
 |
 |
 |
 |
 |
Revisão
|
|
| Sínteses totais das enoquipodinas Total syntheses of enokipodins |
|
Camila B. Nascimento; Fernando Macedo Jr.
Departamento de Química, Universidade Estadual de Londrina, 86057-970 Londrina - PR, Brasil Recebido em 17/09/2013 *e-mail: macedofc@uel.br Enokipodins comprises a family of sesquiterpenes isolated from the mushroom Flammulina velutipes. These substances are attractive synthetic targets due to either their biological potential or structural characteristics. Since their isolation, about a decade ago, a considerable number of successful total syntheses have been published. Both via a racemic or enantioselective approach, these studies describe original and creative synthetic routes, particularly concerning the methodology used for constructing the benzylic quaternary center of these natural products. In this brief review, we discuss the reported total synthesis of enokipodins, focusing on the strategy adopted for the quaternary stereogenic center. INTRODUÇAO Dentre os diversos organismos vivos, os fungos destacam-se por sua versatilidade em produzir compostos naturais complexos, muito dos quais apresentam reconhecida importância farmacológica. Flammulina velutipes é um fungo comestível da família Tricolomataceae (Hymenomycetes, Basidiomycota), popularmente conhecido no Japao como enokitake. Os estudos de compostos do seu corpo frutífero resultaram no isolamento de substâncias com açao antitumoral e imunomodulatória. A maioria destes compostos sao proteínas, polissacarídeos e glicoproteínas.1 Também foram isolados deste fungo substâncias como lectinas, esteróis e monoterpentrióis.2-4 Outras pesquisas demonstraram propriedade antimicrobiana do seu extrato, do qual foram isolados sesquiterpenos oxidados do tipo α-cupareno (Figura 1) denominados enoquipodina A (1), B (2a) C (3) e D (4a).5,6
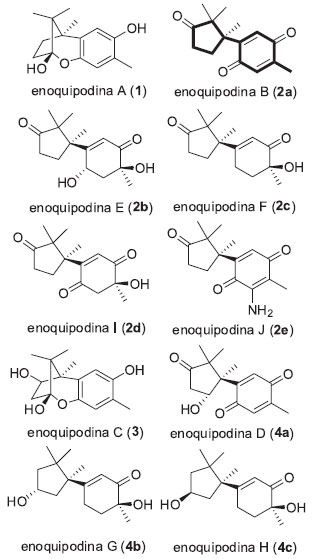 Figura 1. Estruturas químicas das enoquipodinas (núcleo cuparênico em destaque na estrutura 2a)
Estas substâncias apresentaram resultados promissores quanto a sua atividade antifúngica contra Cladosporium herbarum e quanto a sua capacidade de inibiçao do crescimento de bactérias gram-positivas de conhecida patogenicidade para seres humanos, como Bacillus subtilis e Staphylococcus aureus.5,6 A avaliaçao das doses inibitórias mínimas (DIM) mostrou que os valores para as enoquipodinas A e C para B. subtilis foram tao baixas quanto a do antibiótico penicilina G.7,8 Mais recentemente, foram relatados os isolamentos das enoquipodinas E à J.9 As enoquipodinas I (2d) e J (2e) apresentaram atividades antimicrobianas para B. subtilis (IC50 = 164,3 ± 6,2 µM e IC50 = 151,2 ± 4,5 µM, respectivamente), as enoquipodinas F (2c), I (2d) e G (4b) apresentaram atividades antifúngicas para o fungo Aspergillus fumigatus (IC50 = 78,6 ± 8,2 µM, 233,4 ± 3,8 µM e 235,1 ± 4,2 µM). Foram observadas ainda atividades antioxidantes (IC50 = 229,1 ± 3,6 µM para a enoquipodina J (2e) utilizando 2,2-difenil-1-picrilhidrazila (DPPH). Estruturalmente, as enoquipodinas apresentam interessantes desafios sintéticos, particularmente devido à presença de um sistema ciclopentânico contendo um centro estereogênico quarternário benzílico.
