
 |
 |
 |
 |
 |
Artigo
|
|
| Determinação de bromo e iodo em alga marinha comestível por ICP-MS após decomposição por combustão iniciada por micro-ondas Bromine and iodine determination in edible seaweed by ICP-MS after digestion by microwave-induced combustion |
|
Marcia Foster MeskoI,*; Isis Gonçalves TorallesI; Marcelo Guerreiro CrizelI; Vanize Caldeira CostaI; Natanael Rodrigo Xavier PiresII; Claudio Martin Pereira de PereiraI; Rochele Sogari PicolotoIII; Paola Azevedo MelloIII
ICentro de Ciências Químicas, Farmacêuticas e de Alimentos, Universidade Federal de Pelotas, Campus Capão do Leão, 96900-010 Pelotas - RS, Brasil Recebido em 19/08/2013 *e-mail: marcia.mesko@pq.cnpq.br A method based on microwave-induced combustion (MIC) was applied for the decomposition of different types of edible seaweed (Nori, Hijiki and Wakame) for subsequent determination of bromine and iodine by inductively coupled plasma mass spectrometry (ICP-MS). Decomposition of 500 mg of each sample was achieved in less than 30 min. A single and diluted solution (150 mmol L-1 (NH4)2CO3) was used for the absorption of both analytes and a reflux step of 5 min was applied to improve analyte recoveries. Accuracy was evaluated using certified reference materials and agreement was between 103 and 108% for both elements. INTRODUÇÃO Historicamente, as algas marinhas comestíveis sempre fizeram parte da dieta alimentar de países orientais como Japão, China e Coreia do Sul. Contudo, com a migração de povos asiáticos para diferentes países do ocidente, este alimento marinho também passou a ser inserido na culinária ocidental, o que proporcionou um crescimento no consumo de macroalgas em um nível global.1 Como o bromo e o iodo, geralmente, estão presentes em níveis elevados em áreas próximas aos oceanos, as algas marinhas podem ser fontes de ambos os elementos. Assim, é extremamente relevante o estudo da concentração total de Br e I, principalmente em organismos de importância alimentar, pois dependendo da concentração presente no alimento, diferentes riscos à saúde humana podem ser observados.2-9 De um modo geral, é possível encontrar diversos relatos na literatura sobre a determinação de metais em diferentes tipos de algas,10-14 contudo trabalhos envolvendo a determinação de halogênios neste tipo de amostra ainda são escassos e, constituem assim, um desafio atual.15,16 A determinação de halogênios pode ser considerada uma tarefa difícil, em consequência da elevada volatilidade destes analitos e do risco de contaminação durante as etapas de preparo das amostras e de determinação. A possibilidade de perda dos analitos aumenta com a utilização de ácidos inorgânicos na etapa de decomposição, uma vez que o uso de ácidos em combinação com os halogênios pode levar à formação de substâncias voláteis.17,18 Entretanto, cabe salientar que existem trabalhos na literatura descrevendo a utilização de soluções ácidas no preparo de amostras de algas marinhas, previamente à determinação de I, embora nesses estudos não sejam fornecidos detalhes de como foi feita a avaliação da exatidão do método utilizado.19,20 Uma alternativa para o preparo de amostras visando à determinação de halogênios é o emprego de métodos baseados na combustão. Esses métodos possibilitam a conversão do carbono e hidrogênio presentes na amostra em seus respectivos produtos de oxidação e permitem o uso de soluções alcalinas que favorecem a formação de compostos não voláteis com os analitos, minimizando possíveis perdas por volatilização. A temperatura atingida na combustão permite a decomposição de praticamente todas as matrizes orgânicas, possibilitando a obtenção de baixos teores de carbono residual (RCCs). Com a redução do RCC alguns problemas de interferências na etapa de quantificação podem ser minimizados ou eliminados, uma vez que o carbono pode interferir no transporte e na ionização dos analitos quando a determinação é feita por ICP-MS.21-23 Dentre os métodos de combustão, cabe destacar a combustão iniciada por micro-ondas (MIC), uma vez que possibilita uma elevada frequência analítica, o mínimo consumo de reagentes, a obtenção de baixos valores de brancos e baixos limites de detecção (LDs), bem como o emprego de materiais inertes e que suportam elevadas pressões e temperaturas.15 De um modo geral, a MIC envolve a decomposição de amostras orgânicas em frascos fechados e pressurizados com oxigênio, onde a etapa de ignição se dá pela incidência da radiação micro-ondas em um pequeno pedaço de papel filtro, umedecido com uma solução de NH4NO3. Após a combustão da amostra, os analitos são absorvidos em uma solução apropriada, sendo que as soluções alcalinas têm sido descritas como as mais adequadas para o caso dos halogênios.18,24-28 Para a determinação da concentração total de Br e I, técnicas como a cromatografia de íons (IC),27 a espectrometria de emissão óptica com plasma indutivamente acoplado (ICP OES)26,29 e a espectrometria de massa com plasma indutivamente acoplado (ICP-MS)30,31 têm sido relatadas na literatura. No entanto, as técnicas de IC e ICP OES, em muitos casos, não fornecem limites de detecção (LDs) satisfatórios para a determinação de elementos em baixas concentrações.32 Desta forma, apesar da ICP-MS apresentar problemas devido à ocorrência de pronunciado efeito de memória para a determinação de alguns elementos, principalmente para I,33,34 exigindo, assim, que as amostras sejam tratadas com vistas a eliminar este tipo de problema, esta técnica tem sido amplamente aplicada para a determinação destes halogênios, pois apresenta vantagens como elevada sensibilidade, seletividade, capacidade multielementar e rapidez de análise.18,24,25,35,36 Neste sentido, tendo em vista as dificuldades inerentes ao preparo de amostras visando à posterior determinação de halogênios, a pequena quantidade de informações a cerca da concentração de Br e I em alimentos, bem como a carência de métodos que possibilitem a determinação destes elementos, este trabalho teve como objetivo desenvolver um método de preparo de amostra para a subsequente determinação de Br e I em diferentes tipos de algas marinhas comestíveis por ICP-MS.
PARTE EXPERIMENTAL Amostras Amostras de algas marinhas comestíveis dos tipos Nori (Porphyra spp.), Hijiki (Hizikia fusiforme) e Wakame (Undaria pinnatifida) foram escolhidas para este estudo. A alga do tipo Nori foi obtida no comércio de Pelotas/RS, e as algas Hijiki e Wakame foram obtidas no comércio de São Paulo/SP. Para cada tipo de alga, foram adquiridos diferentes lotes, os quais foram cominuídos e homogeneizados previamente ao desenvolvimento do método. Cabe destacar que as algas do tipo Nori e Wakame eram de origem chinesa e a alga do tipo Hijiki de origem japonesa. Instrumentação No procedimento de cominuição das amostras foi utilizado um moinho criogênico, modelo 6750 (Spex CertiPrep, EUA), e para a secagem das mesmas foi empregada uma estufa com circulação de ar, modelo 400/2ND (Nova Ética, Brasil). A água ultrapura, utilizada em todos os procedimentos, foi obtida a partir do equipamento Milli-Q® Gradient (Millipore Corp., USA). Para a decomposição das amostras por MIC foi utilizado um forno de micro-ondas Multiwave 3000® (Anton Paar, Áustria), do tipo cavidade, equipado com um rotor com capacidade para 8 frascos de quartzo, com volume interno de 80 mL. As amostras foram prensadas na forma de comprimidos utilizando uma prensa hidráulica, modelo Manual Hydraulic Press 15 ton (Specac, Inglaterra) e, posteriormente, foram dispostas dentro dos frascos de decomposição, suspensas por um suporte de quartzo. Um banho de ultrassom Odontobrás (modelo Ultrasonic Cleaner 1440D Série Evolution, Brasil) foi utilizado para a descontaminação do papel filtro usado como auxiliar no processo de combustão. O potencial hidrogeniônico (pH) das soluções obtidas após decomposição por MIC foi obtido por medida em pHmetro digital modelo pH21 (Hanna, Brasil). Para a determinação de Br e I foi utilizado um espectrômetro de massa com plasma indutivamente acoplado PerkinElmer SCIEX (modelo ELAN® DRC II, PerkinElmer, EUA), equipado com nebulizador pneumático Meinhard® tipo A e tocha com tubo injetor de 2 mm de d.i. Para a geração do plasma foi utilizado gás argônio com 99,996% de pureza (White Martins, Brasil). As condições utilizadas para a determinação de Br e I por ICP-MS estão descritas na Tabela 1.

