 e 63 ± 7 μm. A diferença é cerca de 5× e 13× maior nos valores de profundidade de pite quando comparados aos valores de perda de espessura estimados pela densidade de carga anódica total. Isso demonstra que uma vez identificada a corrosão localizada (por curvas de polarização anódica, curva de polarização cíclica ou microscopia) não se deve utilizar a equação de Faraday para obter a perda de massa visto que a corrente anódica medida não corresponde à área total ensaiada, mas apenas às regiões associadas à corrosão por pite (corrosão localizada). Logo, a análise por imagens de microscopia óptica ou eletrônica se faz necessária nessas condições, principalmente imagens de seção transversal que proporcionam maior precisão e detalhes sobre a morfologia dos pites, conforme realizado neste trabalho. Cabe ressaltar que os valores de profundidade média dos pites calculados segundo a norma ASTM G46-2132 são aproximadamente iguais aos valores medidos por seção transversal (após remoção dos produtos de corrosão), conforme apresentado nas Figuras 9i e 9j, dado que os pites observados para a liga 1050 são relativamente hemisféricos. Além disso, pelas teorias da cinética de corrosão, as extrapolações das retas de Tafel só podem ser utilizadas para corrosão uniforme e reações sob controle por ativação. Este não é o caso do alumínio que se apresenta passivo em meios neutros e cuja reação catódica de redução de oxigênio é controlada por difusão no Ecorr. A Tabela 1 mostra uma comparação da profundidade de corrosão calculada por extrapolação anódica de retas de Tafel (icorr) e assumindo o tempo médio de varredura de uma curva de polarização anódica (aproximadamente 1500 segundos neste trabalho). Os valores são da ordem de sub-nanômetros, muito aquém dos valores reais de profundidade medidos. Cabe ressaltar que os valores calculados por icorr assumem estado estacionário no Ecorr e não tem uma comparação direta com valores de perda de espessura após ensaios de polarização anódica. Fazendo uma análise estatística da densidade de carga (C cm-2) consumida nas curvas de polarização anódica em meios aerado e semi-desaerado, os valores obtidos foram 21 ± 1 e 15 ± 0,8 C cm-2, respectivamente. Embora a densidade de carga anódica seja menor para a amostra da liga polarizada na condição semi-desaerada, a amostra apresentou pites com maior profundidade e menor densidade de ataque. Isto é, a corrosão ocorreu de forma mais localizada em condição semi-desaerada quando comparada com a condição aerada. A condição aerada resultou em corrosão localizada rasa e distribuída (Figura 7). Como discutido anteriormente, na condição semi-desaerada, a amostra permanece polarizada na região passiva por algum tempo (Figura 6) onde ocorre o espessamento do filme anódico, reduzindo a quantidade de locais para iniciação de pites. No entanto, após iniciados, a propagação desses pites é mais acentuada resultando em pites mais profundos devido à maior razão entre a carga e o número de pites na condição semi-desaerada. Há vários trabalhos na literatura49-51 que discutem o efeito da camada passiva na propagação de pites. Para algumas ligas expostas a meios contendo íons cloreto, pites crescem inicialmente sob uma "tampa" de filme passivo, ou pit cover, o que resulta em propagação acentuada em um meio bastante ocluso enriquecido em espécies agressivas.28,52 O efeito da vazão de gás nitrogênio (N2) durante a etapa de desaeração da solução foi avaliado para obtenção das curvas anódicas e a Figura 10 apresenta esses resultados.
