
 |
 |
 |
 |
 |
Revisão
|
|
| Proteases virais: importantes alvos terapêuticos de compostos peptideomiméticos Viral proteases: important targets of peptidemimetic compounds |
|
Estela Maris Freitas Muri*
Departamento de Tecnologia Farmacêutica, Faculdade de Farmácia, Universidade Federal Fluminense, Rua Mario Viana 523, Santa Rosa, 24241-000 Niterói - RJ, Brasil Recebido em 23/04/2013 *e-mail: estelamuri@yahoo.com.br Proteases catalyze the hydrolysis of peptide bonds of proteins and peptides to produce smaller peptides and free amino acids. These enzymes are involved in physiologic processes such as blood coagulation and cellular death, and are related to life cycle of several viruses, such as hepatitis C, dengue, and AIDS. These features make most of proteases very important therapeutic targets for new pharmaceutical compounds. The development of peptidemimetics with improved pharmacokinetic properties is driving extensive research in the field of viral protease inhibitors. The present paper aims to highlight the design and synthesis of peptidemimetics that are able to inhibit viral proteases related to hepatitis C, dengue, and AIDS. INTRODUÇAO Proteases representam uma classe de enzimas com importantes papéis em processos fisiológicos.1 Além disso, elas possuem aplicaçao comercial, estando entre os três maiores grupos de enzimas industriais, sendo responsáveis por 60% da venda internacional de enzimas.2 Estas enzimas estao envolvidas em processos biológicos essenciais, como a coagulaçao sanguínea, morte celular e diferenciaçao de tecidos, entre outros.3 Várias etapas proteolíticas importantes ocorrem no mecanismo invasivo de tumores,4 assim como no ciclo de infecçao de um grande número de vírus e microrganismos patogênicos.5 Estes fatos tornam as proteases um alvo quimioterápico valioso para o desenvolvimento de novos compostos farmacêuticos. Tradicionalmente as proteases ou peptidases sao enzimas que catalisam a hidrólise de ligaçoes peptídicas presentes em proteínas ou peptídeos, liberando peptídeos de tamanho variável ou aminoácidos livres.6 Na nomenclatura internacional de classificaçao de enzimas (EC), as peptidases pertencem à classe 3 e subclasse 3.4, que ainda está dividida em dois grandes grupos: as exo e as endopeptidases.7 As exopeptidases catalisam a quebra das ligaçoes peptídicas nas extremidades N ou C terminal das cadeias polipeptídicas e, assim, podem ser denominadas de aminopeptidases ou carboxipeptidases, respectivamente. As endopeptidases atuam preferencialmente nas regioes internas das cadeias polipeptídicas.8 As proteases podem ser classificadas também quanto à natureza química do sítio catalítico/mecanismo de açao, sao elas: i) serina proteases; ii) cisteína proteases; iii) aspártico proteases; iv) treonina protease e v) metaloproteases. Cada classe de proteases tem seu conjunto particular de aminoácidos no sítio ativo. As serina proteases apresentam a tríade catalítica composta por Ser, His e Asp, as cisteína proteases mostram os aminoácidos Cys, Asp e His, as aspártico proteases, Asp e Asp e as metaloproteases apresentam um íon metálico no sítio ativo.9 As proteases se ligam de maneira específica ao substrato e este modo de ligaçao é representado pelo sistema de nomenclatura descrito por Schechter e Berger.10 De acordo com este sistema, os resíduos de aminoácidos (ou cadeias laterais) do substrato sao nomeados da terminaçao N para a C, como: Pn......., P3, P2, P1, P1', P2', P3'........Pn' e os correspondentes sítios de ligaçao da enzima sao nomeados como: Sn......., S3, S2, S1, S1', S2', S3'........Sn' (Figura 1). A hidrólise da ligaçao peptídica ocorre entre P1 e P1' e a ligaçao é chamada "ligaçao cindível ou hidrolisável". A numeraçao dos resíduos é também dada a partir da ligaçao cindível e o resíduo P1 do substrato é chamado de resíduo de especificidade primária.10
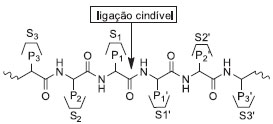 Figura 1. Sistema de nomenclatura de Schechter e Berger
A participaçao crucial de proteases nos ciclos de replicaçao de diversos vírus é hoje amplamente reconhecida, levando ao interesse de grandes indústrias farmacêuticas no desenvolvimento de inibidores destas enzimas.11
DENGUE A dengue é uma das doenças tropicais mais difundidas no mundo. A cada ano, cem milhoes de pessoas infectam-se com o vírus da dengue (também chamado de DENV), principalmente nas cidades e zonas urbanas tropicais, enquanto cerca de 2,5 bilhoes de pessoas sofrem com o risco de infecçao.12 A dengue é uma doença febril aguda, que pode ser de curso benigno ou grave, dependendo da forma como se apresenta: infecçao inaparente, dengue clássica (DC), febre hemorrágica da dengue (FHD) ou síndrome do choque da dengue (SCD). O vírus da dengue (do tipo RNA) é um arbovírus, com quatro sorotipos conhecidos, 1, 2, 3 e 4, sendo que a infecçao com um deles nao confere imunidade à infecçao pelo outro.13 Grandes esforços estao sendo feitos ao redor do mundo para o desenvolvimento de vacinas e fármacos contra o DENV. Desde a década de 1970, a Organizaçao Mundial de Saúde (OMS) tem patrocinado diversos estudos que ampliam os conhecimentos sobre a imunidade dos sorotipos e a fisiopatologia da FHD/SCD, que sao essenciais para o desenvolvimento de vacinas seguras e efetivas contra a dengue. Existem várias dificuldades para o desenvolvimento de uma vacina efetiva contra o DENV, mas o principal problema é que a infecçao por um sorotipo nao confere imunidade contra os demais, ou seja, há necessidade de uma vacina tetravalente, dificultando o seu desenvolvimento.14 Outro problema é a falta de um modelo animal para testes de efetividade das vacinas. Embora os modelos mais utilizados, ratos e macacos, sejam infectados pelo DENV, esses animais nao apresentam uma patologia semelhante à dos humanos, elevando assim os custos das pesquisas devido à dificuldade para se avaliar a proteçao vacinal.15 Devido à ausência de uma vacina ou droga eficaz contra o DENV, o tratamento utilizado até o momento é sintomático contra a febre e a reposiçao de fluido plasmático devido ao extravamento dos capilares nos pacientes com FHD e SCD.16 O ssRNA (RNA fita simples) do DENV codifica, além de diversas proteínas estruturais (C, E, prM), 7 proteínas nao-estruturais (NS1, NS2A, NS2B, NS3, NS4A, NS4B e NS5). Essas proteínas sao extremamente importantes para a formaçao do complexo de replicaçao viral. Além disso, duas regioes nao traduzidas (NTR) presentes em cada extremidade dos genomas sao importantes para as etapas de traduçao e transcriçao do genoma RNA.17 O primeiro evento de infecçao do DENV na célula hospedeira consiste na traduçao do RNA genômico em uma poliproteína. Com o processamento