Reagentes e soluções Todos os reagentes utilizados no trabalho foram de grau analítico (Merck, Alemanha) e todas as soluções foram preparadas com água ultrapura (18,3 MΩ cm). Os sais de iodeto de potássio (KI) e brometo de potássio (KBr) foram utilizados para preparar as soluções das curvas de calibração para a determinação de Br e I por ICP-MS. As faixas de concentração empregadas foram de 1 a 10 µg L-1 para Br e 0,1 a 1 µg L-1 para I. Cabe ressaltar que as curvas de calibração foram preparadas em presença de carbonato de amônio ((NH4)2CO3) em concentração próxima à concentração final das amostras, após serem diluídas. Nitrato de amônio (NH4NO3) 6 mol L-1 foi utilizado para promover a ignição das amostras no procedimento de combustão. Para a decomposição por MIC, os frascos foram pressurizados com oxigênio comercial (White Martins, Brasil) e a solução de (NH4)2CO3 foi utilizada para absorver os analitos. Ácido nítrico (HNO3) 65% m/m foi utilizado na descontaminação dos frascos de quartzo. Os papéis filtro (Black Ribbon Ashless, Schleicher & Schüll GmbH, Alemanha) utilizados no procedimento de combustão foram descontaminados com álcool etílico 20% (v/v), imersos por 20 min em banho de ultrassom e, posteriormente, lavados com água ultrapura e secos em capela de fluxo laminar por 2 h. O material de referência certificado (CRM) BCR 150 Skim milk powder (Community Bureau of Reference, Bélgica) e o material de referência (RM) NIST 8414 Bovine Muscle (National Institute of Standards and Technology, Estados Unidos) foram utilizados para a avaliação da exatidão do método. Preparo das amostras utilizando combustão iniciada por micro-ondas (MIC) Previamente à decomposição por MIC, os três tipos de algas marinhas comestíveis foram primeiramente cominuídos em moinho criogênico e, posteriormente, secas em estufa a 60 ºC, durante 4 h. No procedimento de decomposição por MIC, foi necessário preparar as amostras na forma de comprimidos de 500 mg, com 13 mm de diâmetro, utilizando uma prensa hidráulica (8 ton, durante 1 min). Os comprimidos foram dispostos na base dos suportes de quartzo contendo um disco de papel filtro (15 mm de diâmetro, 12 mg) umedecido com 50 µL de NH4NO3 6 mol L-1.27 Os suportes foram colocados no interior dos frascos de quartzo, que continham em seu interior 6 mL de solução absorvedora. Posteriormente, os frascos de quartzo foram fechados e pressurizados com 20 bar de O2 durante 1 min. O programa de irradiação utilizado para a decomposição das amostras por MIC foi: i) 1400 W, por 5 min (etapa de combustão, seguida de refluxo), ii) 0 W, por 20 min (etapa de resfriamento).22 Depois do procedimento de combustão, as soluções obtidas foram analisadas por ICP-MS. Avaliação da concentração da solução absorvedora para Br e I Neste trabalho, (NH4)2CO3 foi utilizado para o preparo das soluções absorvedoras avaliadas. Este foi selecionado devido ao fato de ter sido empregado para a absorção de Br e I em estudos anteriores, nos quais os autores relatam que recuperações acima de 95% foram obtidas para ambos os elementos.30,31Assim, para a otimização do método, foram avaliadas 5 concentrações de solução absorvedora de (NH4)2CO3: 25, 50, 100, 150 e 200 mmol L-1, mediante a realização de ensaios de recuperação para iodo (40 µg g-1 de I). Cabe salientar que o ensaio de recuperação só foi feito para I, tendo em vista que este elemento apresenta o caráter mais ácido dentre os halogênios e, sob este aspecto, apresenta a condição de estabilidade mais extrema em meio aquoso.37 Estes ensaios foram desenvolvidos utilizando a alga marinha do tipo Nori, dada a maior facilidade de aquisição da mesma. Além disso, para avaliar a exatidão do método proposto foi feita também a decomposição do CRM BCR 150 e do RM NIST 8414, sob as mesmas condições das amostras. A determinação de Br e I por ICP-MS também foi feita nas algas marinhas Hijiki e Wakame utilizando a solução de (NH4)2CO3 na concentração selecionada, a partir dos ensaios realizados com a alga do tipo Nori. Cabe destacar que foi determinado o pH de todas soluções absorvedoras obtidas, após as decomposições. Este procedimento foi realizado com o intuito de relacionar as recuperações de I com possíveis variações deste potencial, tendo em vista que em pH baixo podem ser formadas espécies voláteis com estes analitos.
