
 |
 |
 |
 |
 |
Artigo
| A new approach to evaluate immobilization of β-galactosidase on Eupergit® C: structural, kinetic, and thermal characterization |
|
Anna R. C. BragaI; Marceli F. SilvaII; José V. OliveiraIII; Helen TreichelIV; Susana J. KalilI,*
IEscola de Química e Alimentos, Universidade Federal do Rio Grande, 96203-900 Rio Grande - RS, Brasil Recebido em 27/08/2013 *e-mail: dqmsjk@furg.br This study aimed to evaluate β-galactosidase immobilization. For this purpose, the ionic strength of the buffer, reaction time, amount of the immobilization support, and pH were evaluated by a central composite design. Assay 8, which consisted of 1.5 mol L-1 phosphate buffer (pH 7.5) and a reaction time of 2 h, produced the maximum yield. Eupergitr C (400 mg) was subsequently used as an immobilization support. Immobilization kinetics wereinvestigated, and a significant increase in the yield was obtained after immobilization compared with that obtained from assay 8 (22.0 U mL-1 vs. 15.6 U mL-1). The enzyme efficiency of actuation was evaluated using o-nitrophenyl-β-D-galactopyranoside and lactose, with lactose providing better results. The reuse of β-galactosidase was evaluated, and more than 50% of the initial enzyme activity was maintained after five cycles of use. Enzyme characterization revealed that immobilization improved some aspects of the thermostability of β-galactosidase. INTRODUCTION Enzymes are subject to inactivation by chemical, physical, and biological factors during use or storage. Enzyme immobilization techniques have been used to enhance enzyme stability and facilitate recuperation and reutilization of the enzyme.1 β-Galactosidase (EC 3.2.1.23) is one of the most frequently used enzymes in the food industry because it hydrolyses lactose, releasing glucose and galactose. The reduction in lactose content has nutritional and technological advantages, and it increases the sweetness of dairy products.2 This enzyme can also catalyze transgalactosylation reactions to produce galactosides and oligosaccharides.3 Many immobilization methods, such as physical adsorption, entrapment, and covalent binding methods on different supports, can be used for enzymes. Supports used include a wide variety of materials, such as inorganic matrices (silica, porous glass, and celite), polysaccharides (agarose, alginate, chitosan, and cellulose), and synthetic polymers (acrylic resins).4-6 With regard to the immobilization process, the methods and supports that are currently used for β-galactosidase include immobilization on anion exchange resins, cellulose-gelatin carrier systems, DEAE agarose, glyoxyl/epoxy/BrCN groups, glutaraldehyde, polyelectrolyte surfaces, silicon surface, Sepabeads-epoxy supports partially modified with boronate, iminodiacetic acid, metal chelates, and ethylenediamine. These methodologies can improve the stability and reusage of the enzyme.7 Eupergit® C consists of macroporous beads with diameters of 100-250 µm that are produced by copolymerization of N,N'-methylene-bis-(methacrylamide), glycidyl methacrylate, allyl glycidyl ether, and methacrylamide. An advantage of this support is the net price (including support, activators, and operation time) of the immobilization process compared with other supports.8 Eupergit® C has been used by many researchers as a carrier for the immobilization of various enzymes, and it was reported that this carrier was extremely stable and exhibited good chemical and mechanical properties (e.g., simple immobilization procedure, high binding capacity, low water uptake, high flow rate in column procedures, and excellent performance in stirred bath reactors). Enzyme immobilization on this support is produced through a two-step mechanism. In the first step, rapid and mild physical adsorption of the enzyme on this support is achieved. In the second step, a covalent reaction occurs between the adsorbed enzyme and neighboring epoxide groups.9 The support epoxy groups can react with the amino, sulfhydryl, and hydroxyl groups of the enzyme, forming extremely stable O-C and N-C bonds.10 Immobilized enzymes can be recovered after a batch, guaranteeing their reutilization in further batches and significantly reducing their costs. The use of immobilized enzymes also allows the implementation of continuous production systems with expressive productivity gains. In addition, as enzymes are subject to inactivation because of physical, chemical, or biological factors during storage or use, immobilization confers stability upon the enzymes, mainly concerning pH and temperature, decreasing operating costs and hence opening alternative routes to new biotechnological applications.11 β-Galactosidase immobilization is an effective process for the successful hydrolysis of lactose in that it can overcome the problems associated with the costs of soluble enzymes.4 Enzymatic immobilization is a complex process that must be studied in consideration of the different aspects of the enzyme, support, and the interaction between them. Therefore, this work aimed to evaluate β-galactosidase immobilization in Eupergit® C using a differentiated perspective in addition to the conventional evaluation. The enzyme performance using different substrates and the reuse and characterization of the enzyme after immobilization were also assessed. In addition, the support and immobilized enzyme were characterized structurally by surface areas analysis [Brunauer, Emmett, and Teller (BET) method] and scanning electron microscopy (SEM) analysis.