desta poliproteína viral, proteases da célula hospedeira e viral sao recrutadas. Para hidrólise das ligaçoes peptídicas da poliproteína na junçao entre C-prM, prM-E, E-NS1 e NS4A-NS4B é utilizada a peptidase sinalase encontrada no retículo endoplasmático (RE) das células hospedeiras, e para hidrólise das ligaçoes peptídicas na junçao entre NS2A-NS2B, NS2B-NS3, NS3-NS4A e NS4B-NS5 é utilizada a serina protease viral NS3. Já a hidrólise na junçao NS1-NS2A ocorre pela açao de uma protease ainda desconhecida localizada no RE.18,19 A atividade serina protease na porçao N-terminal da proteína NS3 dos flavivírus foi primeiramente proposta por Bazan e Fletterick20 e Gorbalenya e colaboradores.21 Em 1993, Falgout e colaboradores demonstraram que a atividade proteolítica da NS3 era dependente da proteína NS2B composta de 40 resíduos que funciona como cofator essencial para atividade catalítica. A NS3 protease do DENV, responsável por hidrólise de ligaçoes peptídicas, é considerada essencial ao processo de replicaçao e maturaçao do vírus da dengue. De acordo com a literatura, a NS3 protease é estruturalmente semelhante à tripsina e apresenta no seu sítio catalítico a tríade: His51 - Asp75 - Ser135.22 Hepatite C A hepatite é uma doença inflamatória que compromete os hepatócitos cuja evoluçao pode ser de portador assintomático até cirrose e carcinoma hepatocelular. Ela pode ser classificada como viral (causada por vírus), tóxica (causada pela reaçao ao álcool, drogas ou medicamentos) ou auto-imune, condiçao na qual ocorre a açao de células citotóxicas sobre o tecido hepático. Em 1989, o vírus da hepatite C (HCV) foi reconhecido como agente infeccioso responsável por formas adquiridas de hepatites diferentes de A e B, chamada entao de hepatite C, sendo hoje considerada uma doença silenciosa que infecta cerca de 3 milhoes de indivíduos somente no Brasil.23 O tratamento da hepatite C tem como objetivo a supressao sustentada da replicaçao do vírus, prevenindo a progressao da doença.24 O uso da terapia combinada, interferon-alfa (IFN-alfa) e ribavirina (RBV), trouxe melhores taxas de resposta viral sustentada em comparaçao com a monoterapia com IFN, introduzida nos anos 90.25 Em pacientes com HCV genótipo 1, o tratamento ideal é realizado com IFN peguilado (PEG-IFN) e RBV e nos pacientes com HCV genótipos 2 e 3, o tratamento pode ser realizado com IFN convencional e RBV. O processo de peguilaçao do IFN envolve a adiçao de polietilenoglicol (PEG), produzindo o PEG-IFN, o qual apresenta uma absorçao e eliminaçao mais lentas, permitindo que seja administrado uma vez por semana, em vez de três vezes, como ocorre com o IFN convencional.23 O ssRNA (RNA fita simples) do HCV codifica diversas proteínas estruturais (C, E1, E2 e p7) e seis proteínas nao-estruturais (NS2, NS3, NS4A, NS4B, NS5A e NS5B).26,27 O ciclo do HCV é semelhante ao do DENV, ou seja, após a liberaçao do RNA viral no citoplasma, o mesmo é traduzido em uma grande poliproteína precursora que se insere na membrana do RE. Nesta etapa ocorre a clivagem desta poliproteína pelas proteases do hospedeiro e pelas proteases virais NS2 e NS3/4A.28 A estrutura tridimensional do domínio NS3 protease do HCV, elucidada por cristalografia de raios-X, mostrou que ela possui dois subdomínios em folhas-b formando um barril típico de uma serina protease do tipo quimotripsina. Ela apresenta uma fenda de interaçao ao substrato localizada entre os subdomínios, importante para atividade proteolítica. Para que o processamento seja eficaz, a protease NS3 necessita de um cofator, o polipeptídeo NS4A, que é uma pequena proteína na qual alguns dos resíduos de aminoácidos hidrofóbicos que o compoem têm a funçao de ancorar o complexo enzimático na membrana exterior do RE.29 A NS3/4A apresenta no seu sítio catalítico a tríade formada por His57 - Asp81 - Ser139.30,31
AIDS Desde os primeiros casos descritos em 1981 (EUA), a AIDS espalha-se em sucessivas ondas de infecçao por todas as regioes do globo, apesar dos rápidos avanços científicos que foram alcançados.32 A infecçao pelo HIV resulta em uma doença crônica e progressiva que pode levar à destruiçao do sistema imunológico. A evoluçao da doença é caracterizada por uma elevada taxa de replicaçao viral, que leva à emergência de variantes virais mais virulentas.32 Até o momento, já foram descritos dois tipos de vírus: HIV-1 e HIV-2, sendo o HIV-1 o mais virulento e disseminado pelo mundo. Este apresenta elevada variabilidade genética denominada quasispecies, no qual o vírus é considerado como subpopulaçoes e nao um genoma único cuja variabilidade está em torno de 6%, podendo chegar a 50% entre indivíduos de regioes geográficas diferentes. Já o HIV-2 parece ser menos patogênico e é encontrado, quase que exclusivamente, no oeste da Africa.32 O HIV pertence ao grupo dos retrovírus citopáticos e nao-oncogênicos, o qual necessita, para se multiplicar, de uma enzima denominada transcriptase reversa. Esta enzima é responsável pela transcriçao do RNA viral em uma cópia de DNA, que pode, entao, integrar-se ao genoma do hospedeiro, através da enzima integrase. Possui também uma enzima chamada de HIV protease, fundamental para a quebra de unidades protéicas em pequenas moléculas, permitindo uma montagem viral perfeita.33,34 A protease do HIV-1 é responsável pela clivagem das proteínas precursoras em proteínas estruturais e enzimas que tem um papel essencial na replicaçao e maturaçao viral. Ela é um homodímero constituído de 2 monômeros com 99 resíduos cada, sendo seu sítio catalítico formado pela tríade catalítica: Asp-25, Thr-26 e Gli-27.35
MECANISMO CATALITICO DA SERINA PROTEASE Dentre os diversos tipos de proteases, as serina proteases sao, provavelmente, as mais cuidadosamente investigadas. Estas enzimas empregam a catálise ácido-básica geral e a catálise covalente para a sua açao, onde o mecanismo catalítico inicia-se na tríade catalítica característica, His57 - Asp81 - Ser139, que está localizada no sítio ativo da enzima (Esquema 1).36
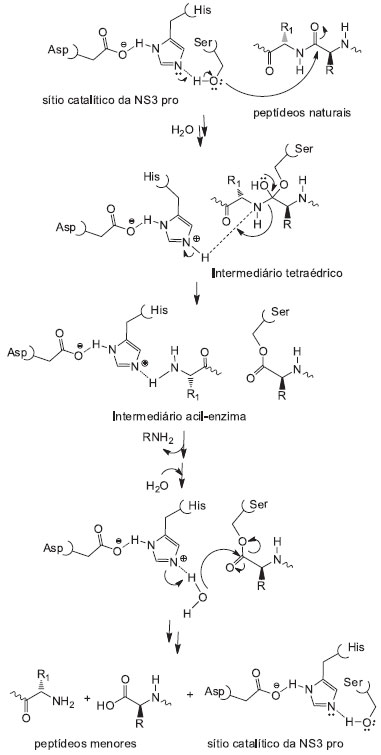 Esquema 1. Mecanismo catalítico da serina protease
A tríade é uma estrutura coordenada que consiste de três aminoácidos: histidina, aspartato e serina, onde cada aminoácido desempenha uma tarefa específica no processo de catálise. A cadeia lateral da Ser139 (-CH2-OH) é ativada por uma catálise básica mediada por Asp81 e His57 através de interaçoes por ligaçao hidrogênio (numeraçao dos resíduos referente à serina protease do HCV). A Ser do sítio ativo (nucleófilo ativado) ataca, entao, a ligaçao amídica do substrato polipeptídico, formando um intermediário oxiânion tetraédrico. Este intermediário se rompe pela eficiente transferência de próton da His para formar uma amina e um intermediário acil-enzima. A liberaçao da porçao N-terminal e a introduçao de uma molécula de H2O levam ao ataque da carboxila do substrato, formando o fragmento C-terminal do substrato e regenerando a enzima (Esquema 1).36
