
 |
 |
 |
 |
 |
Revisão
| Advances in ethanol reforming for the production of hydrogen |
|
Laura Guerrero; Sebastián Castilla; Martha Cobo*
Department of Chemical Engineering, School of Engineering, Universidad de La Sabana, Campus Universitario Puente del común Km 7, Autopista Norte, Bogotá, Colombia Recebido em 13/06/2013 *e-mail: martha.cobo@unisabana.edu.co Catalytic steam reforming of ethanol (SRE) is a promising route for the production of renewable hydrogen (H2). This article reviews the influence of doping supported-catalysts used in SRE on the conversion of ethanol, selectivity for H2, and stability during long reaction periods. In addition, promising new technologies such as membrane reactors and electrochemical reforming for performing SRE are presented. INTRODUCTION Atmospheric pollution and degradation of the air are topics investigated widely because of their direct impact on the quality of life of the population. Significant contribution of fossil fuels to environmental degradation in energy generation processes and the depletion of petroleum reserves make it necessary to find alternative energy sources, which are renewable, sustainable, cost-effective, and safe, to replace the current sources of energy. Hydrogen (H2) has been identified as a suitable fuel for sustainable energy production. It can be used in fuel cells to generate electricity at high efficiency, in addition to producing water as the only byproduct. Demand for H2 from fuel cells will increase considerably in the coming years; however, it is currently produced from a fossil fuel (natural gas).1 Therefore, it is desirable to incorporate renewable sources for its generation to allow for the development of a sustainable H2 economy.2 Reforming alcohols for H2 production has generated great interest because this technique is based on renewable sources, such as bioethanol, which is currently a widely produced and distributed biofuel. One of the most promising methods for H2 production from ethanol is the catalytic steam reforming of ethanol (SRE). The selection and development of a suitable catalyst for SRE is a key aspect, as the catalyst must be stable, active, selective, and maximize hydrogen production while simultanously disfavoring the formation of byproducts (CO and CH4). Various catalysts for SRE including metal oxides, and transition and noble metals supported on oxides (with a wide range of redox and acid-base properties) have been studied. The nature of the metal, type of precursor (or dopant), method of preparation, type of support, additives, and operating conditions have been found to exert a great influence on SRE.3 In addition, the regeneration and reuse of the catalysts are features that must be considered for a more efficient application.4 Various review articles on the design of the catalyst for the SRE have been published.2,5-7 In these articles, the selection of active metals and catalyst support has been analyzed as the critical factor that allows for obtaining catalysts with high activity and selectivity. In contrast to previous review articles, this study compares the systems that report greater ethanol conversion rates and selectivity toward hydrogen, while placing emphasis on the stability of catalysts for periods exceeding 50 h of operation. In addition, the presence or absence of any precursors and their effect on catalytic behavior has been evaluated. The state of progress of the new catalytic systems proposed for hydrogen production based on ethanol reforming, such as monoliths, microreactors, and membrane reactors, is also analyzed. Although they are interesting proposals, all new catalytic system exhibit lower efficiencies than those of the traditional technologies.
STEAM REFORMING OF ETHANOL (SRE) Hydrogen, a clean alternative to current fuels, can be produced from various sources, such as coal, natural gas, light gas oil, propane, methane, gasoline, light diesel, biomass, and water.5 Of these, H2 production from ethanol is convenient for various reasons: (1) ethanol is renewable and its availability has considerably increased in recent years;8 (2) it is easy to transport; (3) it decomposes relatively easily in the presence of water and generates a mixture rich in hydrogen; (4) it is free of sulfur and compounds that poison the catalyst; and (5) the reaction for H2 production with ethanol is thermodynamically feasible.9,10 In addition, the benefits of H2 as a fuel include: (1) reduction in the dependency on fossil fuels, (2) environmentally benign, (3) reduces air pollution, (4) renewable, (5) obtained from various sources, (6) carbon-free fuel source, (7) high energy content per unit mass, and (8) high octane rating.9 Ethanol steam reforming (shown in pathway 1 of Figure 1) occurs through the following reaction:10
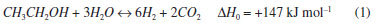
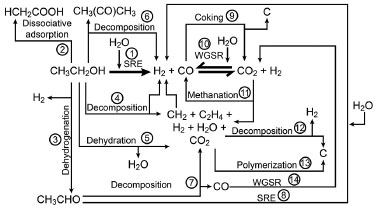 Figure 1. Probable reaction mechanisms occurring during ethanol reforming5
However, when an ethanol-water mixture is in contact with a catalyst at high temperature, several reactions can occur. Figure 1 shows the possible reaction pathways. Pathway 3 indicates dehydrogenation of ethanol to acetaldehyde, which is an intermediate in the process for H2 production with low coke formation.11 The acetaldehyde undergoes steam reforming during H2 and CO production, which is shown in pathway 8.12 In addition, the Water Gas Shift Reaction (WGSR), observed in pathways 10 and 14, allows for the additional production of H2 and decrease in CO yield. This reaction is reversible and is favored at low temperatures. At high temperatures, the equilibrium shifts to the left (pathways 10 and 14), which limits the conversion of CO. Therefore, for bringing about greater conversions of CO and H2, the WGSR reaction is performed in two steps: a high-temperature shift (HTS; 623-643 K) and low-temperature shift (LTS; 473-493 K).5 However, alternate reactions can lead to ethanol decomposition (pathways 2, 4, and 5), which results in a series of undesirable reactions (pathways 6 and 7) that can lead to the production of CH4 and C2H4. The latter favors coal-production pathways, which causes coking of the catalyst (pathways 8, 12, and 13), and consequently hinders hydrogen production.5 In summary, there are desirable and undesirable reaction pathways that need to be judiciously regulated for effective SRE. In general, the preferred routes 1, 3, and 10 are promoted by supported catalysts, which facilitate the adsorption of ethanol and its subsequent dehydrogenation. Intriguingly, catalysts that promote ethanol decomposition generally end with carbon deposits on their surface, thereby, reducing their lifetimes. It has been found that basic supports preferentially promote ethanol dehydration while acidic supports stimulate ethanol decomposition. Regarding WGSR (pathway 10 and 14), a few mixed catalysts that simultaneously promote both SRE and WGSR have been proposed, however, the conditions for their practical applications have not yet been optimized. In the following sections, the nature of the catalyst and the mechanisms of transformation are discussed.