RESULTADOS E DISCUSSÃO Otimização da concentração da solução absorvedora para Br e I Para estabelecer a solução absorvedora mais adequada para a retenção de Br e I foram estudadas as concentrações de 25, 50, 100, 150 e 200 mmol L-1 de (NH4)2CO3 como solução absorvedora. A determinação dos analitos foi feita por ICP-MS e os resultados obtidos a partir deste estudo encontram-se apresentados na Figura 1.
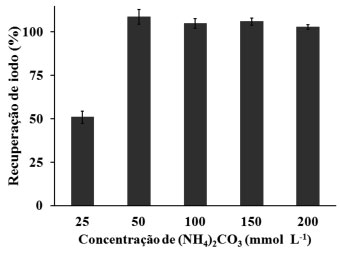 Figura 1. Valores percentuais de recuperação de iodo por ICP-MS em amostras de alga marinha do tipo Nori, utilizando diferentes concentrações de solução de (NH4)2CO3 para a absorção do analito (os valores estão representados pela média ± desvio padrão, n=3)
Como pode ser verificado na Figura 1, com exceção da solução absorvedora na concentração de 25 mmol L-1, todas as concentrações avaliadas, apresentaram recuperações satisfatórias (entre 103 e 109%) para o I, com desvios padrão relativos (RSDs) inferiores a 4%. Na concentração de 25 mmol L-1, a recuperação de I foi de 51% e o RSD de 7%. Esta baixa recuperação para I pode estar associada ao caráter significativamente ácido da solução obtida após decomposição por MIC (pH 3,5), o que não foi verificado para as demais concentrações de solução absorvedora avaliadas (de 50 a 200 mmol L-1), cujos valores de pH obtidos foram sempre maiores que 5,5. Cabe ressaltar que existem relatos na literatura18,25,33,38,39 informando sobre a possibilidade da ocorrência de perdas de I por volatilização na forma de HI e I2, na presença de meio ácido. Além disso, cabe destacar que a utilização de meio alcalino como solução absorvedora contribui para a formação de compostos não-voláteis com o iodo e reduz o efeito de memória comumente observado para este elemento durante as determinações por ICP-MS. O efeito de memória ocorre, principalmente, na câmara de nebulização e nas paredes das tubulações que transportam as soluções até o nebulizador.30,33,40 Embora tenham sido obtidas boas recuperações para I quando utilizadas as soluções de 50 e 100 mmol L-1 de (NH4)2CO3, optou-se por utilizar como solução absorvedora a concentração de 150 mmol L-1, tendo em vista que o RSD obtido para a mesma foi de 2%. Além disso, o pH dos digeridos com esta solução foi em torno de 8,0, o que possivelmente ocasiona uma maior estabilidade do analito em solução e minimiza o efeito de memória durante as determinações por ICP-MS. A partir disto, a concentração de 150 mmol L-1 foi escolhida como a melhor solução absorvedora para Br e I na decomposição por MIC e posterior determinações por ICP-MS. A exatidão do método proposto foi avaliada pela decomposição do CRM BCR 150 e do RM NIST 8414 e subsequente determinação de Br e I por ICP-MS, utilizando (NH4)2CO3 150 mmol L-1 como solução absorvedora. Os resultados obtidos estão apresentados na Tabela 2.