SINTESES DAS ENOQUIPODINAS As peculiaridades estruturais e o potencial biológico associados às enoquipodinas levaram Srikrishna e colaboradores a desenvolver uma proposta para a primeira síntese total de alguns destes sesquiterpenos.10-12 Estes autores descreveram a obtençao da (+/-)-enoquipodina A (1) partindo do 2,5-dimetoxitolueno (5) em 10 etapas (Esquema 1). A construçao do centro quaternário benzílico do intermediário 9 foi alcançada por meio de um rearranjo de Claisen do álcool alílico 8 utilizando éter etil-vinílico na presença de acetato mercúrico. Após a reaçao de Grignard do aldeído 9, o fechamento do anel de cinco membros foi realizado por metátese de ciclizaçao de olefinas resultando na obtençao do intermediário 11. A ciclopentanona 14 foi obtida após a oxidaçao, seguida por alquilaçao e hidrogenaçao de 11. Por fim, o tratamento de 14 com tribrometo de boro forneceu a (+/-)-enoquipodina A com 20% de rendimento global.
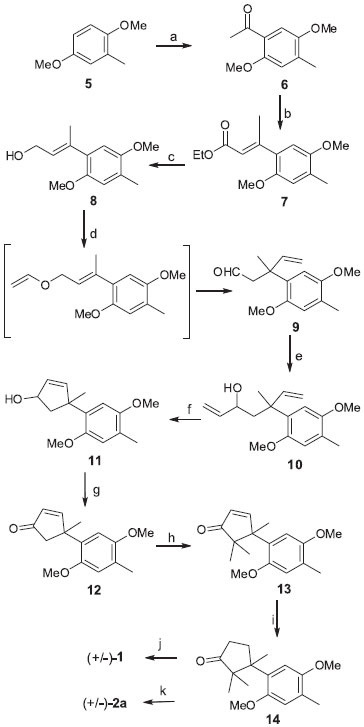 Esquema 1. Rota sintética racêmica para a enoquipodina A proposta por Srikrishna e colaboradores.10-12 Reagentes, condiçoes e rendimentos: (a) AcCl, CH2Cl2, SnCl4, 2 h, 0 ºC, t.a., 78%; (b) (EtO)2P(O)CH2COOEt, NaH, THF, refluxo, 5 h, 88%; (c) LiAlH4, Et2O, -70 °C, t.a., 2 h, 91%; (d) CH2=CHOEt, Hg(OAc)2, tubo selado, 100 °C, 10 h; 175 °C, 48 h, 63%; (e) CH=CHMgBr, THF, t.a., 1 h, 88%; (f) PhCH=Ru(Cl)2(PCy3)2 (5 mol %), CH2Cl2, t.a., 4 h, 95%; (g) PCC, NaOAc, CH2Cl2, t.a., 1 h, 86%; (h) NaH, THF-DMF, MeI, t.a., 12 h, 77%; (i) H2, Pd/C, EtOH, 1 h, 92%; (j) BBr3, CH2Cl2, 0 °C, t.a., 4 h, 78%; (k) NCA, CH3CN, H2O, t.a., 100%
Os autores também propuseram a obtençao da (+/-)-enoquipodina B (2a) através da clivagem oxidativa do intermediário 14 utilizando nitrato cérico de amônio (NCA). Como resultado de estudos sobre a obtençao de derivados ciclobutanóis, intermediários chave na síntese de alguns cuparenos, Secci e colaboradores propuseram uma nova rota sintética para a (+/-)-enoquipodina A (Esquema 2).13 A síntese tem como material de partida sulfeto de ciclopropilfenila 15 que após metalaçao, reage com a 4-metil-2,5-dimetoxiacetofenona fornecendo o carbinol quiral 16. O intermediário chave ciclobutanona 17 foi obtido a partir da reaçao de 16 com ácido p-toluenossulfônico (APTS) em quantidades equimolares. Destaca-se, ainda, uma outra expansao de anel envolvendo o rearranjo semi-pinacol do ciclobutanol 18 que levou à obtençao da ciclopentanona 14, utilizando tanto APTS como eterato de trifluoreto de boro. O produto final (+/-)-1 foi obtido em 5 etapas com 44% de rendimento global.