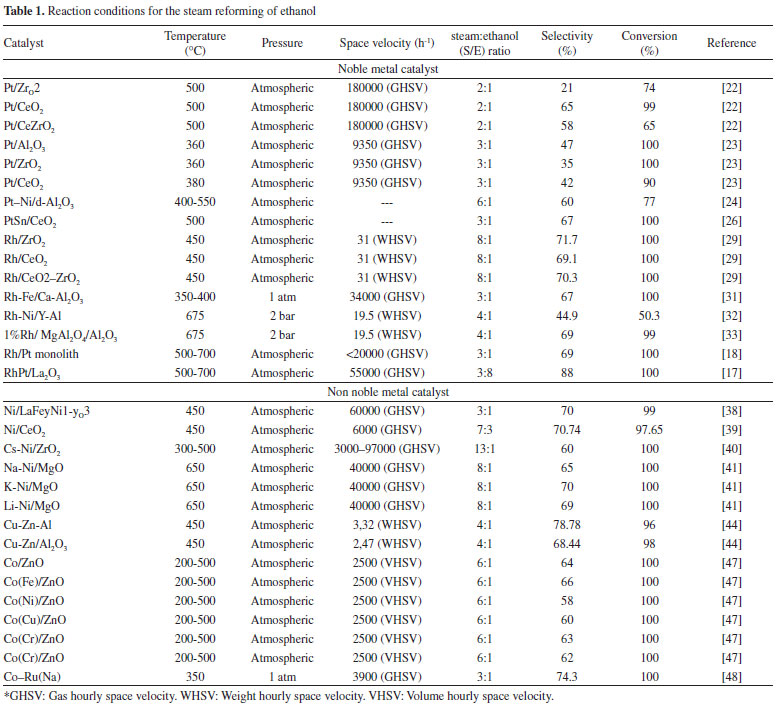
ETHANOL-REFORMING CATALYSTS Studies have been performed with a great number of catalysts for hydrogen production from ethanol. The most widely investigated include the noble metal- and nickel-based catalysts.11,13 As stated previously, a suitable SRE catalyst must be stable, active, selective, and able to maximize hydrogen production while disfavoring the formation of byproducts. Although many metal catalysts are active in this process, they are continuous deactivated by carbon depositions (coke formation); nickel-based catalysts are particularly sensitive to such deactivation (pathways 8, 12, and 13 in Figure 1).14 Due to their high redox capacity, noble metal-based catalysts incur less coke formation than nickel-based catalysts; however, the high cost of cost noble metals demands high strength and activity. The catalytic yield of supported catalysts used for SRE has been greatly influenced by the nature of the support, because the support affects the dispersion and stability of the metal and can participate in the reaction.15 Among the metal-oxide-based supports, those based on alumina are frequently used because of their mechanical and chemical stability under the reaction conditions. However, acidic sites on the support promote ethanol dehydrogenation and lead to formation of ethylene (Figure 1, pathways 8, 12, and 13) which in turn deactivates the catalysts by carbon deposition. Therefore, basic additives or precursors that favor water adsorption and mobility of the OH groups on the Al2O3 surface are frequently added. Alkaline oxides (such as MgO) are widely used in reforming formulations to neutralize the acidity of Al2O3.16 The exclusive use of these alkaline oxides and their mixtures as supports (CeO2, ZrO2, CexZr1-xO2, and La2O2) has been proposed because they favor ethanol dehydrogenation reactions (pathway 3, Figure 1) instead of dehydration.1,17 However, these alkaline oxides have a low surface area, and it is common to find them supported on oxides with greater surface area (such as SiO2).18 In many cases, other metals and compounds have been added to the principal active phase in catalysts with the goal of improving their properties. Here, the properties of another metal are exploited to complement the functioning of the active phase. Furthermore, the addition of the second metal can enhance the features of the catalyst, such as a better dispersion and smaller particle size; the added metal can segregate the particles of the active phase and prevent aggregation by the formation of a system of interpolation between the two metals. These interactions can change the adsorption and redox properties of the active metals.19 Different methods of evaluating the activity of catalysts used in SRE have been proposed. Prominently, the evaluations are based on hydrogen selectivity, ethanol conversion, and stability over time. Haga et al.20 found that the catalysts used in SRE show the following order of selectivity to H2 on alumina supports: Co > Ni > Rh > Pt > Ru > Cu. However, some authors relate the activity to the resistance of coke formation. For example, Frusteri et al.21 found that the activity of metal-based catalysts (Rh > Co > Ni > Pd) is consistent with their resistance to coke formation (spatial velocity: 40000 h-1; duration: 20 h). Catalytic systems with noble metals Catalysts based on Pt Pt supported on alumina, zirconia, and ceria have been proposed as active catalysts in SRE. Among the noble metals, properties that include high activity in C-C bond cleavage, high conversion in WGSR, and temperature-independent reliable activity make Pt an adequate active component for ethanol-reforming reactions.22 Lima et al.23 show that the nature of the support significantly affects the distribution of the products obtained in SRE. Pt/CeO2 and Pt/CeZrO2 have high selectivity for H2 (65% and 58%, respectively), whereas Pt/ZrO2 produces high quantities of undesirable species. Performing SRE in a similar temperature range (<500 ºC), Panagiotopoulou et al.24 found that Pt catalysts have the following order of selectivity for H2: Pt/Al2O3 (47%)>Pt/CeO2 (42%)>Pt/ZrO2 (35%). Moreover, adding metal precursors or dopants affect the activity of Pt catalysts.25 Soyal et al.26 found that the Pt catalyst supported on alumina doped with 15% Ni has a 60% selectivity for H2 at 500 ºC. This behavior was attributed to a better dispersion and abundance of active catalyst in the presence of Ni. Lima et al.27 evaluated the addition of Sn to Pt/CeO2 as a means of eliminating coke formation on the surface of the catalyst. With this addition, an ethanol conversion of 100% and a H2 selectivity of 67% are obtained at 500 ºC. In addition, Sn inhibits the decomposition of intermediate species, thereby increasing the resistance of the catalyst to deactivation. Catalysts based on Rh Rh