MECANISMO CATALITICO DA ASPARTIL PROTEASE Acredita-se que o mecanismo de açao das aspartil proteases também ocorra via catálise ácido-base. Os resíduos Asp 25 e Asp 25' sao responsáveis por catalisar a hidrólise das ligaçoes peptídicas. Um resíduo aspartil auxilia na adiçao de uma molécula de água à carbonila da amida do substrato, formando um intermediário tetraédrico que possui grande afinidade pelo sítio catalítico. Após a formaçao deste intermediário, ocorre a quebra da ligaçao C-N com a formaçao de um ácido carboxílico e uma amina primária.37
PEPTIDEOS E PEPTIDEOMIMÉTICOS Embora muitos peptídeos naturais possuam propriedades farmacocinéticas e farmacodinâmicas que permitam seu uso terapêutico, isto nao é um fenômeno comum nesta classe de compostos. A baixa estabilidade química, a reduzida biodisponibilidade e a suscetibilidade frente a proteases limitam o uso destes peptídeos na clínica médica.38 Outro problema que se adiciona a estes já descritos é a flexibilidade conformacional de fragmentos proteicos, os que atuam via interaçao com uma proteína receptora podem assumir estruturas tridimensionais de baixa afinidade pela mesma. Uma alternativa ao uso dos peptídeos naturais é o desenvolvimento de compostos peptideomiméticos, os quais sao estruturalmente semelhantes ao peptídeo natural, mas que tiveram suas estruturas modificadas de forma a conferir propriedades farmacológicas mais específicas e maior seletividade por um determinado sítio receptor.39 Entretanto, para o planejamento de um peptideomimético é necessário compreender o papel desempenhado por seus diversos componentes estruturais.40 A afinidade pelo receptor, por exemplo, encontra-se fortemente associada às cadeias laterais dos aminoácidos do peptideomimético, que interagem com grupamentos de caráter complementar na estrutura do receptor através de interaçoes de natureza eletrostática, como ligaçoes hidrogênio, dipolo-dipolo e forças de Van der Waals.41 Desta forma, o esqueleto básico de todo peptídeo, constituído por ligaçoes amídicas (peptídicas) intercaladas por uma unidade metilênica ou metínica, pode ser alterado dando origem ao peptideomimético, desde que mantendo as cadeias laterais nas posiçoes adequadas para a interaçao com o receptor. A substituiçao de ligaçoes peptídicas por outras química e enzimaticamente mais estáveis irá conferir maior resistência frente a proteases, o que é crucial para o desenho de inibidores enzimáticos.42,43
PEPTIDEOMIMÉTICOS INIBIDORES DE PROTEASES Protease do DENV Poucos inibidores da NS3 protease do vírus da dengue de natureza peptideomimética têm sido descritos na literatura, dentre eles está a aprotinina (um antifibrinolítico natural), um inibidor padrao de proteases incluindo tripsina, quimotripsina, plasmina, ativador tissular do plasminogênio e calicreína tissular e plasmática. A aprotinina apresentou um IC50=65nM contra serina protease recombinante do DENV-2.44 Algumas estratégias têm sido empregadas na pesquisa de inibidores da NS3/NS2B protease do DENV com vários graus de sucesso, dentre elas a síntese racionalmente planejada de peptideomiméticos e ciclopeptídeos,45 screening virtual baseado na estrutura, screening de produtos naturais, entre outros.46 Entretanto, somente poucos inibidores peptideomiméticos e compostos estruturalmente pequenos sao descritos.47 Na estratégia de síntese racionalmente planejada de peptideomiméticos, Yin e colaboradores descreveram o composto do tipo aldeído 1 (Figura 2), o qual mostrou ser um inibidor reversível e competitivo, com um Ki de inibiçao igual a 5,8 mM contra a NS3 protease do DENV-2.48 Um estudo de relaçao estrutura-atividade (SAR) foi realizado na tentativa de encontrar novos inibidores.49 Neste estudo, o composto 1 serviu de protótipo para o entendimento das interaçoes farmacofóricas inibidor-enzima e desenvolvimento de novas moléculas peptideomiméticas, as quais vieram a confirmar a preferência da enzima por resíduos básicos de Arg na porçao terminal, especialmente na posiçao P2. Como resultado dos estudos de SAR obteve-se o tripeptídeo 2, mais ativo, mostrando um Ki=1,5µM (Figura 2).50,48
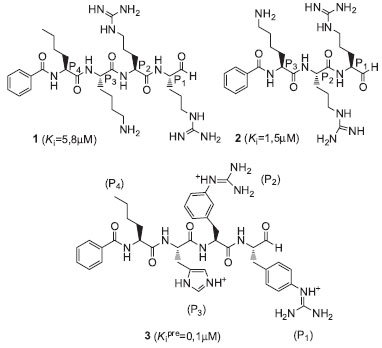 Figura 2. Peptideomiméticos derivados de aldeído como inibidores da NS3/NS2B protease do DENV
Apesar de existirem vários métodos para a síntese em soluçao de peptídeos modificados em sua estrutura original,51 metodologias sintéticas em fase sólida (solid-phase peptide synthesis-SPPS), idealizadas por Bruce Merrifield na década de 1960, tornaram-se bastante atrativas. Através desta metodologia, peptídeos ou peptideomiméticos podem ser sintetizados pela fixaçao de um aminoácido a um suporte polimérico, seguida por uma sequência de reaçoes sobre este aminoácido inicial, de forma a construir uma cadeia peptídica. A fixaçao à matriz polimérica permite reduzir o número de etapas de purificaçao no processo, aumentando assim o rendimento final do peptídeo desejado, além de permitir a automaçao do processo, o que possibilita a construçao de uma biblioteca de compostos a serem testados através de high-throughput screening.52 Devido a essas vantagens a série de compostos do tipo aldeído, incluindo o composto 1 mostrado acima, foi sintetizada em fase sólida utilizando a resina de Weinreb. Nesse método, a funçao aldeído foi liberada na última etapa, após a remoçao em meio ácido do grupo protetor na cadeia lateral, minimizando a condensaçao entre a cadeia lateral guanidina da arginina e a funçao aldeído, que formaria um composto deidro cíclico inativo.53 Na estratégia de screening virtual baseado na estrutura, um recente estudo de modelagem molecular realizado por Frecer e Miertus54 utilizou-se de técnicas combinatórias para desenhar uma biblioteca de compostos peptideomiméticos utilizando a estrutura 3D da NS3/NS2B protease do DENV. Esta biblioteca virtual, composta por mais de 9.200 derivados desenhados tendo como modelo o aldeído 1 e resíduos de aminoácidos nao naturais nas posiçoes P1 à P4, foi testada in silico. O composto 3 (Figura 2), considerado o mais promissor da busca virtual realizada, apresentou um Ki cerca de 50 vezes menor (Kipre=0,1µM) do que o composto modelo 1 e foi analisado em termos de interaçao enzima-inibidor e propriedades físico-químicas relacionadas a adsorçao, distribuiçao, metabolismo e excreçao (ADME). Apesar de alguns inibidores já terem sido descritos