EXPERIMENTAL Microorganism Kluyveromyces marxianus CCT 7082, deposited in the Tropical Culture Collection of the Andre Tosello Foundation (Brazil), was cultivated, and the enzyme obtained was used in the studies below. Inoculum The cultures were grown on a medium containing 10 g of lactose, 5 g of KH2PO4, 1.2 g of (NH4)2SO4, 0.4 g of MgSO4.7H2O, and 1 g of yeast extract in 1 L of 0.2 mol L-1 potassium phosphate buffer, pH 5.5.12 The medium was sterilized at 121 ºC for 15 min, and lactose was sterilized by filtration. Next, 150 mL of the inoculated medium was cultivated in conical flasks (500-mL capacity) for 14 h at 180 rpm and 30 ºC in an orbital shaker (Tecnal TE-420, Brazil).13 Submerged cultivation The enzyme was produced by submerged cultivation using the culture medium optimized by Manera et al.13 containing the following compounds (in g L-1): lactose (28.2), KH2PO4 (5.0), (NH4)2SO4 (8.8), MgSO4.7H2O (0.4), and yeast extract (17.0) in 0.2 mol L-1 potassium phosphate buffer, pH 6.0. The cultivations were started with 10% of the inoculum, and the cultures were incubated for 96 h at 180 rpm and 30 ºC in an orbital shaker (Tecnal TE-420). Enzyme extraction The enzymatic extract was distributed in 50-mL flasks containing 25 mL of a cell suspension (40 mg mL-1 in 50 mmol L-1 phosphate buffer plus 0.1 mmol L-1 MnCl2.4H2O, pH 6.6) and 27.5 g of glass beads (diameter ranging from 0.95 to 1.05) and mixed in a Vortex (Fenix AP 56, Brazil) for 20 min with the temperature maintained at 4 ºC.14 The suspension was then centrifuged at 5200 ×g for 10 min at 4 ºC (Cientec CT-5000R, Brazil),15 and the supernatant was assayed for β-galactosidase activity. Enzyme purification Ammonium sulfate was added to the solution containing β-galactosidase to 70% saturation, maintained overnight, and then centrifuged. The supernatant was discarded, and the β-galactosidase-rich precipitate was dissolved in 0.05 mol L-1 potassium phosphate buffer (pH 6.6) and dialyzed against the same buffer.16 Enzyme immobilization The immobilization essays were conducted in a thermostatic reactor containing Eupergit® C, buffer, and enzyme (10 mL) at a fixed temperature of 10 ºC and agitation of 150 rpm. To evaluate the effects of different factors in β-galactosidase immobilization, a central composite design (CCD) with four replicates at the central point, giving 20 trials, was used. The four variables were selected on the basis of the literature, and the responses were evaluated as the immobilization yield-based activity (%). The experiments were performed with variation in the following factors: amount of the support (200-400 mg), pH (6.5-7.5), ionic strength of the buffer (potassium phosphate) in use (0.5-1.5 mol L-1), and reaction time (2-8 h). After immobilization, the solution was filtered and washed using potassium phosphate buffer (10 mL, 0.1 mol L-1 (pH 6.6), 0.1 mmol L-1 Mg2+). The responses were calculated by the following equation (Eq. 1):
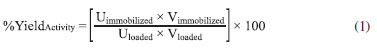
where Uimmobilized is the enzyme activity in the particle (wet basis), Vimmobilized is the particle amount, Uloaded denotes the free enzyme activity, and Vloaded is the enzyme initial volume. The general immobilization yield was calculated using Eq. 2, which provides the percentage of bound enzyme regardless of whether it is bound to the support, as follows:
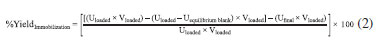
where Uloaded denotes the free enzyme activity, Vloaded is the enzyme initial volume, Uequilibrium blank is the amount of enzyme that was not denatured, and Ufinal is the supernatant activity. The immobilization yield in terms of total binding proteins was also evaluated using Eq. 3, which considers the amount of protein loaded and the amount of immobilized protein, as follows:
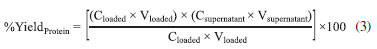
where Cloaded and Csupernatant represent the protein concentrations contained in the initial enzyme solution and the filtrate, respectively, and Vloaded and Vsupernatant are the volumes of protein in the solution and filtrate, respectively. The equations used in the present study were adapted from Manta et al.17 Determination of kinetics binding enzyme to the support The experiments to determine binding kinetics at equilibrium were performed in a reaction mixture containing the support, a buffer of the desired molarity, and the enzyme extract at pH 7.5 at 10 ºC under agitation of 150 rpm for 120 min. Samples were collected at periodic time intervals and filtered to remove the support. The kinetics determination was adapted from Kalil et al.18 Then, enzyme activity was determined in triplicate in the solution until the equilibrium was reached. Experiments were performed without the support to assess enzyme denaturation associated with the buffer molarity. Effect of pH on β-galactosidase activity The influence of pH on enzyme activity at 37 ºC was determined by assaying the activity at different pH values ranging from 2.6 to 9.0 using 0.1 mol L-1 concentrations of the following buffer systems: citrate phosphate (pH 2.6, 4.6, and 6.3), sodium acetate (pH 3.6, 4.6, and 5.6), sodium phosphate (pH 6.3, 7.3, and 8.0), and Tris-HCl (pH 7.3, 8.0, and 9.0).19 The relative activity (%) was expressed as the