is an active and stable catalyst in SRE. Moura et al.28,29 suggest that among the noble metals, Rh is the most active and selective for the hydrogen production. Diagne et al.30 studied Rh/CeO2, Rh/ZrO2, and Rh/CeO2-ZrO2 catalysts in SRE. A 70% selectivity for H2 with complete ethanol conversion at 450 ºC is observed. The CO2/CO ratio is lowest in case of Rh/CeO2. Catalysts based on mixtures of Zr and Ce oxides are more active. The high yield of CO2 in systems that contain Zr is attributed to the amount of oxygen available on the surface. The production of these carbon oxides in Rh-based catalytic systems was studied by Chen et al.31 They show that the selectivity for CO at 350 ºC decreases by 48% upon doping the Rh-based catalyst with Fe. This is attributed to the presence of iron oxides, which minimize CO adsorption in the neighboring Rh sites and transform the adsorbed CO into a formate species to be consumed in the WGSR, thereby resulting in a greater H2 yield, low selectivity for CO, and a stable Rh catalyst. In other words, the addition of a dopant to Rh-based catalysts increases their selectivity for H2 and yield. Moreover, the dopant affects the stability and durability of a catalyst. The addition of Ni to the Rh/Y2O3-Al2O3 catalyst chemically modifies the metallic phase and support to NiAl2O4. The presence of nickel reinforces the amount of Lewis-acid sites, which avoids the deactivation of the catalyst by carbonaceous deposits. In addition, it prevents Rh loss and increases selectivity for H2, as Ni can catalyze the transformation methane to hydrogen.32 Rh catalysts have also been studied in systems with impurities in the gas flow. The presence of these impurities leads to the deactivation of the catalytic system. However, dopants such as Ni increase the stability and resistance of catalysts.33 The yield of H2 and ethanol conversion decreases when the impurities in the input gas flow are alcohols with increasing number of carbons. Moreover, the presence of branched alcohols is particularly detrimental to the catalytic activity; formation of olefins such as C2H4 leads to polymerization and coke-formation, which results in poisoning of the catalyst. This effect increases with the increase in alkyl chain length.33 Catalysts based on Rh-Pt The two sections above demonstrate that satisfactory results in SRE are observed with either Rh- or Pt-based catalysts. Therefore, the design of bimetallic catalysts, based on the properties noble metals offer, has been pursued and has given promising results.17,34,35 The Pt-Rh bimetallic catalyst has a high activity, is less susceptible to coke formation (when compared with nickel-based catalysts), and can easily be regenerated if coke formation does occur. This catalyst is also tolerant to sulfur, which is important for reforming ethanol contaminated with thio-bearing compounds.18 The Rh-Pt/ZrO2 catalyst washcoated on cordierite monoliths has a 100% ethanol conversion and a >65% H2 selectivity at 600 ºC.18 Similar results are reported by Sheng et al.36 using a Rh-Pt/CeO2 catalyst, wherein the H2 selectivity is 60% at temperatures greater than 400 ºC. Cobo et al.17 used Rh-Pt/La2O3 catalyst to effect the complete conversion of ethanol with an H2 selectivity of 88% at 600 ºC; the formation of catalytically active Rh-Pt-Rh2O3 was postulated.17 However, none of the studies demonstrate the capability of Rh-Pt catalysts in promoting the WGHS at temperature >600 ºC. Catalytic systems with other metals (Ni, Cu, Al, Zn, and Co) Catalysts based on Ni Ni is one of the most studied metals in the reforming of fuels and biofuels.2 It is preferred for its low cost and activity.37 In these catalysts, a high dispersion of Ni ensures greater selectivity for hydrogen and decreased selectivity for methane. Moreover, the preparation method intervenes in the catalytic activity because when Ni is in a nanoscopic size the amount of coke in the form of filaments decreases.38 As in the case of noble metal-based catalysts, the addition of a metal dopant, such as Cu, to Ni-based catalytic systems has a great effect on the ethanol conversion and distribution of products. Thus, with the inclusion of 1.5% of a Cu-precursor, the ethanol conversion increases from 18% to 40% and a H2 selectivity of 71% is achieved.39 Other dopants, such as Ce, Li, K, La, and Cs, have also been studied. Akiyama et al.40 found that the Ni/ZrO2 catalyst doped with Cs yields higher levels of H2 when compared with the catalyst that is doped with Li, K, La, or Ce. In similar studies, Frusteri et al.41 prepared a Ni/MgO catalyst and added Li, Na, or K and found that the Li and Na promote NiO reduction but negatively influence the dispersion of the supported catalyst whereas addition of K does not affect the morphology or dispersion significantly. Li and K increase the stability of Ni/MgO by decreasing Ni sintering. Coke