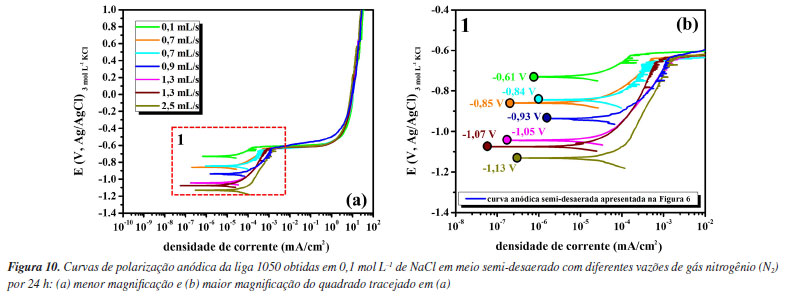
Observa-se que com a variação da vazão de gás N2 houve diminuição significativa no potencial de corrosão devido à diminuição da concentração de O2 no meio. Esses resultados corroboram a discussão anterior referente à curva catódica (Figura 8) considerando que o meio não foi completamente desaerado, mesmo utilizando uma vazão de N2 de 0,9 mL s-1 por 24 h. A completa desaeração do meio (O2 < 10 ppb) é difícil e, em geral, uma célula com boa vedação e pressão positiva é necessária.53,54 Importante ressaltar que o potencial de ruptura do filme passivo, no entanto, permaneceu aproximadamente o mesmo para todas as concentrações de O2 no meio. Os resultados apresentados neste trabalho indicam que a desaeração do meio pode auxiliar na avaliação da susceptibilidade a corrosão localizada do alumínio e suas ligas, especialmente em condições não anodizadas onde o filme passivo formado ao ar é menos espesso e o potencial de ruptura do filme passivo é muito próximo do potencial de corrosão.55-57 A caracterização do potencial de ruptura do filme em meios aerado e desaerado combinado com o monitoramento do potencial de corrosão no meio aerado fornece informações mais completas da susceptibilidade a corrosão localizada do alumínio. Isso é confirmado pelos ensaios de imersão, conforme apresentado na Figura 11.

Ensaios de imersão são comumente utilizados para monitoramento da corrosão em diversas aplicações. Ensaios de polarização, no entanto, fornecem uma avaliação mais rápida e podem ser utilizados para investigar previamente o tipo de corrosão (uniforme ou localizada) e mecanismos de propagação. Após 24 h de imersão (Figuras 11a e 11c), já se observa o início do ataque cristalográfico. Com o aumento do tempo de imersão para 168 h (Figuras 11b e 11d), a nucleação da corrosão por pite com ataque cristalográfico se torna evidente. Assim como observado na Figura 8, durante os testes de imersão, nota-se o ataque cristalográfico do alumínio, caracterizado principalmente pela formação de arestas ao redor da cavidade, como indicado pelas linhas tracejadas azuis (Figuras 11c e 11d). Estes resultados demonstram que os testes de polarização, quando conduzidos e analisados criteriosamente, representam uma ferramenta importante para avaliar o comportamento do alumínio e comparar o desempenho de diferentes ligas (ou tratamentos de superfície) em meios aquosos salinos, uma vez que apresentam uma correlação significativa com os testes de imersão. É importante destacar que é comum encontrar na literatura resultados de curvas de polarização em que as curvas catódicas e anódicas são obtidas em uma mesma varredura de potenciais, a partir de potenciais catódicos, algumas centenas de mV abaixo do potencial de corrosão, e a varredura na direção anódica é realizada até algumas centenas de mV acima do potencial de corrosão. Porém, para alguns metais, como o alumínio e suas ligas, essa metodologia pode resultar em artefatos e avaliações incorretas, visto que o alumínio é um metal anfótero e, como já mencionado, corrói tanto durante a varredura anódica quanto durante a varredura catódica. Além disso, tanto a varredura anódica como a varredura catódica alteram a química e o pH do meio próximo à superfície do metal, o que pode resultar em um meio que não é representativo da condição de uso. Em geral, respostas representativas são obtidas iniciando-se a varredura das curvas de polarização próximo ao potencial de corrosão, ou seja, algumas poucas dezenas de mV acima ou abaixo. Em geral, as reações químicas e o mecanismo de corrosão do metal em um meio devem ser avaliados previamente para a escolha apropriada dos parâmetros da varredura. Por exemplo, iniciar a curva de polarização a partir de potenciais catódicos em direção ao PCA, em meio aerado, pode levar a interpretações errôneas, conforme apresentado na Figura 12.