na literatura com valores de IC50 em uma faixa menor que micromolar, as indesejadas características físico-químicas e alta toxicidade dos mesmos nao permitiram posterior desenvolvimento. O planejamento racional de inibidores específicos da NS3/NS2B protease do DENV tem se mostrado bastante difícil, devido à relativa planaridade do sítio ativo da enzima, fato que requer uma mudança conformacional substancial do fragmento NS2B, cuja estrutura tornou-se disponível muito recentemente, para que ocorra a ligaçao de um inibidor.55 Protease do HCV Desde a sua identificaçao em 1993, a serina protease NS3/4A do HCV tem sido alvo de intensos estudos para a descoberta de inibidores seletivos e potentes para a terapia de pacientes com hepatite C. O sucesso do desenvolvimento de inibidores da HIV protease demonstra que proteases virais podem ser excelentes alvos para o desenho de novas drogas baseadas na estrutura química.56,57 Entretanto, no caso da serina protease do HCV, a busca por pequenas moléculas, potentes, seletivas, que sejam bioativas por via oral, tem sido parcialmente dificultada pelo fato do sítio da ligaçao ao substrato ser razoavelmente plano e marcadamente hidrofóbico. Mesmo assim, progressos significantes têm sido feitos nos últimos anos para a identificaçao de novos inibidores da serina protease NS3/4A.28 Inibidores nao-covalentes Inibidores peptídicos lineares Estudos iniciais na busca por um inibidor potente de HCV protease utilizaram a estratégia de planejamento de drogas baseada na estrutura. Esta estratégia originou-se a partir da inibiçao significativa da enzima pelos produtos peptídicos que sao liberados da clivagem enzimática de vários substratos.48 Desta forma, foram desenvolvidos diversos inibidores peptídicos e peptideomiméticos, como por exemplo, o hexapeptídeo 4, o qual apresentou um Ki=0,04µM. Tendo 4 como protótipo, outras estruturas menores e menos carregadas foram obtidas, tal como o composto tripeptídico 5 com um Ki=0,6µM (Figura 3).48
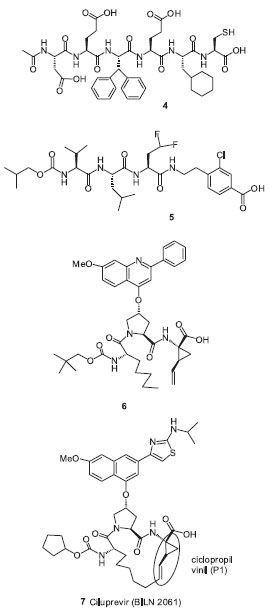 Figura 3. Estruturas de peptideomiméticos inibidores de HCV protease
Inibidores peptídicos macrocíclicos Vários métodos de despeptidizaçao têm sido usados na tentativa de melhorar o perfil farmacocinético de inibidores peptídicos, tais como o uso de aminoácidos nao-naturais, uso de isósteros peptídicos e a macrociclizaçao.30 Os macrociclos, com estrutura e tamanho de anéis apropriados, podem se ligar mais fortemente a uma enzima de uma maneira rígida e pré-organizada, além de serem menos susceptíveis à hidrólise por proteases.58 A estrutura de raios-X da NS3 protease revelou que as porçoes S1 e S3 estao bem próximas. Assim, pela ciclizaçao dos resíduos P1 e P3 dos inibidores peptídicos (ex. o composto 6, figura 3), vários inibidores macrocíclicos foram obtidos, os quais apresentaram bons perfis pré-clínicos levando-os aos testes clínicos. O ciluprevir (7) (BILN 2061 - Boehringer Ingelheim), um macrociclo de 15 membros, foi o primeiro composto inibidor de protease do HCV a entrar em testes clínicos (Figura 3). Os primeiros resultados foram promissores, mostrando um declínio rápido da carga viral plasmática em todos os pacientes com HCV genótipo 1 tratados, mas resultados subsequentes apresentaram certa toxicidade cardíaca em altas doses, além da baixa eficácia contra os genótipos 2 e 3, assim foi retirado de estudo.59 Estudos de SAR mostraram que a porçao ciclopropil vinil na posiçao P1 de 7 apresentou o melhor encaixe com a superfície enzimática, passando a ser elemento fundamental na estrutura de posteriores inibidores. Inibidores covalentes Inibidores macrocíclicos Diversos inibidores macrocíclicos têm sido descritos através da utilizaçao da estratégia de macrociclizaçao, nos quais a cadeia lateral P2 foi ciclizada com o grupamento da extremidade P3, originando os chamados macrociclos P2-P3, bem como aqueles denominados P1-P3, provenientes da ciclizaçao dos aminoácidos nas respectivas posiçoes. Dentre os macrociclos P2-P3 estao os derivados 8 e 9 (Figura 4) que, apesar de apresentarem elevada potência em testes enzimáticos, foram menos potentes em testes celulares com replicon. Já os macrociclos P1-P3 apresentaram melhores perfis de atividade no replicon celular.48,60
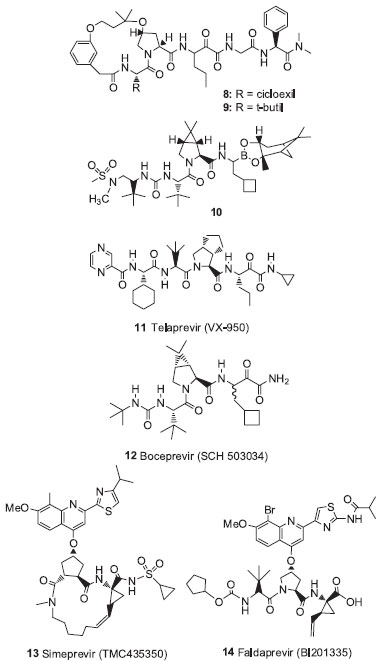 Figura 4. Inibidores da enzima HCV protease
Inibidores lineares Na classe de inibidores covalentes lineares destacam-se os compostos do tipo ácido borônico/boronatos (ex. o composto 10, figura 4) e os derivados do tipo α-cetoamidas. Essas funçoes têm sido investigadas como eletrófilos que, potencialmente, poderiam reagir com a hidroxila nucleofílica da serina do sítio ativo.53 O telaprevir (11) (VX-950 - Vertex Pharmaceuticals, Inc.) (Incivek/Incivor)61 e o boceprevir (12) (SCH503034 - Merck Sharp & Dohme Co., Inc.) (Victrelisr) (Figura 4), representantes da classe das α-cetoamidas, foram aprovados para o tratamento do HCV genótipo 1 pelo FDA, EMA e ANVISA, dentre outras agências, em 2011. Apesar da boa atividade contra o genótipo 1 do HCV, a monoterapia com esses agentes resulta em rápida seleçao de variantes droga-resistentes.62 Estudos de fase II e III mostraram que a combinaçao dessas drogas com PEG-IFN e ribavirina levou a uma substancial reduçao na frequência de mutantes resistentes.63 Assim eles sao empregados como um terceiro medicamento no tratamento de pacientes com hepatite C crônica (infectados com o genótipo 1), junto com o PEG-IFN e a ribavirina, visando a obtençao de maior resposta terapêutica. Vários outros agentes inibidores da protease NS3/4A estao em fases diferentes de testes clínicos, o que aumentará bastante as opçoes de tratamento em um futuro próximo.64 Dentre eles estao o simeprevir (13) (TMC435350),65 um inibidor macrocíclico nao covalente e o faldaprevir (14) (BI201335),66 um inibidor linear nao covalente (Figura 4). Atualmente, ambos estao em estudos clínicos de fase 3. Protease do HIV (HIV PR) Dentre as três patologias em questao neste trabalho, a AIDS é a que mais avançou na comercializaçao de fármacos inibidores da protease. Nas últimas décadas, dez inibidores foram aprovados pelo FDA e outros se encontram em estágio de testes clínicos.56,57 O saquinavir (15), desenvolvido pelo laboratório Hoffmann-La Roche, foi o primeiro inibidor da HIV PR aprovado pelo FDA, em dezembro de 1995. Atualmente outros fármacos sao comercializados, dentre eles estao o ritonavir (16), indinavir (17), nelfinavir (18) e darunavir (19) (Figura 5). Ressalta-se que quase todos os fármacos inibidores da HIV PR sao compostos peptideomiméticos, contendo um grupamento hidroxila planejada para interagir com a díade de resíduos Asp central.