formation on the neat and doped catalyst occurs in small amounts. Previous studies demonstrate an increased catalytic activity on addition of a compound to improve their catalytic properties.42 Catalysts based on other metals Although Ni, in addition to the noble metals, is one of the most studied metals in SRE, catalysts based on Cu, Zn, and Co have also been designed. Cu or bimetallic Cu-Zn catalysts have shown activities of 60% and a H2 selectivity of 70%. The difference in behavior between the mono- and bi-metallic catalysts is based both on the nature of the accompanying metal and the preparation of the catalyst.43 Better results are reported with catalysts prepared by the sol-gel method when compared to those by impregnation method.44 With Cu catalysts, water acts as an oxidizing agent and helps removal of part of copper in the oxidized state CO to rapidly oxidize to CO2 before desorption,45 resulting in a gas more suitable for subsequent use in fuel cells that require CO concentrations below 100 ppm.46 Although, Cu can be inactive in the SRE because it promotes dehydrogenation of ethanol to acetaldehyde, studies have shown that Cu is more reducible than Ni and Co.45 The latter has been studied in catalysts doped with Fe, Ni, Cu, Cr, and Na. Casanova et al.47 found that with incorporation of Cu in a Co/ZnO catalyst, ethanol dehydrogenation increases; however, it inhibits the capacity for subsequent dehydrogenation. High quantities of Co favor the reaction because of its ability to participate in redox mechanisms. Catalysts doped with Na, Cr, and Fe give higher yields of hydrogen, which indicates that these structures are more active and selective for SRE than the Co/ZnO catalyst. In addition, Co-Ru catalysts have a H2 selectivity of 74% and the presence of noble metals,48 such as Ru or Rh, facilitate the reducibility of Co, thereby allowing for the regeneration of the bimetallic catalyst.49 Comparison of the catalytic systems The thermodynamic favorability of the SRE reaction makes the complete conversion of ethanol feasible with a wide variety of catalysts under selective experimental conditions.50-52 In general, the selectivity for H2 is 60%-70%, which is close to that predicted by thermodynamic analysis (~70%) and depends on the reaction conditions. Table 1 lists the reaction conditions that were used to evaluate the activity of catalysts for reforming along with the observed ethanol conversion and selectivity for hydrogen.
The effect of the metal dopant or precursor on the activity of the catalysts used in SRE has been widely studied. Better catalytic activity is observed for catalysts that are doped when compared to those that are not.26-28,41,46,48 The effect of the metal dopant can be related to disposition, area, and dispersion of the active phase, parameters that usually affect changes in the selectivity and conversion.53 Although most investigators seek to obtain better results by using a metal dopant with different characteristics than the catalyst, such enhancements in activity is not always observed. Such is the case with the Rh-Ni/Y-Al catalyst; doping with Ni reduced the selectivity of the reaction catalyzed by the monometallic Rh catalyst.31,30
STABILITY AND DURABILITY TESTS IN SRE CATALYSTS The addition of a metal precursor is known to increase the stability and durability of the catalyst.54 The stability of the catalysts is a crucial factor in their selection. A stable catalyst is one that maintains its activity for the longest period.55 To evaluate the potential of a catalyst to be used in SRE, it is essential to perform durability tests that account for its stability and resistance to poisoning. Occasionally, the particle size and the dispersion of the active phase are the critical parameters that determine the stability and durability of a catalyst. For example, the Ru-based catalyst (1-2 nm), which is derived from clusters as reported,56 is nearly three times more durable than the catalyst produced with commercial Ru because of its particle size. When studied under similar conditions, Ir/CeO2 is stable for over 60 h of testing,57 which is 12 h longer than that of the Ru-based catalyst derived from clusters. After this period, the amount of undesirable products increases rapidly because WGSR and methane reforming are increasingly inhibited with increase in reaction time. Similar results were found with Rh-based catalysts supported on ceria and alumina in microchannel reactors. The activity dropped after 40 h of reaction because of the separation of the layers of the catalyst from the stainless steel plate, sintering, or coke deposition.58 In a similar study,59 the stability of the catalyst in a channel microreactor at 500 ºC was evaluated. In this case, the catalyst was stable for 35 h, after which the conversion decreased gradually (loss of 20% over 25 h). In contrast, Cobo