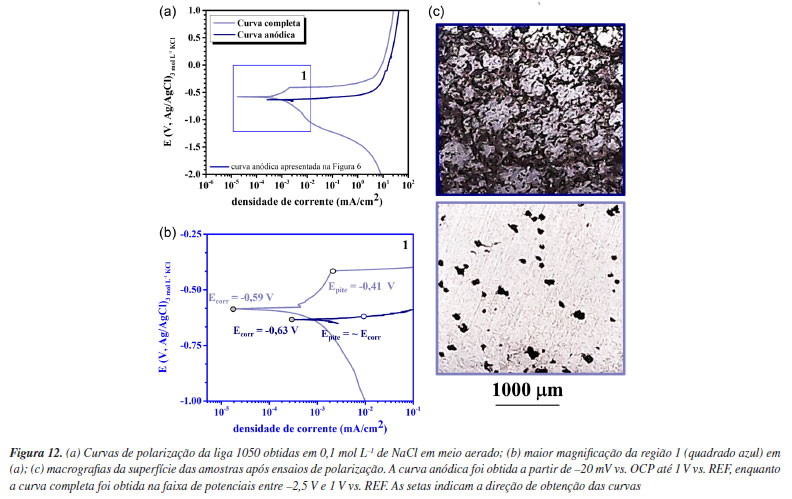
Há diferenças significativas entre as curvas de polarização anódicas obtidas a partir do potencial de corrosão e aquelas obtidas desde centenas de mV abaixo do potencial de corrosão. O potencial de corrosão para a curva com varredura a partir de potenciais catódicos (curva completa) foi de -590 mV, enquanto a obtida a partir do potencial de corrosão, foi de -630 mV. Além disso, houve um aumento no potencial de quebra do filme passivo para a curva com varredura a partir de potenciais catódicos (curva completa). É observado um aumento da "região passiva" para a curva cuja varredura teve início na região catódica. É notado também uma diferença significativa no ataque corrosivo, conforme imagens de microscopia óptica (Figura 12c). As diferenças entre as curvas e a morfologia do ataque devem-se à alcalinização excessiva ocorrida durante a varredura catódica devido às reações de evolução de H2 e redução de O2 causando a precipitação de óxido/hidróxido não protetores na superfície do material. Além disso, observa-se que a curva varrida a partir de potenciais catódicos em direção ao PCA em meio aerado é similar à curva catódica obtida em meio semi-desaerado varrida a partir do PCA em direção a potenciais catódicos (Figura 6). Isso ocorre devido à intensa evolução de hidrogênio na superfície da liga, quando se inicia a varredura de potenciais a partir da região catódica, o que resulta em desaeração local do meio. É importante ressaltar que somente após um tempo de polarização é que se alcança a região de redução de O2. No vídeo 3, disponível no Material Suplementar, pode-se observar as mudanças que ocorrem na superfície do alumínio durante a obtenção da curva de polarização a partir de potenciais catódicos.