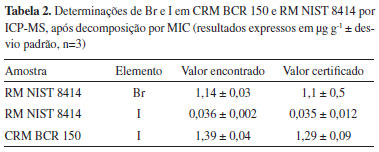
Conforme pode ser observado na Tabela 2, os valores obtidos para as concentrações de Br e I no RM NIST 8414 e de I no CRM BCR 150, após determinações por ICP-MS, foram concordantes com os respectivos valores informados e certificados (104 e 103%, para Br e I, respectivamente, no RM NIST 8414 e de 108% para I no CRM BCR 150), demonstrando que o método apresentou boa exatidão. Além disso, foi possível obter RSDs menores que 6%, o que comprovou que o método proposto apresenta também uma precisão adequada. No entanto, tendo em vista a ausência de um CRM com uma matriz semelhante a da amostra e que possua valores certificados e/ou informados para Br e I, fez-se um estudo adicional com o intuito de complementar a avaliação da exatidão do procedimento de decomposição por MIC utilizando (NH4)2CO3 150 mmol L-1 como solução absorvedora. Para tanto, foi feita a adição do RM NIST 8414 (Bovine muscle) à matriz da amostra para a decomposição por MIC. As recuperações encontradas para Br e I foram superiores a 95%. Os resultados obtidos nesse estudo, em conjunto com resultados da decomposição do CRM e do RM por MIC e dos ensaios de recuperação, realizados anteriormente, indicaram que o método proposto apresenta exatidão satisfatória para decomposição de algas marinhas comestíveis e posterior determinação de Br e I por ICP-MS. Determinação de Br e I em algas marinhas comestíveis Para demonstrar a aplicabilidade do método proposto, o mesmo foi empregado para a decomposição das algas comestíveis do tipo Nori, Wakame e Hijiki e, subsequente determinação de Br e I. As concentrações obtidas para estas amostras estão apresentadas na Tabela 3.

Com base nos resultados apresentados na Tabela 3, pode-se verificar que as concentrações de Br e I, nas diferentes algas marinhas analisadas, variaram de 70,6 a 396 µg g-1 e de 12,5 a 438 µg g-1, respectivamente. Tendo em vista que o pH dos digeridos obtidos a partir das algas avaliadas manteve-se levemente alcalino (7,6), independente da concentração dos analitos, pode-se constatar que o uso da solução absorvedora na concentração de 150 mmol L-1 permitiu que o método desenvolvido fosse aplicado para decomposição de algas comestíveis com diferentes faixas de concentração de Br e I. Ao pesquisar outros trabalhos na literatura,19,20,41-43 verificou-se que, conforme apresentado na Tabela 4, vários autores também encontraram uma ampla faixa de variação para a concentração de Br e I quando avaliados os mesmos tipos de algas marinhas utilizadas neste estudo. Diante disso, cabe ressaltar a importância do desenvolvimento de um método para a quantificação de Br e I em algas marinhas comestíveis, de um modo exato e preciso, em uma ampla faixa de concentração.