et al.17 reported that the Rh-Pt/La2O3 bimetallic catalyst remains active with an ethanol conversion of >99% for 120 h at 600 ºC. However, it must be noted here that the catalyst was regenerated with H2 when changes in product distribution were detected. Although the initial catalytic activity is recovered after regeneration with H2, Cobo et al.17 found that upon regenerating the catalyst in air, the catalytic activity can be completely recovered and the stability as well as the selectivity of the catalyst for H2 is increased. Simson et al.60 used air to regenerate the monolithic Rh-Pt catalyst previously used in the reforming sulfur-contaminated ethanol/gasoline mixtures; however, only a fraction of the active sites on the catalyst are regenerated. Regeneration was performed because after 33 h of reforming, ethanol convers drops from 100% to 84% and hydrogen production rates decrease by 29%. Preventive regeneration with air after every 5 h of reaction time prevents the loss of catalytic activity. The stability of Ni-based catalysts has also been the subject of studies and due of problems associated with its deactivation, the catalysts have been evaluated over shorter reaction times. Barroso et al.61 reported that that Ni/MgAl catalyst has an initial ethanol conversion of 100%, which decreases to 50% after 11 h of reaction at 650 ºC. Palma et al.62 studied the Pt-Ni/CeO2 catalyst at 450 ºC. Although, there are no appreciable changes in the distribution of products after 5 h of reaction, there is a significant drop in the pressure, which indicates coking of the catalyst. Elias et al.63 studied the effect of added Ca on Ni/Al2O3 catalyst. At 500 ºC, there is no noticeable deactivation for over 24 h (a conversion of 99% is maintained) after which a 0.01 mol/g of carbon deposition is reported. With respect to Co-based catalysts, Brum et al.48 obtained a total conversion of ethanol and stable distribution of products for 300 h with a Co-Ru(Na) catalyst. Oxidative activation cycles are performed by subjecting the catalyst to oxygen currents in air for 0.5 h. Co/ZnO doped with Na maintains a 100% conversion of ethanol and a H2 selectivity of 74.3% for 240 h.64 Co-based catalysts were also doped with Na hydrocalcites based on Co, and the tests were conducted at 550 ºC.65 The catalyst remain stable for 300 h while catalyzing the complete conversion of crude bioethanol. Araque et al.66 synthesized a Ce2Zr1.5Co0.47Rh0.07O8-d catalyst and conducted stability tests for 350 h at 550 ºC. During the first 50 h, the ethanol conversion is maintained at 100% and the distribution of products remain constant. However, at 153 h, a change is detected in the distribution of products and at 350 h, ethanol conversion decreases to 65%. The stability studies show that doped Rh-Pt- and Co-based catalysts are resistant to deactivation for periods greater than 100 h and that the processes of reactivation with air can be efficient for maintaining an active catalyst for long periods of time.15,48,65 In contrast, Ni-based catalysts are not stable for more than 10 h of reaction time as they incur coke deposition.61-63 Furthermore, the addition of new metals or alkaline elements is found to improve the properties of the catalyst and make it more stable.48,62,66
NEW TECHNOLOGIES Currently, newer systems for ethanol reforming are being investigated as promising options for H2 production. These are projected as completely new alternative technologies or as a complement for existing systems, such as reforming with steam or oxygen. Electrochemical ethanol reforming is a new technology that is performed in electrolytic cells with proton-exchange membranes. In addition, new reaction systems that include the use of membranes and plasma in the reactors have been implemented. Recently, membranes have been suggested to improve the separation processes.67 Park et al.68 developed a system of oxygen transport membranes called BSCF/Ag (Ba0.5Sr0.5Cu0.2Fe0.8O3-d/Ag) after their constituents. Such membranes supply the oxygen necessary to react with ethanol and produce H2 by autothermal reforming. The optimal conditions with a Rh-based catalyst is determined to be at 600 ºC. One of the most important feature of this system is the supply of high-purity oxygen, which increases ethanol conversion, and reduces coke formation (by increasing the yield of the catalyst). In this study, a conversion of 62% is obtained. However, a large amount of water is produced during the reaction, as part of the H2 produced is consumed by O2. The principal products are hydrogen, water, and acetaldehyde, which suggests that the dehydrogenation of ethanol is the dominant pathway. The functioning of this type of system is illustrated in Figure 2 and shows the entry of air and ethanol towards the membrane (where the reaction occurs) as well as the exit of the reaction products.