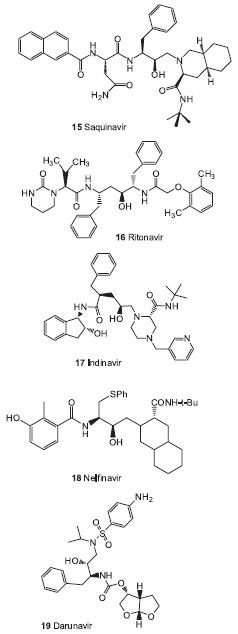 Figura 5. Inibidores da enzima HIV protease
A introduçao da primeira geraçao de inibidores, os fármacos 16, 17 e 18, mudou a natureza da AIDS epidêmica de doença terminal para doença passível de tratamento.67 O darunavir (19) (Prezistar), um dos mais recentes inibidores da HIV-1 PR, foi planejado com o objetivo de maximizar as interaçoes hidrofóbicas e ligaçoes hidrogênio com os resíduos do sítio S2 da protease, pela introduçao do ligante bis-tetraidrofurano (bis-THF).68 Kovalevsky e colaboradores69 mostraram que o composto 19 possui dois sítios de ligaçao na enzima protease: um no próprio sítio ativo e outro em um dos flaps móveis do dímero. A existência deste segundo sítio de ligaçao sugere um mecanismo para a grande efetividade de 19 contra os vírus mutantes que sao resistentes aos demais fármacos da classe. Baseados no sucesso do desenvolvimento do darunavir, Ghosh e colaboradores planejaram novos peptideomiméticos do tipo ciclopentiltetraidrofurano (Cp-THF), como ligantes em P2, substituindo a estrutura bis-THF presente em 19. Os compostos desta série apresentaram valores de Ki na ordem de nM e pM.70 Apesar da existência de vários fármacos no mercado para terapia anti-HIV, que provam inequivocamente que a HIV PR consiste em um alvo terapêutico importante, a terapia anti-HIV PR torna-se limitada devido ao surgimento de cepas virais mutantes resistentes a essas drogas. Esse fato justifica a necessidade da busca constante de novas estruturas capazes de superar e inibir as mutaçoes enzimáticas.71
PLANEJAMENTO E SINTESE DE NOVOS INIBIDORES DE SERINA PROTEASE A necessidade do desenvolvimento de fármacos mais eficazes para a terapia de infecçoes virais de grande impacto no sistema de saúde mundial, como a dengue, a hepatite C e AIDS, tem justificado o empenho de pesquisadores na área de síntese de moléculas peptideomiméticas voltadas para tais patologias virais. Durante as últimas décadas muitos esforços têm sido feitos no sentido de desenvolver estratégias de planejamento e síntese de moléculas peptídicas e peptideomiméticas com conformaçoes específicas, tais como: α-hélice, folhas β ou estendida, uma vez que a incorporaçao de aminoácidos com ângulos corretos de torçao pode ser fundamental para a atividade e seletividade.40 Recentemente, o enfoque maior tem se voltado para o planejamento, síntese e utilizaçao de aminoácidos nao-naturais mono ou bicíclicos, conformacionalmente restritos, para incorporaçao dos mesmos nas estruturas de inibidores enzimáticos peptideomiméticos.72 Neste contexto, Muri e colaboradores vêm trabalhando na pesquisa de compostos peptideomiméticos como potenciais inibidores de serina protease do HCV e DENV. Eles utilizam, como cerne estrutural, o composto chamado isomanídeo (20) (Figura 6) devido à sua estrutura peculiar de éter bicíclico conformacionalmente restrita do tipo em "U", com uma estereoquímica definida, o qual apresenta características estruturais úteis em compostos peptideomiméticos, devido a sua capacidade de agir como um indutor do tipo folha β e reter o padrao de ligaçoes hidrogênio da cadeia lateral peptídica.73,74
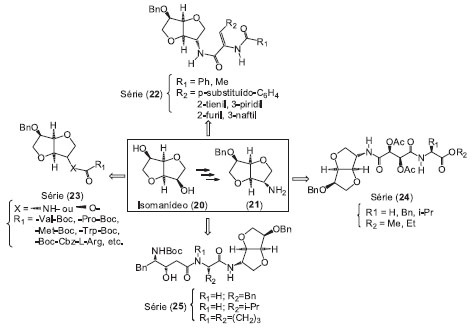 Figura 6. Séries de peptideomiméticos derivados do isomanídeo (20)
O isomanídeo (20) é um carboidrato quiral, comercialmente disponível, o qual pode ser obtido industrialmente pela desidrataçao do D-manitol.75 A estrutura simétrica, bicíclica, apresentando duas hidroxilas na posiçao endo o faz um bloco de construçao atrativo para aplicaçoes sintéticas.76 O isomanídeo e seus derivados já vêm sendo utilizados por vários grupos de pesquisa, como catalisadores de transferência de fases em síntese assimétrica, como ligantes e auxiliares quirais, na síntese de líquidos iônicos quirais, bem como produtos de partida para a síntese de compostos de interesse farmacêutico.77 Um dos primeiros trabalhos descritos por Muri e colaboradores consistiu na síntese da série de compostos (22) (Figura 6), a qual foi obtida pela reaçao de abertura de diversas oxazolonas com a mono-amina benzilada (21), obtida a partir do isomanídeo, em 4 etapas. Os derivados sintetizados foram testados farmacologicamente em modelo celulares do tipo replicon de HCV, no qual o composto com os substituintes, R1=Ph e R2=2-tienil, apresentou o melhor perfil de inibiçao com EC50 = 35 µM.78 Com o intuito de otimizar o perfil farmacológico dos peptideomiméticos obtidos, o mesmo grupo de pesquisas planejou e sintetizou ésteres e amidas (série 23, Figura 6) pela introduçao de cadeias laterais provenientes de diversos aminoácidos e dipeptídeos, com o intuito de verificar o grau de interaçao das mesmas com a protease do HCV.79 O derivado com o substituinte X=-NH e cadeia lateral derivada do aminoácido arginina, a N-Boc-N-di-CBz-L-Arg, apresentou uma inibiçao de 45% da atividade protease da enzima em uma concentraçao de 100mM. Este composto mais ativo, o derivado da arginina, foi ancorado (estudos de docking) no sítio ativo da enzima NS3/4A protease do HCV, mostrando-se localizado na fenda hidrofóbica S1, onde o resíduo arginina interagiu com os resíduos IIe132, Leu135, Lis136, Gli137, Ser139 e Phe154. Neste estudo, também se observou a presença de seis ligaçoes hidrogênio, sendo quatro do resíduo arginina com os aminoácidos His57, Arg155 e Ala157 da enzima e duas do anel isomanídeo.80 Baseados na vasta literatura sobre o uso do ácido tartárico como cerne estrutural nao-hidrolisável para o planejamento de inibidores de proteases, novas estruturas peptideomiméticas derivadas do isomanídeo foram planejadas, contendo esqueletos oriundos do ácido D e L-tartárico, representada pela série (24) (Figura 6). No teste de inibiçao enzimática frente à NS3/4A protease do HCV, o composto derivado do aminoácido L-Val e ácido D-tatárico apresentou o melhor perfil de inibiçao igual a 63%.81 A série de compostos (25) foi planejada pela introduçao do cerne nao-hidrolisável da estatina e L-aminoácidos, na cadeia lateral do isomanídeo. Esta série encontra-se em fase de testes biológicos.82
CONCLUSOES O presente trabalho mostra uma ampla revisao da literatura sobre compostos peptideomiméticos como inibidores de serina e aspartil proteases, enfatizando a necessidade do desenvolvimento de fármacos mais eficazes para a terapia de infecçoes virais, uma vez que poucos inibidores da HCV protease e dez inibidores da HIV protease estao disponíveis comercialmente. Esta necessidade é reforçada pela existência de diversas variantes virais e emergência de cepas mutantes, além das dificuldades para o desenvolvimento de uma vacina efetiva. Diversos pontos da replicaçao do HIV e HCV, além das proteases, podem ser considerados alvos potenciais para desenvolvimento de novos agentes para terapia farmacológica, ampliando assim as possibilidades de obtençao de um novo fármaco para tais patologias de interesse mundial.