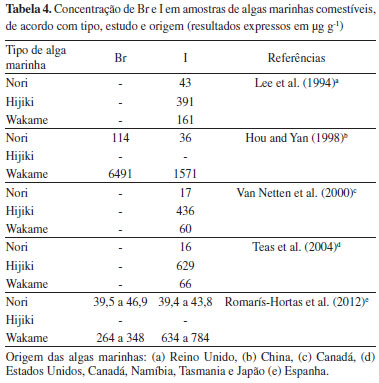
No trabalho de Lee et al.,41 as variações de concentração obtidas para I foram justificadas em função da idade, condição, parte da planta analisada e origem da alga. Provavelmente, as variações das concentrações encontradas para Br também sejam decorrentes dos mesmos fatores citados por estes autores, visto que Br e I possuem características químicas semelhantes. Adicionalmente, cabe destacar que, de um modo geral, as concentrações obtidas nos estudos citados na Tabela 4, podem estar relacionadas, em parte, com os métodos usados para o preparo das amostras para a determinação de halogênios, tendo em vista que muitos não apresentam dados referentes à avaliação da exatidão dos resultados. Dentre os trabalhos citados, apenas o desenvolvido por Romarís-Hortas et al.43 utilizou solução alcalina no procedimento de preparo das amostras (extração alcalina assistida por radiação micro-ondas empregando hidróxido de tetrametilamônio - TMAH) e posterior determinação por ICP-MS. Cabe enfatizar que existe uma grande dificuldade em utilizar os procedimentos de extração alcalina para amostras orgânicas e posterior determinação de halogênios por ICP-MS, tendo em vista que parte da amostra permanece nas soluções obtidas, podendo causar interferências durante a etapa de determinação. Assim, previamente à determinação faz-se necessária a realização de diluições com o intuito de evitar possíveis interferências no transporte e ionização dos analitos, o que pode comprometer o LD do método. O elevado RCC nas amostras ainda pode ocasionar a necessidade de uma limpeza da interface do equipamento e que a calibração tenha de ser feita com maior frequência. Contudo, é importante destacar que usando a MIC para decomposição de algas marinhas comestíveis foi possível utilizar uma solução em torno de 10 vezes mais diluída ((NH4)2CO3 150 mmol L-1) quando comparada com a empregada para a extração dos analitos no trabalho citado anteriormente (TMAH 1,4 mol L-1). Cabe mencionar ainda que com a MIC foi possível utilizar uma massa de amostra pelo menos 5 vezes maior na decomposição (aproximadamente 500 mg) quando comparada com as massas utilizadas em outros trabalhos reportados na literatura (50 a 100 mg).19,42,43 Os LDs e limites de quantificação (LQs) do método proposto foram mensurados e, para tanto, foram consideradas as relações 3s (n=10), e 10s (n=10), respectivamente.44 Os LDs obtidos para Br e I foram 0,011 e 0,010 µg g-1, respectivamente. Por sua vez, os LQs obtidos foram 0,025 µg g-1 para Br e 0,014 µg g-1 para I. Os LDs e LQs obtidos pelo método proposto foram menores que os relatados na literatura por outros métodos empregados para a determinação de Br e I em algas comestíveis, assim, o mesmo mostra-se mais adequado para a determinação dos analitos em nível de traços.19,20,41-43 É importante destacar que para facilitar as determinações de Br e I nas diferentes algas por ICP-MS utilizou-se diferentes diluições previamente as análises das amostras (Nori: 200 vezes, Hijiki: 400 vezes e Wakame: 1000 vezes). Com estas diluições foi possível utilizar a mesma curva de calibração para as diferentes amostras e minimizar o efeito de memória, reduzindo assim o tempo de lavagem do sistema entre as determinações.
CONCLUSÕES De uma maneira geral, a MIC mostrou-se adequada para a decomposição de diferentes tipos de algas marinhas para posterior determinação de Br e I por ICP-MS. Com a MIC foi possível obter recuperações quantitativas utilizando 150 mmol L-1 de (NH4)2CO3, como solução absorvedora para os analitos. Desta forma, a MIC possibilitou a utilização de soluções diluídas como solução absorvedora para Br e I, bem como a diminuição de quantidades excessivas de reagentes, reduzindo os valores dos brancos e quantidade de resíduos, conforme preconizado pela química verde. Com o uso da MIC foi possível decompor até 500 mg de algas marinhas comestíveis com uma frequência analítica adequada (8 decomposições simultâneas), com um tempo total de decomposição de 25 min (5 min de decomposição + 20 min de resfriamento do sistema), o que torna esse método adequado para posterior determinação de Br e I em alga marinha comestível por ICP-MS.
AGRADECIMENTOS Os autores agradecem à FAPERGS, a CAPES, ao CNPq, a L'Oreal-Unesco-ABC (Prêmio para Mulheres na Ciência) pelo apoio financeiro para desenvolvimento do projeto e ao Laboratório de Análises Químicas Industriais e Ambientais, da Universidade Federal de Santa Maria - Santa Maria, RS, pela disponibilidade de desenvolvimento de parte das análises realizadas neste trabalho.