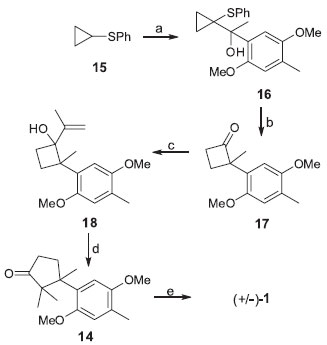 Esquema 2. Rota sintética racêmica para a enoquipodina A proposta por Secci e colaboradores.13 Reagentes, condiçoes e rendimentos: (a) i .n-BuLi, THF, -20 ºC; ii. 4-metil-2,5-dimetoxiacetofenona, THF, -20 ºC, t.a., 92%; (b) APTS, C6H6, refluxo, 91%; (c) brometo de isopropenilmagnésio, THF, -40 ºC, 71%; (d) APTS, C6H6, refluxo, 94% ou BF3 .OEt2, , CH2Cl2, -40 ºC, 91%; (e) BBr3, CH2Cl2, 0 °C a t.a., 4 h, 78%
Luján-Montelongo e Avila-Zágarra descreveram a obtençao da (+/-)-enoquipodina A (1) em 5 etapas (Esquema 3) baseando-se em estudos de reaçoes de carbociclizaçao regio e estereosseletivas de α-aril-δ-epoxinitrilas.14
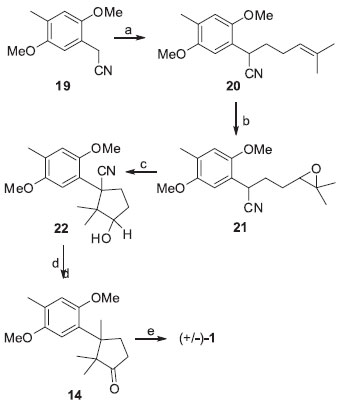 Esquema 3. Rota sintética racêmica para a enoquipodina A proposta por Luján-Montelongo e Avila-Zágarra.14 Reagentes, condiçoes e rendimentos: (a) i. HMDSLi, THF, -78 ºC, 30 min; ii. 5-iodo-2-metilpent-2-eno, THF, -78ºC, 3 h, 95%; (b) AMCPB, CH2Cl2, 0ºC, 3 h, 95%; (c) HMDSK, PdCl2, tolueno, 0-110 ºC, 1 h, 71%; (d) i. DIBAL-H, benzeno, 0ºC, 4 h; ii. HCl 10%, 30 min; iii. N2H4, H2O, EtOH, trietilenoglicol, 90-120 ºC, 45 min; iv. KOH, 150-220 ºC, 97%, 2,5 h; v. PCC, CH2Cl2, 0 ºC, 91%, 1,5 h; (e) iodeto de cicloexila, DMF, 150 ºC, 5 h, 93%
A síntese parte da 4-metil-2,5-dimetoxifenilacetonitrila (19) que, após alquilaçao com hexametildissilazana de lítio (HMDSLi) e 5-iodo-2-metilpent-2-eno, é convertida no intermediário com o centro quaternário benzílico 20. A α-aril-δ-epoxinitrila 21 foi obtida a partir da oxidaçao de 20 com ácido m-cloroperbenzóico. O derivado ciano-ciclopentílico 22 foi formado por meio da ciclizaçao regiosseletiva de 21 catalisada por paládio. A ciclopentanona 14 foi obtida após a reduçao de 22 com hidreto de diisobutilalumínio e oxidaçao de Dess Martin. O tratamento de 14 com iodeto de cicloexila forneceu a (+/-)-enoquipodina A (1) com 58% de rendimento global. Em 2004, Kuwahara e Saito propuseram a primeira rota de síntese total enantiosseletiva para as enoquipodinas A (1) e B (2a) (Esquema 4).15 A partir do 4-metil-2,5-dimetoxibenzaldeído (23), o intermediário quiral 26 foi sintetizado por meio de 7 etapas. Por sua vez, o centro estereogênico foi criado a partir de uma alquilaçao diastereosseletiva de 26 utilizando sec-butil-lítio e iodeto de metila. Com a remoçao do auxiliar quiral derivado do (S)-valinol, o intermediário 28 formado foi convertido na ciclopentenona 12 por meio de uma condensaçao aldólica intramolecular. A enoquipodina B (2a) foi obtida após dimetilaçao, seguida da hidrogenaçao e oxidaçao de 12. A reduçao de 2ª, utilizando hidrossulfito de sódio, permitiu ainda a obtençao da enoquipodina A (1) com 15% de rendimento global.