ratio of the β-galactosidase activity efficiency obtained at a certain pH to the maximum activity efficiency obtained over the given pH range. Effect of temperature on β-galactosidase activity The effect of temperature on the β-galactosidase activity efficiency was determined by performing the standard enzyme assay procedure at different temperatures ranging from 25 to 60 ºC, whereas pH was fixed according to the buffer used in the analysis (0.1 mol L-1 potassium phosphate buffer, pH 6.6). The substrate (o-nitrophenyl-β-D-galactopyranoside; ONPG) was pre-incubated at the respective temperatures for 5 min. The relative activity (%) was expressed as the ratio of the β-galactosidase activity efficiency obtained at a certain temperature to the maximum activity efficiency obtained over the given temperature range.19 Kinetics of thermal deactivation and estimation of the deactivation energy To investigate the thermal deactivation kinetics of β-galactosidase, the enzyme was incubated at different temperatures (37, 40, 45, 50, 55, and 60 ºC) in the absence of substrate. Aliquots were withdrawn at periodic intervals and cooled in an ice bath prior to the assay. The residual activity was expressed as a percentage of the initial activity efficiency. From a semi-natural logarithmic plot of the residual activity versus time, the deactivation rate constant (Kd) was calculated, and the half-life was estimated using Eq. 4. The half-life (t1/2) is defined as the time at which the residual activity reaches 50%.20

The temperature dependence of Kd was analyzed through the Arrhenius plot. The deactivation energy was calculated from the Arrhenius equation as follows:
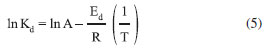
where Ed is the deactivation energy of the transition state of enzyme deactivation, A is a constant, and R the universal gas constant. Ed was estimated from the intercept of the plot of ln (Kd) against 1/T. Determination of kinetic constants The kinetics of most reactions catalyzed by enzymes follows the Michaelis-Menten equation (Eq. 6).

Km was determined using a double-reciprocal (1/V versus 1/S) Lineweaver-Burk plot (Equation 7) with different substrate concentrations using ONPG as the substrate (1-10 mmol L-1).
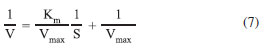
β-Galactosidase assay β-Galactosidase activity was evaluated using two substrates: ONPG and lactose. β-Galactosidase activity was measured spectrophotometrically using ONPG.21 One unit of enzyme activity (U) was defined as the quantity of enzyme that liberates 1 µmol of o-nitrophenol per min under the conditions of the assay. When lactose was used as the substrate, β-galactosidase activity was assessed on the basis of the amount of liberated glucose quantified using an enzymatic-colorimetric kit, Glucose PAP Liquiform (Labtest®, Brazil) with an absorbance reading of 505 nm. The conversion to glucose concentration was performed using a previously determined calibration curve. An enzyme activity unit was defined as the amount of enzyme required to produce 1 µmol of glucose per minute under trial conditions.22 Protein content The protein content of each sample was determined using the Qubit Fluorometer (Kit Quant-iT Protein Assay) following the methodology proposed by the manufacturer. The kit provides concentrated assay reagent, dilution buffer, and pre-diluted BSA standards. The reagent was diluted to 1:200, after which 200 mL of the diluted reagent was added to the wells of a microplate. Next, 1-20-mL sample volumes were added and mixed, and fluorescence was measured. The assay is highly selective for protein. In the range of 0.25-5 mg of protein, the response curve is sigmoidal (pseudolinear from 0.5-4 mg), and it exhibits low protein-to-protein variation. The assay was performed at room temperature, and the signal was stable for 3 h. Common contaminants, such as salts, solvents, or DNA, but not detergents, are well tolerated in the assay. Reuse of immobilized β-galactosidase Immobilized enzyme was added to a 200 mmol L-1 lactose solution (150 rpm for 30 min at 35 ºC), and this preparation was divided into triplicates to assay the enzyme activity. After each run, the immobilized enzyme was removed from assay tubes and washed with 0.1 mol L-1 potassium phosphate buffer, and the process was successively repeated for eight cycles. The first activity determined was considered 100% for the calculation of the remaining percent activity in subsequent runs. Characterization of the support and immobilized β-galactosidase The textural analysis of immobilized catalysts was conducted from isotherms of the adsorption/desorption of N2. The specific superficial area, average porous volume, and porous diameter were determined using Autosorb-1 (Quantachrome, Nova 2200e model) equipment. Before analysis, samples were completely dried under vacuum at 100 ºC. Measurements were conducted using liquid N2 (-196 ºC). The superficial specific area was determined by the BET method, and the average porous diameter was determined by the Barret, Joynere, and Halenda method in the adsorption limit. For SEM performed using Shimadzu SZ 550, the samples were coated with a gold film and analyzed using JEOL JSM-6390LV at 20 kV acceleration voltage.