REFERÊNCIAS 1. Mchugh, D. J.; A guide to the seaweed industry, FAO: Roma, 2003. 2. Wuilloud, R.; Altamirano, J. Em Handbook of elemental speciation II - Species in the environment, food, medicine and occupational health; Cornelis, R. Caruso, J.; Crews, H.; Heumann, K., eds.; John Wiley & Sons, Ltd: England, 2005, cap. 2.24. 3. Kurokawa, Y.; Maekawa, A.; Takahashi, M.; Hayashi, Y.; Environ. Health Perspectives 1990, 87, 309. 4. Vobecky, M.; Babicky, A.; Lener, J.; Biol. Trace Elem. Res. 1996, 55, 215. 5. Van Dokkum, W.; De Vos, R. H.; Muys, T.H.; Wesstra, J.A.; Br. J. Nutr. 1989, 61, 7. 6. Chaney, S. G. Em Manual de Bioquímica com Correlações Clínicas; Chaney, S. G.; Devlin, T. M., ed.; Blücher: São Paulo, 2007, cap. 28. 7. Czajka-Narins, D. M. Em Krause: alimentos, nutrição & dietoterapia; Czajka-Narins, D. M.; Kathleen, M. L.; Escott-Stump, S., eds.; Roca: São Paulo, 1998, cap. 7. 8. Granner, D. K. Em Haper: Bioquímica; Murray, R. K.; Granner, D. K.; Mayes, P. A.; Rodwell, V. W., eds.; Atheneu: São Paulo, 1998, cap. 46. 9. World Health Organization. Trace elements in human nutrition and health. WHO: Geneva, 1996. 10. Domínguez-González, R.; Moreda-Piñeiro, A.; Bermejo-Barrera, A.; Bermejo-Barrera, P.; Talanta 2005, 66, 937. 11. Besada, V.; Andrade, J. M.; Schultze, F.; González, J. J.; J. Marine Syst. 2009, 75, 305. 12. Domínguez-González, R.; Romarís-Hortas, V.; García-Sartal, C.; Moreda-Piñeiro A.; Barciela-Alonso, M. C.; Bermejo-Barrera, P.; Talanta 2010, 82, 1668. 13. Brito, G. B.; Souza, T. L.; Bressy, F. C.; Moura, C W. N.; Korn, M. G. A.; Mar. Pollut. Bull. 2012, 64, 2238. 14. Souza, P. O.; Ferreira, L. R.; Pires, N. R. X.; Filho, P. J. S.; Duarte, F. A.; Pereira, C. M. P.; Mesko, M. F.; Braz. J. Pharmacogn. 2012, 22, 825. 15. Magalhães, C. E. C.; Flores, E. M. M.; Krug, F. J.; Barin, J. S.; Mesko, M. F. Em Métodos de preparo de amostras: Fundamentos Sobre Preparo de Amostras Orgânicas e Inorgânicas para Análise Elementar; Krug, F. J., ed.; CENA/USP: Piracicaba, 2010, cap. 6. 16. Krug, F. J.; Nóbrega, J. A. Em Fundamentos Sobre Preparo de Amostras Orgânicas e Inorgânicas para Análise Elementar; Krug, F. J., ed.; CENA/USP: Piracicaba, 2010, cap. 1. 17. Sulcek, Z.; Povondra, P.; Methods of Decomposition in Inorganic Analysis, 1ª Ed, CRC Press: Boca Raton, 1989. 18. Knapp, G.; Maichin, B.; Fecher, P.; Hasse, S.; Schramel, P.; Fresenius J. Anal. Chem. 1998, 362, 508. 19. Teas, J.; Pino, S.; Critchley, A.; Braverman, L. E.; Thyroid 2004, 14, 836. 20. Van Netten, C.; Cann, S. A. H.; Morley, D. R.; Van Netten, J. P.; Sci. Total Environ. 