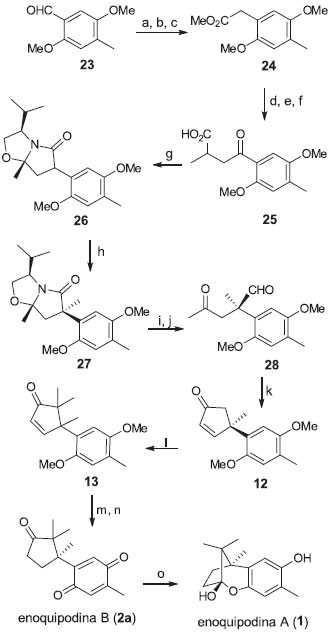 Esquema 4. Rota sintética enantiosseletiva para as enoquipodinas A e B proposta por Kuwahara e Saito.15,16 Reagentes, condiçoes e rendimentos: (a) C6H5C(=O)NHCH2CO2H, (CH3CO)2O, CH3COONa, 100 ºC, 2 h, 88%; (b) i. NaOH 10%, refluxo, 12 h, 88%, ii. NaOH 40%, H2O2 15%, 0 a 25 ºC, 12 h, 99%; (c) MeLi, NH4Cl, refluxo, 1 h, 53%; (d) DAL, 3-cloro-2-metilpropeno, NaI, THF-HMPA (7:1), -78 ºC, 3.5 h, 98%; (e) OsO4, NOM, NaIO4, THF-H2O (3:1), t.a., 18 h, 98%; (f) KOH(aq), THF, 50 ºC, 12 h, 98%; (g) (S)-valinol, tolueno, refluxo, 15 h, 85%; (h) s-BuLi, MeI, THF, -78 ºC, 2 h, 88%; (i) Red-Al, THF, 0 ºC a t.a., 1.5 h; (j) (n-Bu)4NH2PO4(aq) 1 mol L-1, EtOH, t.a., 24 h; (k) K2CO3, t-BuOH, refluxo, 4.5 h, 82%; (l) NaH, MeI, THF-HMPA (5:1), t.a., 17 h, 61%; (m) H2, Pd/C, EtOAc, t.a., 16 h; (n) NCA, CH3CN-H2O (3:2), t.a., 30 min, 79%; (o) Na2S2O4, EtOH-H2O, t.a., 30 min, 55%
Os mesmos autores relataram a obtençao das enoquipodinas C (3) e D (4a) em cinco e quatro etapas, respectivamente (Esquema 5).16 O epóxido 29 foi obtido a partir de uma epoxidaçao estereosseletiva de 13 utilizando peróxido de hidrogênio em meio alcalino. O composto 29 foi convertido em 30 a partir de uma clivagem redutiva utilizando disseleneto de difenila e boroidreto de sódio. Assim, a oxidaçao de 30 com nitrato cérico de amonio (NCA) levou à síntese da enoquipodina D (4a) com 4% de rendimento global. A enoquipodina C (3), por sua vez, foi sintetizada com 3,5% de rendimento global a partir da reduçao de 4a.
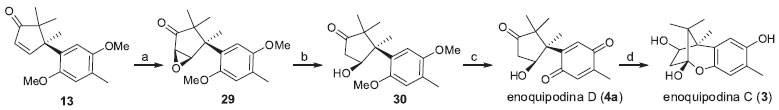 Esquema 5. Rota sintética enantiosseletiva para a enoquipodina C e D proposta por Kuwahara e Saito.16 Reagentes, condiçoes e rendimentos: (a) H2O2 30% aq., NaOH, MeOH, 60 ºC, 6,5 h, 51%; (b) PhSeSePh, NaBH4, EtOH, AcOH, t.a., 1 h, 68%; (c) NCA, CH3CN-H2O (3:2), t.a., 39 min, 78%; (d) Na2S2O4, EtOH-H2O, t.a., 30 min, 89%
Em 2009, Yoshida e colaboradores estabeleceram uma nova síntese enantiosseletiva para a obtençao das enoquipodinas A (1) e B (2a) (Esquema 6) a partir do 2,5-dimetoxitolueno.17 Destaca-se nesta abordagem a construçao do centro quartenário por meio da adiçao do ácido arilborônico 32 a um aleno quiral, catalisada por paládio para a obtençao de 33. Após um rearranjo de Enchenmoser-Claisen, o álcool alílico 33 foi convertido na amida 