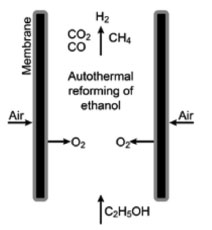 Figure 2. BSCF/Ag oxygen transport membranes
Using a similar approach, Seelam et al.69 explored the SRE in a reactor with a platinum-based membrane supported on stainless steel. The experiment was performed with two catalysts: Ni/ZrO2 and Co/Al2O3. They found that the optimal results are obtained with the cobalt catalyst at a pressure of 12 bar and a gas hourly space velocity (GHSV) of 800 h-1, which results in a H2 yield of 45% (95% purity of permeated H2) with a bioethanol conversion rate of 4%. The membrane activity was evaluated at 400 ºC with permeability tests at 2-4 bars of retentate pressure, which produces a flow of pure H2 of 18 mL/min for 1 h. Lim et al.70 studied the effect of pressure and H2 permeability in SRE with membranes based on palladium and silica with a Na-Co/ZnO catalyst. The experiments were performed at 623 K and pressures of 1, 5, and 10 atm in both packed-bed reactors (PBR) and membrane reactors (MR) for the sake of comparison. With an increase in the pressure, the conversion of ethanol decreases and molar flow of hydrogen increases in both systems. These conversion rates exceed those reported by other studies as at pressures of 1, 5, and 10 atm, they are 50%, 31%, and 23% in the PBR and 63%, 40%, and 34% in the MR, respectively. 66 In this study, deactivation of the Na-Co/ZnO catalyst is not detected; the ethanol conversion and molar flow of stable products is maintained for 6 h at each tested pressure. Electrochemical reforming is another novel technique developed for the production of hydrogen from ethanol. Caravaca et al.71 reported a study performed using an electrolysis cell with a proton-exchange membrane (PEM). The anode was a bimetallic Pt-Ru-based catalyst and the cathode was a Pt-based material. The optimum conditions for the process were determined to be 80 ºC and 6 M of ethanol (in a water-ethanol solution). It was determined that H2 production is assured with a voltage between 0.4 V and 0.9 V; this is less than that needed for electrolysis of water because H2 production is strictly a result of ethanol reforming. An important result of this system is that the only gaseous product detected at the cathode is H2; neither CO nor CO2 are detected in the cell. The low activity of Pt-Ru catalysts towards C-C bond cleavage at the tested reaction temperatures is the likely reason for the absence of carbon oxides in the cell. In addition, H2 production in the cell increases with temperature and electrical current. Different modifications to the reactor to improve its performance have been proposed. Structured catalysts or washcoated structures are most widely used. Bruschi et al.72 utilized a parallel-plate reactor with square channels (500-2000 µm) that are coated with a Pd catalyst. They installed a flue-gas heating system (GC) in co- and counter-current. Their results show that the yield of H2 is mainly linked to the supply of heat to the process by the GC, and the system in counter-current is most suitable when ethanol is fed in at low temperatures. The reactor yield decreases with increase in the wall thickness because of the decrease in thermal exchange. The ethanol conversion and hydrogen yield increases with inlet temperature. Finally, in the co-current configuration, an ethanol conversion of 28% is obtained, and the hydrogen yields are negligible for the counter-current configuration. This type of reactor is comparable to that utilized by Divins et al.,73 which consists of a micro-monolithic reactor that is 7 mm in diameter and is based on silicon that is coated with a Rh-Pd/CeO2 catalyst. In this reactor, complete conversion is observed and a mole of ethanol yields 3.8 moles of H2 at 600 ºC. These results surpass those with the conventional monolith of cordierite because of the increase in contact area per unit volume. Finally, Hu et al.74 investigated plasma reforming of ethanol for H2 production. The experiment was performed in a plasma discharge reactor with a dielectric barrier packed with 2-mm quartz beads that improved the inlet power. In this study, it was found that the reforming efficiency decreases with an increase in the flow rate. For the system with a power of 100 W at a flow rate of 5 mL/min in a 2.0-mm quartz bed, a 45% conversion is observed with 75% ethanol at the inlet. The new modifications proposed in ethanol reforming for H2 production mainly seek to decrease the production of undesirable byproducts, such as CO, in the outlet current and increase the energy efficiency of the reaction. However, these systems have not yet matched the conversion rates and yields of conventional systems, and require further studies to foster their effectiveness against complementary conventional methods.
CONCLUSIONS The catalytic steam reforming of ethanol is a promising route for the sustainable production of hydrogen for use in fuel cells. For this process, the catalyst must be stable, active, selective, and maximize hydrogen production while minimizing the generation of byproducts, such as CO and CH4. Addition of a metal dopant or precursor improves the activity of catalysts in most cases. These enhancements result from the effect of the dopant on the arrangement, area, and dispersion of the active phase, which often influences the selectivity and conversion in a catalytic reaction. Among the active metals, bimetallic (Rh-Pt) and Co-based catalysts supported on basic oxides (of Zr, Ce, and La) are stable (>100 h of reaction time) and have a high selectivity for H2, as these catalysts promote dehydrogenation instead dehydration. Ni-based catalysts are active, but due to the lower redox capacity of Ni when compared to that of noble metals, they are rapidly deactivated by the formation of coke. The new reactors for SRE, such as plasma, electrochemical, and membrane reactors as well as microreactors are not as efficient as traditional systems in the conversions of ethanol or the selectivity for H2. However, the low production of byproducts, leading to the production of H2-rich streams, positions them as a promising alternatives in SRE.