2000, 255, 169. 21. Barin, J. S.; Flores, E. M. M.; Knapp, G. Em Trends in Sample Preparation; Arruda, M. A. Z., ed.; Nova Science Publishers: New York, 2006, cap. 3. 22. Flores, E. M. M.; Barin, J. S.; Mesko, M. F.; Knapp, G.; Spectrochim. Acta, Part B. 2007, 62, 1051. 23. Grindlay, G.; Mora, J.; Loos-Vollebregt, M.; Vanhaecke, F.; Spectrochim. Acta, Part B. 2013, 86, 42. 24. Vanhoe, H.; Allemeersch, F.; Versieck, J.; Dams, R.; Analyst 1993, 118, 1015. 25. Gélinas, Y.; Krushevska, A.; Barnes, R. M.; Anal. Chem. 1998, 70, 1021. 26. Oliveira, A. A; Nóbrega, J. A.; Pereira-Filho, E. R.; Trevizan, L. C.; Quim. Nova 2012, 35, 1299. 27. Flores, E. M. M.; Mesko, M. F.; De Moraes, D. P.; Pereira, J. S. F.; Mello, P. A.; Barin, J. S.; Knapp, G.; Anal. Chem. 2008, 80, 1865. 28. Nóbrega, J. A.; Santos, M. C.; Sousa, R. A.; Cadore, S.; Barnes, R. M.;Tatro, M.; Spectrochim. Acta Part B. 2006, 61, 465. 29. Naozuka, J.; Da Veiga, M. A. M. S.; Oliveira, P. V.; Oliveira, E.; J. Anal. Atom. Spectrom. 2003, 18, 917. 30. Mesko, M. F.; Mello, P. A.; Bizzi, C. A.; Dressler; V. L.; Knapp, G.; Flores, E. M. M.; Anal. Bioanal. Chem. 2010, 398, 1125. 31. Müller, A. H.; Mello, P. A; Mesko, M. F.; Duarte, F. A.; Dressler, V. L; Müller, E. I.; Flores, E. M. M.; J. Anal. At. Spectrom. 2012, 27, 1889. 32. Shelor, C. P; Dasgupta, P. K.; Anal. Chim. Acta 2011, 702, 16. 33. Al-Ammar, A.; Reitznerová, E.; Barnes, R. M.; Fresenius J. Anal. Chem. 2001, 370, 479. 34. Todolí, J. L.; Mermet, J. M.; Spectrochim. Acta Part B. 1999, 54, 895. 35. Divjak, B.; Novic, M.; Goessler, W.; J. Chromatogr. 1999, 862, 39. 36. Rose, M.; Miller, P.; Baxter, M.; Aplleton, G.; Crews, H.; Croasdalle, M.; J. Environ. Monit. 2001, 3, 361. 37. Kotz, J. C.; Treichel, P. M.; Weaver, G. C.; Química Geral e Reações Químicas, 6ª Ed., Cengage Learning: São Paulo, 2012. 38. Fecher, P. A.; Goldmann, I.; Nagengast, A.; J. Anal. At. Spectrom. 1998, 13, 977. 39. Pereira, J. S. F.; Mello, P. A.; Moraes, D. P.; Duarte, F. A.; Dressler, V. L.; Knapp, G.; Flores, E. M. M.; Spectrochim. Acta, Part B. 2009, 64, 554. 40. Nóbrega, J. A.; Santos, M. C.; Sousa, R. A.; Cadore, S.; Barnes, R. M.; Tatro, M.; Spectrochim. Acta, Part B. 2006, 61, 465. 41. Lee, S.; Lewis, J.; Buss, D. H.; B. J. Nutr. 1994, 72, 435. 42. Hou, X.; Yan, X.; Sci. Total Environ. 1998, 222, 141. 43. Romarís-Hortas, V.; Bermejo-Barrera, P.; Moreda-Piñero, J.; Moreda-Piñero, A.; Anal. Chim. Acta 2012, 745, 24. 44. Taverniers, I.; De Loose, M.; Bockstaele, E. V.; Trends in Anal. Chem. 2004, 23, 535. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access