34. A reaçao de 34 com metil-lítio seguida pela clivagem oxidativa da ligaçao dupla forneceu o cetoaldeído 36, que foi transformado no intermediário ciclopentenona 12 a partir de uma condensaçao aldólica intramolecular. As enoquipodina A (1) e B (2a) foram obtidas a partir de 12 com 6% e 10% de rendimento global, respectivamente, utilizando a mesma metodologia descrita por Kuwahara e Saito.15,16
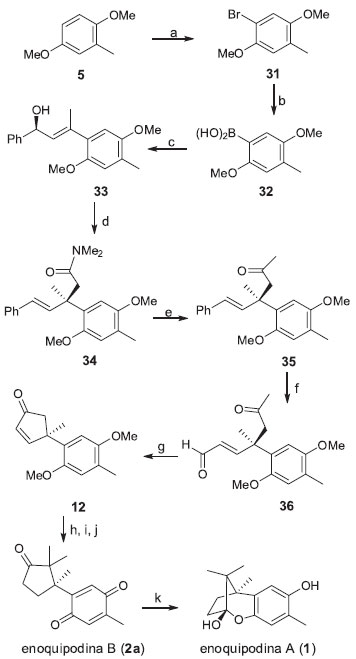 Esquema 6. Rota sintética enantiosseletiva para a enoquipodina A proposta por Yoshida e colaboradores.17 Reagentes, condiçoes e rendimentos: (a) Br2, NaOAc, AcOH, 1 h, 95%; (b) n-BuLi, B(OiPr)3, THF, -78 ºC, 30 min, 78%; (c) (S)-1-fenilbuta-2,3-dien-1-ol, [Pd2(OH)2(PPh3)4][BF4]2 (5 mol %) Et3N, dioxano/H2O (20/1), 60 °C, 30 min, 68%; (d) MeC(OMe)2NMe2, xileno, refluxo, 48 h. 80%, 91% e.e.; (e) MeLi; Et2O, -78 ºC, 1 h, 75%; (f) i. OsO4, NOM, acetona/H2O, t.a., 20 min; ii. Pb(OAc)4, CH2Cl2, 0 ºC, 2 h, 70%; (g) K2CO3, iBuOH, refluxo, 4 h, quant.; (h) NaH, MeI, THF-HMPA , t.a., 1 h, 57%; (i) H2, Pd/C, EtOAc, t.a., 22h, quant.; (j) NCA, CH3CN-H2O, 0 ºC, 20 min, 76%; (k) Na2S2O4, EtOH-H2O, t.a., 30 min, 58%
Recentemente, Frontier e colaboradores relataram a obtençao da enoquipodina B (2a) em apenas seis etapas a partir do 3-metoxi-4-metilbenzaldeído com 38% de rendimento global (Esquema 7).18 Os centros quaternários vicinais foram criados em uma única etapa por meio de um processo sequencial envolvendo uma ciclizaçao de Nazarov e um rearranjo de Wagner-Meerwein quimiosseletivo para a obtençao de 38. A transformaçao é mediada por um complexo de cobre (II) e um ligante quiral bidentado, cuja estrutura mostrou influenciar tanto os excessos enantioméricos da ciclizaçao quanto a quimiosseletividade do rearranjo catiônico. Embora baixos excessos enantioméricos tenham acompanhado a ciclizaçao (20% e.e.), a utilizaçao do ligante mostrado no esquema abaixo forneceu uma boa quimiosseletividade para a migraçao do grupo aromático em detrimento à migraçao de hidreto. O grupo carbometóxi foi removido pela reaçao de 38 com iodeto de sódio e ácido acético em diglima sob refluxo para a obtençao de 39. A desmetilaçao de 39 com tribrometo de boro proporciona a obtençao do intermediário 40 que foi facilmente convertido para enoquipodina B (2a) por oxidaçao com sal de Fremy.