CONCLUSÕES As curvas de polarização anódica do alumínio 1050 em meio neutro com cloreto, sob condições aeradas, devem ser utilizadas apenas para avaliações qualitativas. Para uma interpretação crítica dos resultados, é essencial realizar a caracterização morfológica do ataque após os ensaios de polarização anódica. Em meios aerados, não foi observada uma região passiva ou um potencial de ruptura do filme nas curvas de polarização, no entanto, a presença de ataque localizado foi confirmada por microscopia óptica após os ensaios. Em contraste, em meios com baixo teor de oxigênio, uma região passiva e um potencial de ruptura do filme foram identificados nas curvas de polarização anódicas. Verificou-se que o ataque localizado foi mais profundo em superfícies testadas em meio com baixo teor de oxigênio em comparação aos ensaios em meios aerados. Além disso, a liga de alumínio 1050, tanto nos ensaios de imersão no PCA quanto após os ensaios de polarização anódica, apresentou corrosão com morfologia de ataque localizado e cristalográfico. Observou-se também que o potencial de corrosão (Ecorr) tende a diminuir enquanto a faixa passiva aumenta com o incremento da vazão de gás nitrogênio (N2) durante o processo de desaeração da solução. Por fim, as curvas de polarização iniciadas a partir de potenciais catódicos em direção ao PCA, seguidas de varredura na direção anódica, podem induzir a interpretações equivocadas sobre a resposta eletroquímica do alumínio.
MATERIAL SUPLEMENTAR Imagens complementares às medidas eletroquímicas deste trabalho estão disponíveis em http://quimicanova.sbq.org.br, em formato de arquivo PDF, com acesso livre. Além disso, vídeos reproduzidos com velocidade de 50× mostrando a superfície do alumínio durante os ensaios de polarização em meio aerado também estão disponíveis.
AGRADECIMENTOS Os autores expressam sua gratidão à Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) pela concessão de bolsa a João Victor de Sousa Araujo, por meio dos processos 2019/18388-1 e 2022/06935-0.
REFERÊNCIAS 1. Vargel, C. Em Corrosion of Aluminium, 1st ed.; Varge, C., ed.; Elsevier: London, 2004, p. 113. [Crossref] 2. Donatus, U.; Bodunrin, M.; Olayinka, A.; Milagre, M.; Oloyede, O.; Aribo, S.; Araujo, J.; Carvalho, C.; Costa, I. Em Advanced Aluminium Composites and Alloys; Dobrzański, L. A., ed.; IntechOpen: London, 2021, ch. 5. [Crossref] 3. McCafferty, E.; Introduction to Corrosion Science, 1st ed.; Springer: New York, 2010. [Crossref] 4. Pedeferri, P.; Corrosion Science and Engineering, 1st ed.; Springer: Switzerland, 2018. [Crossref] 5. Costa, I. Em Química Tecnológica, 1a ed.; Hilsdorf, J.; de Barros, N. D.; Tassinari, C. A.; Costa, I., eds.; Cengage Learning: Brasil, 2004, cap. 5. 6. Lefrou, C.; Nogueira, R.; Huet, F.; Takenouti, H. Em Shreir's Corrosion, vol. 1, 1st ed.; Cottis, B.; Graham, M.; Lindsay, R.; Lyon, S.; Richardson, T.; Scantlebury, D.; Stott, H., eds.; Elsevier: New York, 2010, p. 13-51. [Crossref] 7. Pourbaix, M.; Atlas of Electrochemical Equilibria in Aqueous Solutions; National Association of Corrosion Engineers: Houston, USA, 1966. [Link] acessado em outubro 2024. 