
 |
 |
 |
 |
 |
Artigo
|
|
| Estudo químico e atividades antiproliferativa, tripanocida e leishmanicida de Maxillaria picta Chemical study and antiproliferative, trypanocidal and leishmanicidal activities of Maxillaria picta |
|
Thiago L. de AlmeidaI; Josiane A. MonteiroI; Greice K. P. LopesI; Lucas U. R. ChiavelliI; Silvana M. de O. SantinI; Cleuza C. da SilvaI; Vanessa KaplumII; Débora B. ScariotII; Celso V. NakamuraII; Ana L. T. G. RuizIII; Joao E. CarvalhoIII; Ricardo T. de FariaIV; Armando M. PominiI,*
IDepartamento de Química, Universidade Estadual de Maringá, Av. Colombo 5790, 87020-900 Maringá - PR, Brasil Recebido em 27/11/2013 * e-mail: ampomini@uem.com ABSTRACT The chemical study of the orchid Maxillaria picta resulted in the isolation of the bioactive stilbenes phoyunbene B and phoyunbene C, in addition to four phenolic acids, one xanthone, steroidal compounds and two triterpenes. Crude extract, fractions, subfractions and the isolated xanthone were evaluated for anticancer activity against human tumor cell lines and against evolutionary forms of T. cruzi and L. amazonensis. The structures of the compounds were determined by GC-MS, and 1H NMR, 13C NMR spectral methods as well as bidimensional techniques. INTRODUÇÃO Plantas da família Orchidaceae têm se tornado alvo de estudos fitoquímicos sendo uma rica e variada fonte de compostos que apresentam diversos tipos de atividades biológicas. Já foram relatados os isolamentos de triterpenos,1 alcaloides,2 fenantrenos,3 flavonoides,4 além de estilbenos e ácidos fenólicos,5,6 muitos deles demonstrando propriedades farmacológicas, tais como antioxidante, anticancerígena e analgésica.2,4,5 Pouco se sabe, porém, sobre substâncias com potencial biológico de orquídeas do gênero Maxillaria. Estudos químicos e biológicos foram realizados apenas com a espécie M. densa, nativa do México, utilizada popularmente como relaxante muscular,7 a partir da qual foram identificados fenantrenos e estilbenoides com atividades antifúngicas.8,9 A espécie M. picta é endêmica do bioma Mata Atlântica, no Sul e Sudeste do Brasil, principalmente sobre a Serra do Mar, sendo comumente encontrada em modo epifítico sobre perobas. A espécie é facilmente identificada pela presença de seus pseudobulbos bifoliados, sulcados a canaliculados e pelas suas flores campanuladas, duradouras e sem recompensas florais aos polinizadores,10 mas que apresentam fragrâncias e colorações que atraem abelhas operárias sem ferrão do gênero Trigona.11 Alguns trabalhos realizados com M. picta descrevem os componentes florais voláteis que se caracterizam por altas concentrações do monoterpeno linalol.12 O presente trabalho descreve o estudo químico e farmacológico do extrato bruto e frações de M. picta, com a realização de ensaios para atividade antiproliferativa contra diversas linhagens de células cancerosas humanas bem como ensaios tripanocida e leishmanicida, agentes causadores de doenças tropicais negligenciadas de considerável importância, tendo em vista a atual defasagem medicamentosa para o tratamento dessas enfermidades.