RESULTS AND DISCUSSION Table 1 presents the matrix of CCD (real and coded values) with responses after β-galactosidase immobilization with Eupergit® C. The immobilization yield was based on the active enzyme bound to the particle, and it ranged from 2.9% to 15.6%. The best result presented (15.6%) refers to trial 8 (Table 1), corresponding to the following conditions: support amount of 400 mg (level +1), ionic strength of 1.5 mol L-1 (level +1), pH of 7.5 (level +1), and reaction time of 2 h (level -1).
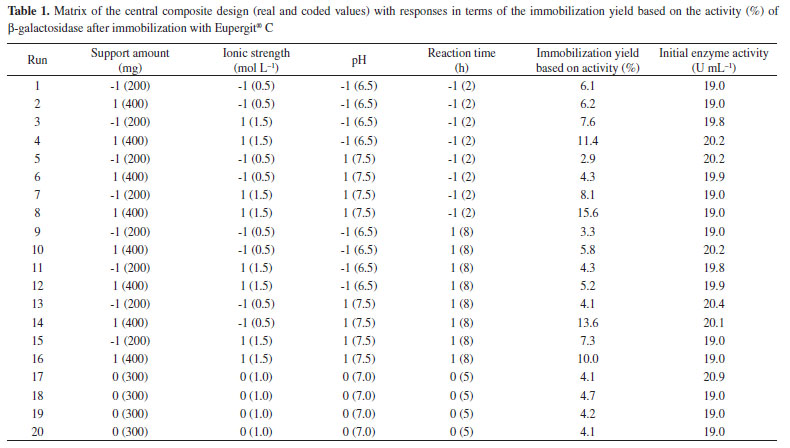
The main effect of the variables on the immobilization yield based on activity was obtained via statistical analysis of the results using a significance level of 95% (p < 0.05) (Table 2). It can be observed that immobilization was influenced by the support amount, ionic strength, pH, and reaction time, hence demonstrating that the variables were well chosen to evaluate the immobilization process.
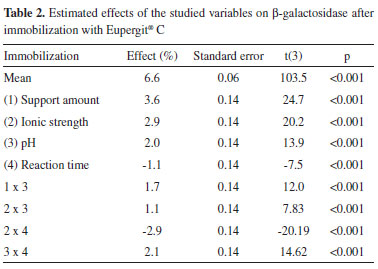
The R2 value, which provided a measure of how much the variability in the observed response values could be explained by the experimental factors and their interactions, was 0.86. A satisfactory performance of the F value (4.6) was obtained, which corresponds to approximately two times the tabulated F (3.1) for β-galactosidase after immobilization with Eupergit® C with a confidence level of 95%. The model for yield based on β-galactosidase activity was used to construct the contour diagrams depicted in Figure 1 to allow better interpretation of the interactions between the variables and the contribution of each variable for maximizing β-galactosidase immobilization.