8. Panossian, Z.; dos Santos, C. A. L.; Cardoso, J. L.; da Silva, A. L. N.; Camargo, R. A.; Anais do 34º Congresso Brasileiro de Corrosão; Fortaleza, Brasil, 2014. [Link] acessado em outubro 2024. 9. Callister, D.; William, D.; Rethwisch, W.; Materials Science and Engineering - An Introduction, 10th ed.; Wiley: USA, 2012. 10. Ul-Hamid, A.; A Beginners' Guide to Scanning Electron Microscopy, 1st ed.; Springer: Cham, 2018. [Crossref] 11. Evans, U.; An Introduction to Metallic Corrosion, 1st ed.; Edward Arnold: London, 1948. 12. Evans, U.; The Corrosion of Metals, 1st ed.; Edward Arnold: London, 1926. 13. Frankel, G.; J. Electrochem. Soc. 1998, 145, 2186. [Crossref] 14. Lalgudi, V.; Ramanathan; Corrosão e Seu Controle, 3a ed.; Hemus: Brasil, 1997. 15. Moreto, J.; Gamboni, O.; Rocha, L.; Corrosão e Protecção de Materiais, 2012, 31, 60. 16. Moreto, J. A.; Marino, C. E. B.; Bose Filho, W. W.; Rocha, L. A.; Fernandes, J. C. S.; Corros. Sci. 2014, 84, 30. [Crossref] 17. Moreto, J. A.; dos Santos, M. S.; Ferreira, M. O. A.; Carvalho, G. S.; Gelamo, R. V.; Aoki, I. V.; Taryba, M.; Bose Filho, W. W.; Fernandes, J. C. S.; Corros. Sci. 2021, 182, 109253. [Crossref] 18. Moreto, J. A.; Broday, E. E.; Rossino, L. S.; Fernandes, J. C. S.; Bose Filho, W. W.; J. Mater. Eng. Perform. 2018, 27, 1917. [Crossref] 19. Araujo, J. V. S.; Bugarin, A. F. S.; Donatus, U.; Machado, C. S. C.; Queiroz, F. M.; Terada, M.; Astarita, A.; Costa, I.; Corros. Eng., Sci. Technol. 2019, 54, 575. [Crossref] 20. Araujo, J. V. S.; da Silva, R. M. P.; Viveiros, B. V.; Milagre, M. X.; Machado, C. S. C.; Costa, I.; Quim. Nova 2022, 45, 680. [Crossref] 21. Araujo, J. V. S.; da Silva, R. M. P.; Milagre, M. X.; Machado, C. S. C.; Costa, I.; Quim Nova 2020, 43, 1058. [Crossref] 22. da Silva, R. M. P.; Milagre, M. X.; Araujo, J. V. S.; Ramirez, O. M. P.; Machado, C. S. C.; Antunes, R. A.; Costa, I.; Quim Nova 2023, 46, 307. [Crossref] 23. Isaacs, H. S.; Adzic, G.; Jeffcoate, C. S.; Corrosion 2000, 56, 971. [Crossref] 24. Moreto, J. A.; Rossino, L. S.; Bose Filho, W. W.; Marino, C. E. B.; Ferreira, M. C.; Taryba, M.; Fernandes, J. C. S.; Mater. Res. 2018, 22, e20180280. [Crossref] 25. Ferreira, M. O. A.; Gelamo, R. V.; Marino, C. E. B.; da Silva, B. P.; Aoki, I. V.; da Luz, M. S.; Alexopoulos, N. D.; Leite, N. B.; Moreto, J. A.; Materialia 2022, 22, 101407. [Crossref] 26. Freitas, L. R.; Gelamo, R. V.; Marino, C. E. B.; Nascimento, J. P. L.; Figueiredo, J. M. A.; Fernandes, J. C. S.; Moreto, J. A.; Surf. Coat. Technol. 2022, 434, 128197. [Crossref] 27. Ferreira, M. O. A.; Nascimento, J. P. L.; Leite, N. B.; de Siervo, A.; Fernandes, G. L.; Vaz, A. R.; Gelamo, R. V.; Aoki, I. V.; Moreto, J. A.; Mater. Res. 2023, 26, e20220453. [Crossref] 28. Araujo, J. V. S.; Milagre, M. X.; Zhou, X.; Costa, I.; Mater. Corros. 