REFERENCIAS 1. Hedstrom, L.; Chem. Rev. 2002, 102, 4501. 2. Rawling, N. D.; Barret, A.; Methods Enzymol. 1994, 244, 18. 3. Sałaga, M.; Sobczak, M.; Fichna, J.; Drug Discovery Today (2013), doi: http://dx.doi.org/10.1016/j.drudis.2013.03.004. 4. Koblinski, J. E.; Ahram, M.; Sloane, B. F.; Clin. Chim. Acta 2000, 291, 113. 5. Rufino, R.; Silva, J. R. L.; J. Bras. Pneumol. 2006, 32, 241. 6. Lopez-Otin, C.; Overall, C. H.; Nat. Rev. Mol. Cell Biol. 2002, 3, 509. 7. IUBMB Enzyme Nomenclature: recommendations of the nomenclature commited of the international union of biochemistry and molecular biology. Academic Press: San Diego, 1992. 8. de Simone, S. G.; Silva Junior, F. P.; Biotecnologia Ciência & Desenvolvimento 2001, 22, 12. 9. Barret, A. J.; Methods Enzymol. 1994, 244, 1; Barret, A. J.; Rawlings, N. D.; Arch. Biochem. Biophys. 1995, 318, 247; da Silva-López, R. E.; Quim. Nova 2010, 33, 1541. 10. Schechter, I.; Berger, A.; Biochem. Biophys. Res. Commun. 1967, 27, 157. 11. De Souza, M. V. N.; Acta Farm. Bonaerense 2005, 24, 291. 12. Nogueira, R. M.; Miagostovich, M. P.; Cunha, R. V.; Zagne, S. M.; Gomes, F. P.; Nicol, A. F.; Coelho, J. C.; Schatzmayr, H.G.; Trans. R. Soc. Trop. Med. Hyg. 1999, 93, 418. 13. Melino, S.; Paci, M.; FEBS J. 2007, 274, 2986; Srichaikul, T.; Nimmannitya, S.; Best Pract. Res. Clin. Haematol. 2000, 13, 261. 14. Innis, B. L.; Eckels, K. H.; Am. J. Trop. Med. Hyg. 2003, 69, 1. 15. Gibbons, R. V.; Vaughn, D. W.; BMJ 2002, 324, 1563. 16. Halstead, S. B.; Lancet 2007, 370, 1644. 17. Chanprapaph, S.; Saparpakorn, P.; Sangma, C.; Niyomrattanakit, P.; Hannongbua, S.; Angsuthanasombat, C.; Katzenmeier, G.; Biochem. Biophys. Res. Commun. 2005, 330, 1237; Fowell, A. J.; Nash, K. L.; Adv. Ther. 2010, 27, 512; Natarajan, S.; Genet. Mol. Biol. 2010, 33, 214. 18. Falgout, B.; Markoff, L.; J. Virol. 1995, 69, 7232. 19. Chanprapaph, S.; Saparpakorn, P.; Sangma, C.; Niyomrattanakit, P.; Hannongbua, S.; Angsuthanasombat, C.; Katzenmeier, G.; Biochem. Biophys. Res. Commun. 2005, 330, 1237. 20. Bazan, J. F.; Fletterick, R. J.; Virology 1989, 171, 637; Bazan, J. F.; Fletterick, R. J.; Semin. Virol. 1990, 1, 311. 21. Gorbalenya, A. E.; Donchenko, A. P.; Koonin, E. V.; Blinov, V. M.; Nucleic Acids Res. 1989, 17, 3889. 22. Falgout, B.; Miller, R. H.; Lai, C. J.; J. Virol. 1993, 67, 2034; Arias, C. F.; Preugschat, F.; Strauss, J. H.; Virology 1993, 193, 888. 23. http://www.saude.rs.gov.br/dados/1316779614748protocolo_hepatitec_marc_pdf_29595.pdf, acessada em Dezembro 2012. 24. Ruiz, F. J. G.; Zylbergeldd, N. D.; Rev. Bras. Med. 2004, 61, 719. 25. Hayashi, N.; Takehara T.; J. Gastroenterol. 2006, 41, 17. 26. Fowell, A. J.; Nash, K. L.; Adv. Ther. 2010, 27, 512. 27. Natarajan, S.; Genet. Mol. Biol. 2010, 33, 214. 28. Kupfer, B. Em Short Guide to Hepatitis C; Mauss, S; Berg, T.; Rockstroh, J.; Sarrazin, C.; Wedemeyer, H.eds.; Flying Publisher: Germany, 2012, cap. 2. 29. Thompson, A.; Patel, K.; Tillman, H.; Mchutchison, J. G.; J. Hepatol. 2009, 50, 184. 30. De Francesco, R.; Carfí, A.; Adv. Drug Delivery Rev. 2007, 59, 1242. 31. Chen, K. X.; Njoroge, F. G.; Curr. Opin. Invest. Drugs 2009, 10, 821. 32. Pope, M.; Hasse, A.; Nat. Med. 2003, 9, 847. 33. Oliveira, L. H. S.; Livro de Virologia Humana, 1ª ed., Cultura Médica: Rio de Janeiro, 1994. 34. Grinsztejn, B.; Nguyen, B.-Y.; Katlama, C.; Gatell, J. M.; Lazzarin, A.; Vittecoq, D.; Gonzalez, C. J.; Chen, J.; Harvey, C. M.; Isaac, R. D.; Lancet 2007, 369, 1261. 35. Hornak, V.; Simmerling, C.; Drug Discovery Today 2007, 12, 132. 36. Drag, M. E; Salvesen, G. S.; Nat. Rev. Drug Discovery 2010, 9, 690; Raney, K. D.; Sharma, S. D.; Moustafa, I. M.; Cameron, C. E.; J. Biol. Chem. 2010, 285, 22725. 37. Leung, D.; Abbenante, G.; Fairlie, P. D.; J. Med. Chem. 2000, 43, 305; Cunico, W.; Gomes, C. R. B.; Junior, W. T. V.; Quim. Nova 2008, 31, 2111. 38. Frokjaer, S.; Otzen, D. E. Nat. Rev. Drug Discovery 2005, 4, 298. 39. Picchi, D. G.; Altei, W. F.; Saito, M. S.; Bolzani, V. S.; Cilli, E. M.; Quim. Nova 2009, 32, 1262. 40. Vagner, J.; Qu, H.; Hruby, V. J.; Curr. Opin. Chem. Biol. 2008, 12, 292; Stefanucci, A.; Pinnen, F.; Feliciani, F.; Cacciatore, I.; Lucente, G.; Mollica, A.; Int. J. Mol. Sci. 2011, 12, 2853. 