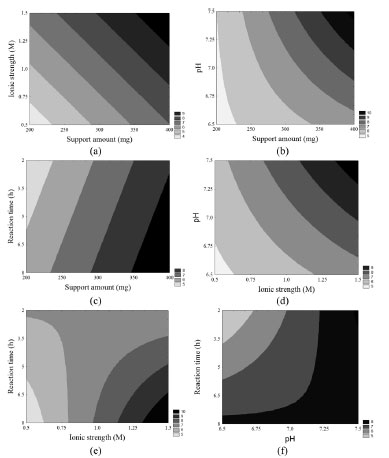 Figure 1. Contour diagrams for the immobilization yield based on β-galactosidase activity as a function of ionic strength and amount of the support (a), pH and amount of the support (b), reaction time and amount of the support (c), pH and ionic strength (d), reaction time and ionic strength (e), and reaction time and pH (f)
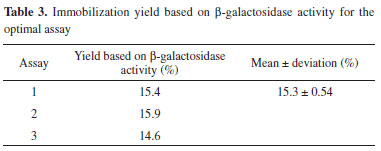
Considering all contour diagrams, the evaluated response displayed an increment when the support amount was maximal, perhaps because the availability of the support to bind the enzyme increases when the amount of the support is higher (Figure 1a-c), therefore resulting in an increment in the immobilization yield based on β-galactosidase activity (Table 1 and Figure 1d and e). An increase in the salt concentration from 0.5 mol L-1 to 1.5 mol L-1 resulted in an increased in the response from 4.3% to 15.6%. Such a positive effect probably occurred due to the increase in the strength of hydrophobic interactions caused by immobilization. According to Alptekin et al.9, the recommended immobilization conditions on this support include the use of a high ionic strength (to force the hydrophobic adsorption of the proteins) because of the fairly hydrophobic nature of the support. However, a high ionic strength can cause protein denaturation, and thus, the range of this variable must be carefully chosen to obtain an adequate balance between maximum binding between the enzyme and support and minimal protein denaturation. The amount of enzyme bound to Eupergit® C randomly changed depending on the pH of the medium because Eupergit® C binds proteins via their epoxide groups, which may react with different nucleophiles on the protein as a function of pH.9 In addition, the pH evaluation in this study accounted for enzyme denaturation, which is higher in the presence of extreme pH values. It was observed that an increase in pH resulted in higher efficiencies in activity (Table 1 and Figure 1f). Although many studies in the literature investigated the immobilization of different enzymes by Eupergit® C,9,23,24 only a few studies evaluated the immobilization of β-galactosidase using this support.25,26 In addition, another difficulty in comparing the results of prior research is that enzymes from different sources have different microbial characteristics and properties. To prove that the experimental immobilization conditions can be reproduced, the best assay was repeated using a new batch of enzyme. Table 3 shows the average results for the immobilization yield based on activity, performed in triplicate, using the immobilization conditions described in the best trial.
Higher amounts of support in the immobilization system resulted in a larger removal of free enzyme from the reaction solution; therefore, a superior immobilization yield was also obtained. Moreover, because of the relatively high cost of Eupergit® C, most of the studies involving this support used approximately 400 mg of Eupergit® C.9,25,26 Furthermore, β-galactosidase from yeast has a narrow working pH range, restricting this parameter to the previously studied range (pH between 6.5 and 7.5). In light of these findings, ionic strength is the most important variable to assess. In this sense, experiments were conducted to investigate the kinetics of immobilization with different buffer concentrations to determine the equilibrium time and behavior of enzyme binding to the support. Therefore, from the best trial and considering the best enzyme-support interaction, binding kinetics were evaluated to increase the immobilization yield based on β-galactosidase activity. Figure 2 shows the time course of the activity of β-galactosidase immobilized by Eupergit® C in experiments conducted at 10 ºC. The initial activities in those assays ranged between 8.5 and 11.5 U mL-1.
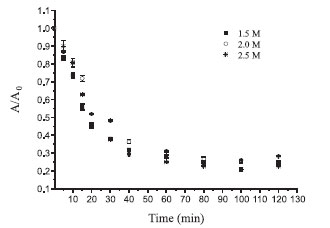 Figure 2. Binding kinetics of the immobilization yield based on β-galactosidase activity
All experiments reached equilibrium after 80 min, at which point no changes in activity were observed in solution. Thus, the immobilization process may be completed at this time. The curves profiles demonstrate that after 15 min, approximately 50% of the enzyme was immobilized (Figure 2). Table 4 shows the results of the immobilization yield based on β-galactosidase activity (%), the yield based on immobilization (%), and the immobilization yield based on protein (%), considering the different molarities evaluated.