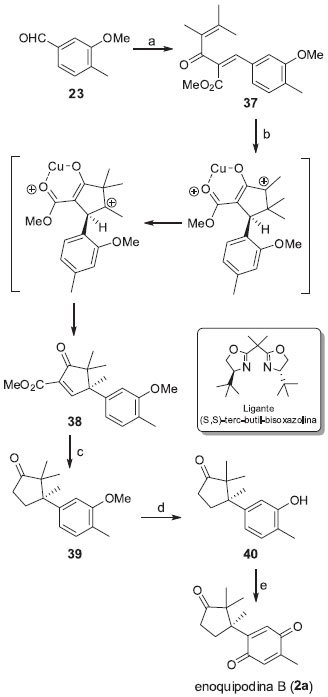 Esquema 7. Rota sintética enantiosseletiva para a enoquipodina B proposta por Frontier e colaboradores.18 Reagentes, condiçoes e rendimentos: (a) 3,4-dimetil-pent-3-en-2-ona, CH3COOH, piperidina, benzeno, refluxo, 92%; (b) [tBuBox-Cu[SbF6]2], CH2Cl2, refluxo, 72% (5:1, 20% e.e.); (c) NaI, CH-3COOH, diglima, refluxo, 30 min, quant.; (d) BBr3, CH2Cl2, 0 °C a t.a., 12 h, 64% (2 etapas); (e) Sal de Fremy, acetona, H2O, t.a., 12 h, 91%
CONCLUSAO As enoquipodinas sao substâncias importantes devido aos seus potenciais biológicos pouco explorados. Estruturalmente, esta classe de produtos naturais impoe desafios sintéticos, como a construçao de anéis ciclopentanos com um centro quaternário benzílico/alílico incorporado. Dentre as sínteses totais racêmicas das enoquipodinas, diferentes estratégias foram adotadas para a construçao de centros quaternários benzílicos/alílicos. Foram empregadas tanto reaçoes clássicas, como o rearranjo de Claisen proposto por Srikrishna e colaboradores10-12 e a alquilaçao da 4-metil-2,5-dimetoxifenilacetonitrila (Luján-Montelongo e Avila-Zágarra),14 quanto a metodologia descrita por Secci e colaboradores13 para a obtençao de ciclobutanóis envolvendo a reaçao de sulfeto de ciclopropilfenila com derivados da acetofenona. As abordagens enantiosseletivas bem sucedidas até o momento consistem na criaçao do centro quaternário por meio de alquilaçao diastereosseletiva (Kuwahara e Saito)15,16 ou pela adiçao de um ácido arilborônico a um aleno quiral catalisada por paládio (Yoshida e colaboradores).17 Neste contexto, destaca-se ainda como uma metodologia promissora a reaçao sequencial mediada por complexos quirais de cobre (ciclizaçao de Nazarov seguida pelo rearranjo de Wagner-Meerwein).18 Sejam racêmicas ou enantiosseletivas, as estratégias descritas até entao para a síntese das enoquipodinas inspiram o desenvolvimento de novas metodologias para a obtençao destas substâncias e convidam para o aperfeiçoamento de métodos de síntese de outros centros quaternários estruturalmente similares.
REFERENCIAS 1. Wasser, S. P.; Weis A. L.; Crit. Rev. Immunol. 1999, 19, 65. 2. Wang, H.; Ng, T. B.; Ooi, V. E.; Mycol. Res. 1998, 102, 897. 3. Yaoita, Y.; Matsuki, K.; Iijima, T; Nakano, S; Kakuda, R.; Machida, K.; Kikushi, M.; Chem. Pharm. Bull. 1998, 46, 944. 4. Hirai, Y.; Ikeda, M.; Murayama, T.; Ohata, T.; Biosci. Biotechnol. Biochem. 1998, 62, 1364. 5. Ishikawa, N. K.; Fukushi, Y.; Yamaji, K.; Tahara, S.; Takahashi, K.; J. Nat. Prod. 2001, 64, 932. 6. Ishikawa, N. K.; Yamaji, K.; Tahara, S.; Fukushi, Y.; Takahashi, K.; Phytochemistry 2000, 54, 777. 7. Ishikawa, N. K.; Yamaji, K.; Ishimoto, H.; Miura, K.; Fukushi, Y.; Takahashi, K.; Mycoscience 2005, 46, 39. 8. Melo, M. R.; Paccola-Meirelles, L. D.; Faria, T. J.; Ishikawa, N. K.; Mycoscience 2009, 50, 78. 9. Wang, Y.; Bao, L.; Yang, X.; Gao, H.; Li, L.; Li, S.; Gao, H.; Yao, X.; Liu, H.; Wen, H.; Food Chem. 2012, 132, 1346. 10. Srikrishna, A.; Rao, M. S.; Synlett 2004, 2, 374. 11. Srikrishna, A.; Lakshmi, B. V.; Ravikumar, P. C.; Tetrahedron Lett. 2006, 47, 1277. 12. Srikrishna, A.; Rao, M. S.; Indian J. Chem. 2010, 49B, 1363. 13. Secci, F.; Frongia, A.; Ollivier, J.; Piras, P. P.; Synthesis 2007, 7, 999. 14. Luján-Montelongo, J. A.; Avila-Zárraga, J. G.; Tetrahedron Lett. 2010, 51, 2232. 15. Kuwahara, S.; Saito, M.; Tetrahedron Lett. 2004, 45, 5047. 16. Saito, M.; Kuwahara, S.; Biosci. Biotechnol. Biochem. 2005, 69, 374. 17. Yoshida, M.; Shoji, Y.; Shishido, K.; Org. Lett. 2009, 11, 1441. 18. Lebœf, D.; Wright, C. M.; Frontier, A. J.; Chem. Eur. J. 2013, 19, 4835. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access