2024, 75, 599. [Crossref] 29. Schneider, M.; Fürbeth, W.; Mater. Corros. 2022, 73, 1752. [Crossref] 30. ASM International Committee; ASM Handbook, vol. 4; ASM International: USA, 1991. [Link] acessado em novembro 2024 31. Ma, Y.; Zhou, X.; Huang, W.; Thompson, G. E.; Zhang, X.; Luo, C.; Sun, Z.; Mater. Chem. Phys. 2015, 161, 201. [Crossref] 32. ASTM G46-21: Standard Guide for Examination and Evaluation of Pitting Corrosion; ASTM International: West Conshohocken, 2021. [Link] acessado em outubro 2024 33. EC-Lab®, version 4; Biologic Inc., Seyssinet-Pariset, France, 2023. 34. Buchanan, R.; Stansbury, E. Em Handbook of Environmental Degradation of Materials, 2nd ed.; Elsevier: London, 2012, p. 87. [Crossref] 35. Muñoz, A. g.; Bessone, J. B.; Corros. Sci. 1999, 41, 1447. [Crossref] 36. Szklarska-Smialowska, Z.; Corros. Sci. 1999, 41, 1743. [Crossref] 37. Amin, M. A.; El Rehim, S. S. A.; El-Lithy, A. S.; Corros. Sci. 2010, 52, 3099. [Crossref] 38. McCafferty, E.; Corros. Sci. 1995, 37, 481. [Crossref] 39. Baumgärtner, M.; Kaesche, H.; Corros. Sci. 1990, 31, 231. [Crossref] 40. Curioni, M.; Scenini, F.; Electrochim. Acta 2015, 180, 712. [Crossref] 41. Donatus, U.; Berbel, L. O.; Costa, I.; Mater. Corros. 2018, 69, 1375. [Crossref] 42. Frankel, G. S.; Fajardo, S.; Lynch, B. M.; Faraday Discuss. 2015, 180, 11. [Crossref] 43. Grilli, R.; Baker, M. A.; Castle, J. E.; Dunn, B.; Watts, J. F.; Corros. Sci. 2010, 52, 2855. [Crossref] 44. Sahu, S.; Swanson, O. J.; Li, T.; Gerard, A. Y.; Scully, J. R.; Frankel, G. S.; Electrochim. Acta 2020, 354, 136749. [Crossref] 45. Revie, R. W.; Uhlig, H. H.; Corrosion and Corrosion Control: An Introduction to Corrosion Science and Engineering, 1st ed.; John Wiley & Sons: EUA, 2008. [Crossref] 46. Seo, J. H.; Ryu, J.-H.; Lee, D. N.; J.Electrochem. Soc. 2023, 150, B433. [Crossref] 47. Huang, J.; Feng, S.; Li, S.; Wu, C.; Chen, J.; J. Alloys Compd. 2021, 861, 158588. [Crossref] 48. MicroArt; Metallogr., Microstruct., Anal. 2023, 12, 733. [Crossref] 49. Frankel, G. S.; Corros. Sci. 1990, 30, 1203. [Crossref] 50. Macdonald, D. D.; J. Electrochem. Soc. 1992, 139, 3434. [Crossref] 51. Macdonald, D. D.; Electrochim. Acta 2011, 56, 1761. [Crossref] 52. Ren, J.; Zuo, Y.; Surf. Coat. Technol. 2005, 191, 311. [Crossref] 53. Hikmat, N. A.; Farhan, A. M.; Majed, R. A.; Engineering and Technology Journal 2009, 27, 1532. [Crossref] 54. DiBari, G. A.: The Electrochemical and Corrosion Behavior of Aluminum; Technical Memorandum, University Park, Nottingham, United Kingdom, 1970. [Link] acessado em outubro 2024 55. Araujo, J. V.; Milagre, M. X.; Costa, I.; Crit. Rev. Solid State Mater. Sci. 2024, 49, 521. [Crossref] 56. Araujo, J. V. S.; da Silva, R. M. P.; Klumpp, R.; Costa, I.; Quim. Nova 2021, 44, 999. [Crossref] 57. Araujo, J.; Chen, J.; Zhou, X.; Costa, I.; Anais do 459 EFC's Annu. Conf. Brussels, 2023. [Link] acessado em outubro 2024.
Editor Associado responsável pelo artigo: Lucia Mascaro |

On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access