The evaluation of the immobilization yield based on β-galactosidase activity illustrates how much of the bound enzyme is active, i.e., capable of catalyzing the reaction. Conversely, the yield based on immobilization indicates the amount of enzyme that is bound to the support but is not necessarily capable of catalyzing the reaction. The evaluation of the difference between these values demonstrated that not all of the enzyme bound to the support is biologically active, which may be due to steric hindrance or enzyme denaturation.8 A higher ionic strength can increase enzyme denaturation. In Table 4, it can be noticed that there was no significant difference in denaturation when the buffer molarity was increased from 1.5 mol L-1 to 2.0 mol L-1. However, a higher molarity of 2.5 mol L-1 led to greater enzyme denaturation. With ONPG, the optimal yields based on β-galactosidase activity (22%) and based on immobilization (69.3%) were obtained when 2.0 mol L-1 buffer was used. The results considering protein retention also corroborated the finding that a molarity of 2.0 mol L-1 was optimal among those evaluated in this study (88.3%) (Table 4). All responses presented are important and complementary in consideration of enzyme immobilization. The percentage of enzyme denaturation caused by the ionic strength of the buffer was relatively low at approximately 11%. Approximately 70% of the enzyme was bound to the support, but only 22% of the enzyme reacted with the substrate. Therefore, some effects over the enzyme are altered when it binds to the support, including the possibility of steric effects and the prevention of catalysis by the immobilized enzyme due to the unavailability of the active site. These phenomena appeared to have occurred, as could be observed when comparing the responses evaluated. Manta et al.17 evaluated the immobilized β-galactosidase in a manner similar to that of our study. The authors considered the amount of protein bound according to the amount of enzyme particles immobilized and the difference in the yield. This broader assessment generates more precise information, improving the understanding of the immobilization process. The most commonly used substrate for β-galactosidase for industrial purposes is lactose; however, in terms of methods for determining enzyme activity, ONPG is most frequently used,17,27,28 although some studies used lactose as the substrate to determinate β-galactosidase activity. However, to our knowledge, there are no studies that evaluated immobilization efficiency using both differences in yield and the amount of enzyme particles immobilized, in addition to comparing the differences between ONPG and lactose, to determinate β-galactosidase activity. For immobilization, it is extremely important to determine these differences, primarily because of the difficulty of binding between the immobilized enzyme and substrate, as it is not soluble in the reaction medium. To better understand the effects of immobilization on enzyme activity, this parameter was evaluated using lactose as the substrate (Table 5). The experiments were conducted in triplicate, and the mean immobilization yield based on β-galactosidase activity with lactose as the substrate was 49.8%. Compared with the value obtained using ONPG as the substrate (22%), greater amounts of the enzyme were available to catalyze lactose hydrolysis after immobilization. In terms of the yield based on immobilization, there was practically no difference between the two substrates, as the value was 69.3% for ONPG and 64.5% for lactose. This finding was expected because the amount of enzyme bound is independent of the substrate, as these results were obtained by determining the difference in enzyme activity in the solution. Based on these results, it is evident that immobilized β-galactosidase more efficiently catalyzes the hydrolysis of lactose than of ONPG.

Considering the finding of a higher yield with lactose as the substrate, several factors could influence these results. The main aspects to be considered are the steric effects, initial concentrations of the substrate (ONPG and lactose), inhibition due to the substrate or products, and kinetic models.29 Therefore, it is extremely important for this type of evaluation to compare the yield based on β-galactosidase activity and that based on immobilization when immobilization processes are developed. Considering the literature, the studies that obtained higher percentages of immobilization with commercial enzymes, which are usually purified and stabilized, used lactose as a substrate to measure the activity26 or made use of spacers.17 Moreover, Ates and Mehmetoglu immobilized commercial β-galactosidase with calcium alginate and cobalt chloride and obtained a greater efficiency of enzyme immobilization (83%); however, the use of Co2+ as the gel-forming agent limits enzyme use in the food industry.30 Enzyme reuse is an important factor to be considered in the immobilization process, especially for enzymes applied industrially. β-Galactosidase reutilization after immobilization with Eupergit® C was studied under optimal conditions (support amount: 400 mg, ionic strength: 2.0 mol L-1, pH: 7.5, and reaction time: 2 h), which revealed that the initially loaded enzyme maintained 50% of its initial activity after five cycles of use. Furthermore, it can be used for three cycles consecutively in the industrial process without a loss of activity (Figure 3). This result is extremely important because of the complexity of industrial β-galactosidase use resulting from its instability in reaction media owing to its sensitivity to process changes.
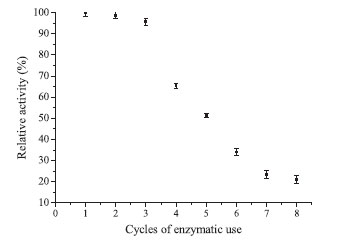 Figure 3. β-Galactosidase reutilization after immobilization by Eupergit® C
The immobilization of β-galactosidase from K. lactis was evaluated by Giacomini et al.28 considering two different chemically modified supports (silica and agarose) and the reuse of the immobilized enzyme. After one cycle of reuse, the enzyme retained only 25% of its initial activity, thus emphasizing the importance of the data obtained in the present study. β-Galactosidase immobilized by Eupergit® C was characterized, revealing the optimal temperature to be 50 ºC. Heidtmann et al.16 performed a kinetic and thermodynamic characterization of β-galactosidase from K. marxianus CCT 7082 fractionated with ammonium sulfate and identified an optimal temperature of 45-50 ºC. These data illustrate that immobilization did not change the optimal temperature of the enzyme. The optimal pH of immobilized β-galactosidase in this study was 7.3, which agreed with the findings of several studies in the literature.31-33 The thermal denaturation constant (Kd) and half-life are reported in Table 6.