41. Hruby, V. J.; Biopolymers 1993, 33, 1073. 42. Fear, G.; Komarnytsky, S.; Raskin, I.; Pharmacol. Ther. 2007, 113, 354. 43. Kharb, R.; Rana, M.; Sharma, P. C.; Yar, M. S.; J. Chem. Pharm. Res. 2011, 3, 173. 44. Leung, D.; Schroder, K.; White, H.; Fang, N.-X.; Stoermer, M. J.; Abbenante, G.; Martin, J. L.; Young, P. R.; And Fairlie, D. P.; J. Biol. Chem. 2001, 276, 45762. 45. Xu, S.; Li, H.; Shao, X.; Fan, C.; Ericksen, B.; Liu, J.; Chi, C.; Wang, C.; J. Med. Chem. 2012, 55, 6881. 46. Vasudevan, S. G.; Keller, T. H.; Bioorg. Med. Chem. Lett. 2006, 16, 36; Lescar, J.; Luo, D.; Xu, T.; Sampath, A.; Lim, S. P.; Canard, B.; Vasudevan, S. G.; Antiviral Res. 2008, 80, 94; Gao, Y.; Cui, T.; Lam, Y.; Bioorg. Med. Chem. 2010, 18, 1331; Noble, C. G.; Chen, Y.-L.; Dong, H.; Gu, F.; Lim, S. P.; Schul, W.; Wang, Q.-Y.; Shi, P.-Y.; Antivir. Res. 2010, 85, 450; Yin, Z.; Patel, S. J.; Wang, W.-L.; Wang, G.; Chan, W.-L.; Rao, K. R. R.; Alam, J.; Jeyaraj, D. A.; Ngew, X.; Patel, V.; Beer, D.; Lim, S. P.; Yang, C.-C.; Hsieh, Y.-C.; Lee, S.-J.; Wu, S.-H.; Liao, C.-L.; Tsao, C.-H.; Chao, Y.-S.; Chern, J.-H.; Wu, C.-P.; Yueh, A.; Antimicrob. Agents Chemother. 2011, 55, 229. 47. Noble, C. G.; Chen, Y.-L.; Dong, H.; Gu, F.; Lim, S. P.; Schul, W.; Wang, Q.-Y.; Shi, P.-Y.; Antivir. Res. 2010, 85, 450; Deng, J.; Li, N.; Liu, H.; Zuo, Z.; Liew, O. W.; Xu, W.; Chen, G.; Tong, X.; Tang, W.; Zhu, J.; Zuo, J.; Jiang, H.; Yang, C. G.; Li, J.; Zhu, W.; J. Med. Chem. 2012, 55, 6278. 48. Chen, K. X.; Njoroge, F. G.; Curr. Opin. Invest. Drugs 2009, 10, 821. 49. Parkinson T.; Pryde, D. C.; Fut. Med. Chem. 2010, 2, 1181. 50. Yin, Z.; Patel, S. J.; Wang, W.-L.; Wang, G.; Chan, W.-L.; Rao, K. R. R.; Alam, J.; Jeyaraj, D. A.; Ngew, X.; Patel, V.; Beer, D.; Lim, S. P.; Vasudevan, S. G.; Keller, T. H.; Bioorg. Med. Chem. Lett. 2006, 16, 40. 51. Machado, A.; Liria, C. W.; Proti, P. B.; Remuzgo, C.; Miranda, M. T. M.; Quim. Nova 2004, 27, 781; Freitas, L. B. de O.; Ruela, F. A; Pereira, G. R.; Alves, R. B.; de Freitas, R. P.; dos Santos, L. J.; Quim. Nova 2011, 34, 1791. 52. Fehrentz, J.-A.; Paris, M.; Heitz, A.; Velek, J.; Liu, C.-F.; Winternitz, F.; Martinez, J.; Tetrahedron Lett. 1995, 36, 7871. 53. Venkatraman, S.; Wu, W.; Prongay, A.; Girijavallabhan, V.; Njoroge, F. G.; Bioorg. Med. Chem. Lett. 2009, 19, 180. 54. Frecer, V.; Miertus, S.; J. Comput.-Aided Mol. Des. 2010, 24, 195. 55. Noble, C. G.; Seh, C. C.; Chao, Alexander T.; Shi, P. Y.; J. Virol. 2012, 86, 438. 56. Peçanha, E. P.; Antunes, O. A. C.; Tanuri, A.; Quim. Nova 2002, 25, 1108. 57. Cunico, W.; Gomes, C. R. B.; Junior, W. T. V.; Quim. Nova 2008, 31, 2111. 58. Tyndall, J. D.; Fairlie, D. P.; Curr. Med. Chem. 2001, 8, 893. 59. Lamarre, D.; Anderson, P. C.; Bailey, M.; Beaulieu, P.; Bolger, G.; Bonneau, P.; Bos, M.; Cameron, D. R.; Cartier, M.; Cordinggley, M. G.; Faucher, A.-M.; Goudreau, N.; Kawai, S. H.; Kukolj, G.; Lagacé, L.; LaPlante, S.R.; Narjes, H.; Poupart, M.-A.; Rancourt, J.; Sentjens, R. E.; Nature 2003, 426, 186; Naggie, S.; Patel, K.; Mchutchison, J.; J. Antimicrob. Chemother. 2010, 65, 2063. 60. Chen, K. X.; Njoroge, F. G.; Arasappan, A.; Venkatraman, S.; Vibulbhan, B.; Yang, W.; Parekh, T. N.; Pichardo, J.; Prongay, A.; Cheng, K. C.; Butkiewicz, N.; Yao, N; Madison, V.; Girijavallabhan, V.; J. Med. Chem. 2006, 49, 995. 61. Perni, R. B.; Almquist, S. J.; Byrn, R. A.; Chandorkar, G. Pravin, R.; Chaturved, P. R.; Courtney, L. F.; Decker, C. J.; Dinehart, K.; Gates, C. A.; Harbeson, S. L.; Heiser, A.; Kalkeri, G.; Kolaczkowski, E.; Lin, K.; Luong, Y.-P.; Rao, B. G.; Taylor, W. P.; Thomson, J. A.; Tung, R. D.; Wei, Y.; Kwong, A. D.; Lin, C.; Antimicrob. Agents Chemother. 2006, 50, 899. 62. Wyles, D. L.; J. Infect. Dis. 2013, 207(S1), S33. 63. Hézode, C.; Forestier, N.; Dusheiko, G.; Ferenci, P.; Pol, S.; Goeser, T.; Bronowicki, J. P.; Bourlière, M.; Gharakhanian, S.; Bengtsson, L.; McNair, L.; George, S.; Kieffer, T.; Kwong, A.; Kauffman, R. S.; Alam, J.; Pawlotsky, J. M.; Zeuzem, S.; N. Engl. J. Med. 2009, 360, 1839; Bacon, B. R.; Gordon, S. C.; Lawitz, E.; Marcellin, P.; Vierling, J. M.; Zeuzem, S.; Poordad, F.; Goodman, Z. D.; Sings, H. L.; Boparai, N.; Burroughs, M.; Brass, C. A.; Albrecht, J. K.; Esteban, R.; N. Engl. J. Med. 2011, 364, 1207. 