REFERENCES 1. Birot, A.; Epron, F.; Descorme, C.; Duprez, D.; Appl. Catal., B 2007, 79, 17. 2. Ni, M.; Leung, D.; Leung, M.; Int. J. Hydrogen Energy 2007, 32, 3238. 3. Costa-Serra, J. D.; Navarro, M.; Rey, F.; Int. J. Hydrogen Energy 2011, 37, 7101. 4. Pereira, A. L. S.; da Silva, C. N.; Afonso, J. C.; Quim. Nova 2011, 34, 145. 5. Haryanto, A.; Fernando, S.; Murali, N.; Adhikari, S.; Energy Fuels 2005, 19, 2098. 6. Vaidya, P. D.; Rodrigues, A.; Chem. Eng. J. 2005, 117, 39. 7. Ramírez de la Piscina, P.; Homs, N.; Chem. Soc. Rev. 2008, 37, 2459. 8. Santos, F. A.; Queiróz, J. H.; Colodette, J. L.; Fernandes, S. A.; Guimarães, V. M.; Rezende S. T.; Quim. Nova 2012, 35, 1004. 9. Tayade, P.; Sapkal, V.; Rode, C.; Sapkal, R.; International Journal of Advances in Engineering & Technology 2012, 3, 436. 10. Mariño, F.; Boveri, M.; Baronetti, G.; Laborde, M.; Int. J. Hydrogen Energy 2004, 29, 67. 11. Akande, A. J.; Idem, R. O.; Dalai, A. K.; Appl. Catal., A 2005, 287, 159. 12. Oliveira, R. L.; Passos, F. B.; Quim. Nova 2013, 36, 375. 13. Aupretre, F.; Descorme, C.; Duprez, D.; Casanave, D.; Uzio. D.; J. Catal. 2005, 233, 464. 14. Freni, S.; Cavallaro, S.; Mondello, N.; Spadaro, L.; Frusteri, F.; Catal. Commun. 2003, 4, 259. 15. Liu, S.; Chen, D.; Zhang, K.; Li, J.; Zhao, N; Int. J. Hydrogen Energy 2008, 33, 3736. 16. Frusteri, F.; Freni, S.; Chiodo, V.; Donato, D.; Bonura, G.; Cavallaro, S.; Int. J. Hydrogen Energy 2006, 31, 2193. 17. Cobo, M.; Pieruccini, D.; Abello, R.; Ariza, L.; Córdoba, L. F.; Conesa, J. A.; Int. J. Hydrogen Energy 2013, In press. 18. Simson, A.; Waterman, E.; Farrauto, R.; Castaldi, M.; Appl. Catal., B. 2009, 89, 58. 19. Llorca, J.; Homs, N.; Sales, J.; Ramírez de la Piscina, P.; J. Catal. 2002, 209, 306. 20. Haga, F.; Nakajima, T.; Yamashita, K.; Mishima, S.; React. Kinet. Catal. Lett. 1997, 63, 253. 21. Frusteri, F.; Freni, S.; Spadaro, L.; Chiodo, V.; Bonura, G.; Donato, S.; Cavallaro, S.; Catal. Commun. 2004, 5, 611. 22. Ciambelli, P.; Palma, V.; Ruggiero, A.; Appl. Catal., B. 2010, 96, 18. 23. De Lima, S. M.; Silva, A. M.; Da Cruz, I. O.; Jacobs, G.; Davis, B. H.; Mattos, L. V.; Noronha, F. B.; Catal. Today 2008, 138, 162. 24. Panagiotopoulou, P.; Velikios, X. E.; Int. J. Hydrogen Energy 2012, 37, 16333. 25. Gomes, W. S.; Silva, U. L. V.; Souza J. P. I.; Quim. Nova. 2013, 36, 507. 26. Soyal-Baltacıoğlu, F.; Aksoylu, A. E.; Önsan, Z. I.; Catal. Today 2008, 138, 183. 27. De Lima, S. M.; Da Silva, A. M.; Jacobs, G.; Davis, B. H.; Mattos, L. V.; Noronha, F. B.; Appl. Catal., B 2010, 96, 387. 28. Moura, J. S.; Souza, M. O. G.; Bellido, J. D. A.; Assaf, E. M.; Opportus, M.; Reyes, P.; Rangel, M.; Int. J. Hydrogen Energy 2012, 37, 3213. 29. Gutierrez, A.; Karinen, R.; Airaksinen, S.; Kaila, R.; Krause, A.O.I.; Int. J. Hydrogen Energy 2011, 36, 8967. 30. Diagne, C.; Idriss, H.; Pearson, K.; Gómez, M. A.; Kiennemann, A; C. R. Chim. 2004, 7, 617. 31. Chen, L.; Shin Choong, C. K.; Zhong, Z.; Huang, L.; Peng Ang, T.; Hong, L.; Lin, J.; J. Catal. 2010, 276, 197. 32. Le Valant, A.; Bion, N.; Can, F.; Duprez, D.; Epron, F.; Appl. Catal., B 2010, 97, 72. 33. Le Valant, A.; GBarron, A.; Bion, N.; Duprez, D.; Epron, F.; Int. J. Hydrogen Energy 2011, 36, 311. 34. Gutierrez, A.; Karinen, R.; Airaksinen, S.; Kaila, R.; Krause, A. O. I.; Int. J. Hydrogen Energy 2011, 36, 8967. 35. Bi, J.; Hong, Y.; Lee, C.; Yeh, C.; Wang, C.; Catal. Today 2007, 129, 322. 36. Sheng, P. Y.; Chiu, W. W.; Yee, A.; Morrison, S. J.; Idriss, H.; Catal. Today 2007, 129, 313. 37. Trane, R.; Dahl, S.; Skjøth-Rasmussen, M. S.; Jensen, A. D.; Int. J. Hydrogen Energy 2012, 37, 6447. 38. Chen, S. Q.; Liu, Y.; Int. J. Hydrogen Energy 2009, 34, 4735. 39. Liu, Q.; Liu, Z.; Zhou, X.; Li, C.; Ding, J.; J. Rare Earths 2011, 29, 872. 