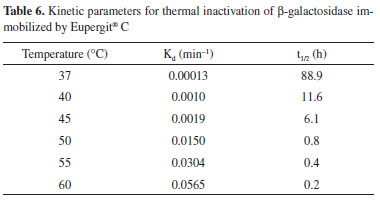
The results presented in Table 6 illustrate that, unlike the data obtained for the optimal temperature and pH, which did not differ between the free and immobilized enzyme, the Kd and half-life of the enzyme were altered by immobilization. The half-life values obtained for the immobilized enzyme were higher at all temperatures than those presented by Heidtmann et al.16 for the partially purified free enzyme. These results demonstrate that the enzyme displays greater thermal stability after immobilization than the free enzyme. The kinetic parameter Km of the immobilized enzyme was 5.2 mmol L-1 with ONPG as the substrate. Heidtmann et al.7 obtained a Km of 3.7 mmol L-1 using the free form of the same enzyme. Thus, in the present study, the immobilized enzyme displayed a reduced specificity for the substrate. In general, there is an increase in Km after β-galactosidase immobilization.28,34,35 These results are in agreement with our findings, and they may be explained by the fact that immobilized enzymes are less accessible to the substrate than free enzymes. Therefore, a higher substrate concentration is required to increase its interaction with the immobilized enzyme than with the free form. Furthermore, some of the active centers are blocked after immobilization, which reduces the reaction rate. After thermal characterization, partial characterization of the particle was performed. A comparison of the superficial areas of the support (25.16 m2 g-1) and immobilized enzyme (15.1 m2 g-1) revealed a significant reduction in this parameter, indicating the adherence of the enzyme to the cavities of the support. An increase in the pore diameter was observed after immobilization (21.3 and 22.4 Å for the support and immobilized enzyme, respectively), probably due to the entrance of the enzyme into the structure of the clay. These results are similar to those obtained by Coghetto et al.,36 who used natural montmorillonite as the support for the immobilization of inulinase from K. marxianus NRRL Y-7571. Figure 4 presents the SEM micrographs of the support (Figure 4a, c, and e) and immobilized enzyme (Figure b, d, and f). The SEM images indicated that β-galactosidase was aggregated on the surface of the support. After immobilization, the micrographs displayed a more cloudy appearance (Figure 4b, 4d, and 4f), suggesting that the enzyme has immobilized both within the pores and on the surface of the support. Regardless, the immobilization process confers notable advantages, and it has the potential for application throughout industry.
 Figure 4. Scanning electron microscopic images of the support Eupergit® C before (a, c, and e) and after (b, d, and f) β-galactosidase immobilization
CONCLUSIONS β-Galactosidase immobilization conferred promising results under conditions of 400 mg of Eupergit® C, an ionic strength of 2.0 mol L-1, a pH of 7.5, and a reaction time of 2 h. Under these conditions, the maximum immobilization efficiency was 22% (ONPG) or 49.8% (lactose), revealing a possible steric impediment depending on the substrate used to measure enzyme activity. The enzyme maintained 50% of its initial activity after five cycles of use, and the data suggested the potential of the enzyme to be used for three consecutive cycles in industrial processes without a loss in activity. The results reported here are of great industrial interest, as only a few studies on this subject (immobilization and stabilization evaluated in a combined manner) are available in the open literature.
ACKNOWLEDGMENTS The authors thank FAPERGS, CAPES, and CNPq for financial support of this study and scholarships. In addition, we would like to thank D. L. Boschetto and the Laboratório Central de Microscopia Eletrônica - UFSC for SEM analysis.