64. Kiser, J. J.; Flexner, C.; Annu. Rev. Pharmacol. Toxicol. 2013, 53, 427. 65. Raboisson, P.; de Kock, H.; Rosenquist, Å.; Nilsson, M.; Salvador-Oden, L.; Lin, T. I.; Roue, N.; Ivanov, V.; Wä hling, H.; Wickström, K.; Hamelink, E.; Edlund, M.; Vrang, L.; Vendeville, S.; Van de Vreken, W.; McGowan, D.; Tahri, A.; Hu, L. L.; Boutton, C.; Lenz, O.; Delouvroy, F.; Pille, G.; Surleraux, D.; Wigerinck, P.; Samuelsson, B.; Simmen, K.; Bioorg. Med. Chem. Lett. 2008, 18, 4853. 66. White, P. W.; Llinas-Brunet, M.; Amad, M.; Bethell, R. C.; Bolger, G.; Cordingley, M. G.; Duan, J. M.; Garneau, M.; Lagace, L.; Thibeault, D.; Kukolj, G.; Antimicrob. Agents Chemother. 2010, 54, 4611. 67. de Souza, M. V. N.; de Almeida, N. V.; Quim. Nova 2003, 26, 366. 68. Ghosh, A. K.; Takayama, J.; Tetrahedron Lett. 2008, 49, 3409; Ghosh, A. K.; Dawson, Z. L.; Mitsuya, H.; Bioorg. Med. Chem. 2007, 15, 7576. 69. Kovalevsky, A. Y.; Liu, F.; Leshchenko, S.; Ghosh, A. K.; Louis, J. M.; Harrison, R. W.; Weber, I. T.; J. Mol. Biol. 2006, 363, 161. 70. Ghosh, A. K.; Sridhar, P. R.; Leshchenko, S.; Hussain, A. K.; Li, J. F.; Kovalevsky, A. Y.; Walters, D. E.; Wedekind, J. E.; Grum-Tokars, V.; Das, D.; Koh, Y.; Maeda, K.; Gatanaga, H.; Weber, I. T.; Mitsuya, H.; J. Med. Chem. 2006, 49, 5252; Ghosh, A. K.; Leshchenko-Yashchuk, S.; Anderson, D. D.; Baldridge, A.; Noetzel, M.; Miller, H. B.; Tie, Y. F.; Wang, Y. F.; Koh, Y.; Weber, I. T.; Mitsuya, H.; J. Med. Chem. 2009, 52, 3902. 71. Ohtaka H.; Freire E. Progr. Biophys. Mol. Biol. 2005, 88, 193; Melo, E. B.; Bruni, A. T.; Ferreira, M. M. C.; Quim. Nova 2006, 29, 555; Qiu, X.; Liu, Z.-P.; Curr. Med. Chem. 2011, 18, 4513. 72. Trabocchi, A.; Scarpi, D.; Guarna, A. Amino Acids 2008, 34, 1; Hanessian, S.; Auzzas, L.; Acc. Chem. Res. 2008, 41, 1241. 73. Hanessian, S.; McNaughton-Smith, G.; Lombart, H. G.; Lubell, W. G.; Tetrahedron 1997, 53, 12789. 74. Gising, J; Belfrage, A. K.; Alogheli, H.; Ehrenberg, A.; Åkerblom, E.; Svensson, R.; Artursson, P.; Karlén, A.; Danielson, U. H.; Larhed, M.; Sandström, A.; J. Med. Chem. (2013), dx.doi.org/10.1021/jm301887f. 75. Fauconnier, A. C. R.; Acad. Sci. 1882, 95, 991; Fauconnier, A.; Bull. Soc. Chim. Fr. 1884, 41, 18; Wiggins, L. F.; J. Chem. Soc. 1945, 4. 76. Kagan, H. B.; Riant, O.; Chem. Rev. 1992, 92, 1007. 77. Carcedo, C.; Dervisi, A.; Fallis, I. A.; Ooi, L.; Malik, K. M. A.; Chem. Commun. 2004, 1236; Muri, E. M. F.; Gomes Jr, M.; Costa, J. S.; Alencar, F. L.; Sales Jr., A.; Bastos, M. L.; Hernandez-Valdes, R.; Albuquerque, M. G.; Cunha, E. F. F.; Alencastro, R. B.; Williamson, J. S.; Antunes, O. A. C.; Amino Acids 2004, 27, 153; Muri, E. M. F.; Gomes Jr., M.; Albuquerque, M. G.; Cunha, E. F. F.; Alencastro, R. B.; Williamson, J. S.; Antunes, O. A. C.; Amino Acids 2005, 28, 413; Antunes, O. A. C.; Quim. Nova 2005, 28, S64; Muri, E. M. F.; Abrahim, B. A.; Barros, T. G.; Williamson, J. S.; Antunes, O. A. C.; Mini-Reviews in Organic Chemistry 2010, 7, 75. 78. Barros, T. G.; Pinheiro, S.; Williamson, J. S.; Tanuri, A.; Pereira, H. S.; Brindeiro, R. M.; Neto, J. B. A.; Antunes, O. A. C.; Muri, E. M. F.; Synthesis 2009, 4, 620; Barros, T. G.; Pinheiro, S.; Williamson, J. S.; Tanuri, A.; Gomes Jr, M.; Pereira, H. S.; Brindeiro, R. M.; Neto, J. B. A.; Antunes, O. A. C.; Muri, E. M. F.; Amino Acids 2010, 38, 701. 79. Luiz, J.; Quim. Nova 1990, 13, 176. 80. Barros, T. G.; Zorzanelli, B. C.; Pinheiro, S.; Brito, M. A.; Tanuri, A.; Costa, E. C. B.; Mohana-Borges, R. S.; Rodrigues, C. R.; Souza, A. T. M.; Ferreira, V. F.; Muri, E. M. F.; Lett. Org. Chem. 2012, 9, 239. 81. Tanuri, A.; da Costa, E. C. B.; Muri, E. M. F.; Mohana-Borges, R. S.; Pinheiro, S.; Ventura, G. T.; Capaccia, A. N.; Abrahim-Vieira, B. A.; Azevedo, P. H. R. A.; Portela, A. C.; Br 10 2012 020638 2, 2012. 82. Zorzanelli, B. C. Dissertaçao de Mestrado, Universidade Federal Fluminense, Brasil, 2013. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access