40. Akiyama, M.; Oki, Y.; Nagai, M.; Catal. Today 2012, 181, 4. 41. Frusteri, F.; Freni, S.; Chiodo, V.; Spadaro, L.; Di Blasi, O.; Bonura, G.; Cavallaro, S.; Appl. Catal., A 2004, 270, 1. 42. Profeti, L. P. R.; Habitzheuter, F.; Assaf, E. M.; Quim. Nova. 2012, 35, 510. 43. Roy, B.; Martinez, U.; Loganathan, K.; Datye, A. K.; Leclerc, C. A.; Int. J. Hydrogen Energy 2012, 37, 8143. 44. Sau, G. S.; Bianco, F.; Lanchi, M.; Liberatore, R.; Mazzocchia, C.; Spadoni, A.; Int. J. Hydrogen Energy 2010, 35, 7280. 45. Finocchio, E.; Rossetti, I.; Ramis, G.; Int. J. Hydrogen Energy 2013, 48, 3213. 46. María Koper, M. T.; García, G.; An. R. Soc. Esp. Fis. Quim. 2010, 106, 22. 47. Casanovas, A.; Roig, M.; De Leitenburg, C.; Trovarelli, A.; Llorca, J.; Int. J. Hydrogen Energy 2010, 35, 7690. 48. Brum Pereira, E.; Ramírez de la Piscina, P.; Homs, N.; Bioresour. Technol. 2011, 102, 3419. 49. Brum Pereira, E.; Ramírez de la Piscina, P.; Martí, N. Homs, S.; Energy Environ. Sci. 2010, 3, 487. 50. Liu, S.; Zhang, K.; Fang, L.; Li, Y.; Energy Fuels. 2008, 22, 1365. 51. Rass-Hansen, J.; Johansson, R.; Møller, M.; Hviid Christensen, C.; Int. J. Hydrogen Energy 2008, 33, 4547. 52. László, G.; Erdőhelyi, A.; In Catalysis for Alternative Energy Generation; Erdőhelyi, A., eds.; Springer: Budapest, 2012, p. 129. 53. Ayastuy, J. L.; Fernández-Puertas, E.; González-Marcos, M.P.; Gutiérrez-Ortiz, M.A.; Int. J. Hydrogen Energy 2012, 37, 7385. 54. Augusto, B. L.; Costa, L. O. O.; Noronha, F. B.; Colman, R. C.; Mattos, L. V.; Int. J. Hydrogen Energy 2012, 37, 12258. 55. Ecopetrol S. A.; Innova 2012, 8. 56. Koh, A. C. W.; Chen, L.; Kee Leong, W.; Peng Ang, T.; Johnson, B. F. G.; Khimyak, T.; Lin, J.; Int. J. Hydrogen Energy 2009, 31, 5691. 57. Cai, W.; Wang, F.; Daniel, C.; van Veen, A. C.; Schuurman, Y.; Descorme, C.; Provendier, H.; Shen, W.; Mirodatos, C.; J. Catal. 2012, 286, 137. 58. Rao Peela, N.; Kunzru, D.; Int. J. Hydrogen Energy 2011, 36, 3384. 59. Rao Peela, N.; Mubayi, A.; Kunzru, D.; Chem. Eng. J. 2011, 167, 578. 60. Simson, A.; Farrauto, R.; Castaldi, M.; Appl. Catal., B 2011, 106, 295. 61. Barroso, M. N.; Galetti, A. E.; Gomez, M. F.; Arrúa, L. A.; Abello, M. C.; Chem. Eng. J. 2013, 222, 142. 62. Palma, V.; Castaldo, F.; Ciambelli, P.; Iaquaniello, G.; Appl. Catal., B 2013, In press. 63. Elias, K. F. M.; Lucredio, A. F.; Assaf, E. M.; Int. J. Hydrogen Energy 2013, 38, 4407. 64. Llorca, J.; Homs, N.; Sales, J.; Fierro, J. L. G.; Ramírez de la Piscina, P.; J. Catal. 2004. 222, 470. 65. Espinal, R.; Taboada, E.; Molins, E.; Chimentao, R. J.; Medinac, F.; Llorca, J.; Appl. Catal., B 2012, 127, 59. 66. Araque, M.; Vargas, J. C.; Zimmermann, Y.; Roger, A. C.; Int. J. Hydrogen Energy 2011, 36, 1491. 67. Nunes, S. P.; Peinemann, K. V.; In Membrane technology in the chemical industry, Pereira, S.; Peinemann, K., eds.; Wiley-VCH: Cambridge, 2007, p. 3. 68. Park, C. Y.; Lee, T. H.; Dorris, S. E.; Park, J. H.; Balachandran, U.; J. Power Sources 2012, 214, 337. 69. Seelam, P. K.; Liguori, S.; Iulianelli, A.; Pinacci, P.; Calabrò, V.; Huuhtanen, M.; Keiski, R.; Piemonte, V.; Tosti, S.; De Falco, M.; Basile, A.; Catal. Today 2012, 193, 42. 70. Lim, H.; Gu, Y.; Oyama, S. T.; J. Membr. Sci. 2012, 396, 119. 71. Caravaca, A.; Sapountzi, F. M.; De Lucas Consuegra, A.; Molina Mora, C.; Dorado, F.; Valverde, J. L.; Int. J. Hydrogen Energy 2012, 37, 9504. 72. Bruschi, Y. M.; López, E.; Schbib, N. S.; Pedernera, M. N.; Borio, D. O.; Int. J. Hydrogen Energy 2012, 37, 14887. 73. Divins, N. J.; Lopez, E.; Rodriguez, A.; Vega, D.; Llorca, J.; Chem. Eng. Process. 2013, 64, 31. 74. Hu, Y. P.; Li, G.; Yang, Y.; Gao, X.; Lu, Z.; Int. J. Hydrogen Energy 2012, 37, 5691. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access