REFERENCES 1. Velazquez, J. B.; Food Bioprocess Technol. 2011, 4, 831. 2. Gekas, V.; López-Leiva, M.; Process Biochem. 1985, 20, 2. 3. Hsu, C. A.; Lee, S. L.; Chou, C. C.; J. Agric. Food Chem. 2007, 55, 2225. 4. Panesar, P. S.; Kumari, S.; Panesar, R.; Enzyme Res. 2010, 2010, 1. 5. Alptekin, O.; Seyhan, T. S.; Yildirim, D.; Alagoz, D.; Enzyme Microb. Technol. 2011, 49, 547. 6. Gurdas, S.; Guleç, H. A.; Mutlu, M.; Food Bioprocess Technol. 2012, 5, 904. 7. Asraf, S. S.; Gunasekaran, P.; Current Research, Technology and Education Topics in Applied Microbiology and Microbial Biotechnology. 2010, 2, 880. 8. Katchalski-Katzir, E.; Kraemer, D. M.; J. Mol. Catal. B: Enzym. 2000, 10, 157. 9. Alptekin, O.; Tukel, S. S.; Yildirim, D.; Alagoz, D.; J. Mol. Catal. B: Enzym. 2010, 64, 177. 10. Cho, Y.-J.; Park, O. J.; Shin, H. J.; Enzyme Microb. Technol. 2006, 39, 108. 11. Elnashar, M. M. M.; Yassin, M. A.; J. Appl. Polym. Sci. 2009, 114, 17. 12. Pinheiro, R.; Belo, I.; Mota, M.; Lett. Appl. Microbiol. 2003, 37, 438. 13. Manera, A. P.; Ores, J. C.; Ribeiro, V. A.; Burkert, C. A. V.; Kalil, S. J.; Food Technol. Biotechnol. 2008, 46, 66. 14. Medeiros, F. O.; Burkert, C. A. V.; Kalil, S. J.; Chem. Eng. Technol. 2012, 35, 911. 15. Medeiros, F. O.; Alves, F. G.; Lisboa, C. R.; de Souza Martins, D.; Burkert, C. A. V.; Kalil, S. J.; Quim. Nova 2008, 31, 336. 16. Heidtmann, R. B.; Duarte, S. H.; Pereira, L. P.; Braga, A. R. C.; Kalil, S. J.; Braz. J. Food. Technol. 2012, 15, 41. 17. Manta, C.; Ferraz, N.; Betancor, L.; Antunes, G.; Batista-Viera, F.; Carlsson, J.; Caldwell, K.; Enzyme Microb. Technol. 2003, 33, 890. 18. Kalil, S. J.; Barbosa, M.; Maugeri, F.; Rodrigues, M. I.; Braz. J. Food. Technol. 2006, 9, 223. 19. Braga, A. R. C.; Manera, A. P.; Ores, J. C.; Sala, L.; Maugeri-Filho, F.; Kalil, S. J.; Food Technol. Biotechnol. 2013, 51, 45. 20. Braga, A. R. C.; Silva, M. S.; Oliveira, J. V.; Treichel, H.; Kalil, S. J.; Bioproc. Biosyst. Eng. 2012, 35, 1541. 21. Inchaurrondo, V. A.; Yantorno, O. M.; Voget, C. E.; Process Biochem. 1994, 29, 47. 22. Szczodrak, J.; J. Mol. Catal. B: Enzym. 2000, 10, 631. 23. Alagoz, D.; Yildirim, D.; Alptekin, O.; Tukel, S. S.; J. Biotechnol. 2010, 150, Supplement, 380. 24. Knezevic, Z.; Milosavic, N.; Bezbradica, D.; Jakovljevic, Z.; Prodanovic, R.; Biochem. Eng. J. 2006, 30, 269. 25. Torres, P.; Batista-Viera, F.; J. Mol. Catal. B: Enzym. 2012, 83, 57. 26. Campello, G. S.; Trindade, R. A.; Rêgo, T. V.; Burkert, J. F. D.; Burkert, C. A. V.; Int. J. Food Eng. 2012, 8, 1. 27. Ansari, S. A.; Husain, Q.; Food Bioprod. Process. 2012, 90, 351. 28. Giacomini, C.; Villarino, A.; Franco-Fraguas, L.; Batista-Viera, F.; J. Mol. Catal. B: Enzym. 1998, 4, 313. 29. Ladero, M.; Santos, A.; García, J. L.; García-Ochoa, F.; Enzyme Microb. Technol. 2001, 29, 181. 30. Ates, S.; Mehmetoglu, U.; Process Biochem. 1997, 32, 433. 31. Burin, L.; Buera, M. D.; Enzyme Microb. Technol. 2002, 30, 367. 32. Ladero, M.; Ferrero, R.; Vian, A.; Santos, A.; Garcia-Ochoa, F.; Enzyme Microb. Technol. 2005, 37, 505. 33. Ladero, M.; Santos, A.; García-Ochoa, F.; Enzyme Microb. Technol. 2000, 27, 583. 34. Haider, T.; Husain, Q.; Chem. Eng. Process.: Process Intensif. 2009, 48, 576. 35. Zhou, Q. Z. K.; Chen, X. D.; Biochem. Eng. J. 2001, 9, 33. 36. Coghetto, C. C.; Scherer, R. P.; Silva, M. F.; Golunski, S.; Pergher, S. B. C.; Oliveira, D.; Oliveira, J. V.; Biocatal. Agric. Biotechnol. 2012, 1, 284. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access