PARTE EXPERIMENTAL Procedimentos gerais Os espectros de RMN de 1H e de RMN de 13C foram obtidos em espectrômetros Varian Mercury Plus, operando a 300,06 MHz para 1H e 75,45 MHz para 13C. Também foram obtidos os espectros pela técnica DEPT e correlações bidimensionais a partir dos mapas de contornos COSY, HMBC e HSQC. Os deslocamentos químicos foram dados em ppm, tendo como referência interna o TMS. Os solventes utilizados foram: CDCl3, CD3OD, D2O e DMSO-d6, Aldrich ou Isotec. Os espectros de massas de baixa resolução foram obtidos com um cromatógrafo em fase gasosa acoplado a um espectrômetro de massas modelo Focus GC (Thermo-Finnigan). As cromatografias em coluna foram realizadas utilizando sílica gel 60 e as filtrações foram feitas em Sephadex LH 20. Os diâmetros das colunas utilizadas variaram de acordo com a massa do material a ser tratado. O acompanhamento das cromatografias em coluna foi feito por análises de cromatografia em camada delgada (CCD). As placas cromatográficas foram preparadas com fase estacionária sílica-gel 60 G e sílica-gel 60 GF, com 0,25 mm de espessura. As substâncias separadas nas placas cromatográficas foram visualizadas através do revelador anisaldeído. Material vegetal A espécie vegetal M. picta foi coletada no campus da Universidade Estadual de Londrina (UEL) e identificada pelo Prof. Dr. Ricardo Tadeu de Faria, em março de 2012. Um espécime é mantido vivo no orquidário da UEL e uma exsicata é mantida sob código FUEL22314. Isolamento dos constituintes químicos Aproximadamente 6,0 kg do material vegetal (raízes, folhas e pseudobulbos) foram moídos e submetidos a uma extração exaustiva com uma mistura de CHCl3/MeOH (3:7) (16 L × 3) a frio. Após a evaporação sob pressão reduzida, obteve-se um extrato bruto (188,0 g). Parte do extrato bruto clorofórmico-metanólico (142,0 g) foi solubilizado em 500 mL de MeOH:H2O 1:1 e submetido à partição em hexano, CHCl3, AcOEt e n-BuOH, resultando nas frações hexânica (MPH, 12,8 g), CHCl3 (MPC, 30,7 g), AcOEt (MPA, 21,5 g), n-BuOH (MPB, 7,0 g) e a remanescente MeOH/H2O (MPM, 67,2 g). A fração MPC foi solubilizada sucessivamente com hexano (MPCH, 5,5 g), CHCl3 (MPCC, 12,1 g), AcOEt (MPCA, 6,4 g) e MeOH (MPCM, 0,8 g), resultando nas suas respectivas subfrações. A fração CHCl3-AcOEt (523,1 mg) foi submetida a uma separação em cromatografia em Sephadex LH 20 (MeOH), resultando em 71 frações. Após análise por CCD as frações foram agrupadas de acordo com sua similaridade cromatográfica. A subfração CA-1 (102,1 mg) foi submetida a uma cromatografia em coluna de Sephadex LH 20 (MeOH). Esta separação resultou no isolamento das substâncias 1 e 2 na forma de mistura (22,3 mg), enquanto que a fração CA-2 mostrou conter a substância 3 (21,0 mg). A fração CHCl3-Hexânica (3,60 g) foi submetida a cromatografia em coluna em gel de sílica, eluida com hexano, CHCl3, AcOEt e MeOH em gradiente crescente de polaridade. A subfração CH-1 (10,3 mg) resultou no isolamento da mistura 4 e 5 (14,3 mg), enquanto que a subfração CH-2 (123,6 mg) também foi submetida a cromatografia em coluna em gel de sílica, resultando na identificação da mistura de substâncias 6, 7 e 8 de massa igual a 30,3 mg. A fração CHCl3-CHCl3 (7,11 g) foi submetida a cromatografia em coluna em gel de sílica com hexano, CHCl3, AcOEt e MeOH em gradiente crescente de polaridade. A fração CC-1 (741,0 mg) foi submetida ao mesmo processo, resultando novamente na mistura 1 e 2 (13,3 mg). A fração hexânica (5,86 g) foi também submetida a cromatografia em coluna em gel de sílica utilizando como eluentes hexano, CHCl3 e MeOH, em gradiente crescente de polaridade. As subfrações H-1 (376,8 g) e H-2 (380,3 g) foram submetidas a um fracionamento em coluna de sílica-gel resultando no isolamento da mistura de compostos 4 e 5 (22,9 mg) e 6, 7 e 8 (30,3 mg), respectivamente. A fração AcOEt (566,0 mg) foi submetida a uma filtração em Sephadex LH 20 somente com MeOH, resultando em 126 frações. A fração A-1 (68,1 mg), submetida a uma filtração em Sephadex LH 20, levou ao isolamento da substância 9 (4,8 mg), enquanto que a fração A-2 (115,7 mg) realizada nas mesmas condições, levou ao isolamento das substâncias 10 (4,1 mg), 11 (10,3 mg) e da mistura 11 e 12 (12,7 mg). A filtração em Sephadex LH 20 da fração A-3 (20,7 mg), resultou no isolamento da mistura das substâncias 11 e 13 (3,1 mg). Parte da fração MeOH/H2O (601,8 mg) foi submetida a um fracionamento em Sephadex LH-20 usando como eluentes H2O e MeOH em gradiente decrescente de polaridade, resultando no isolamento da substância 9 (26,0 mg). Além disso, a substância 9 foi obtida em grande quantidade (2,54 g) a partir da precipitação dessa fração. Avaliação da atividade antiproliferativa A avaliação da atividade antiproliferativa do extrato bruto, frações e dos compostos isolados foi realizada contra diversas linhagens de células tumorais humanas. Linhagens celulares Para a realização da triagem in vitro foram utilizadas as linhagens de células tumorais humanas [rim (786-0), melanona (UACC-62), leucemia (K562) mama (MCF-7), ovário (OVCAR-3), próstata (PC-3), cólon (HT29), pulmão (NCI-H460), glioma (U251) e ovário resistente (NCI-ADR/RES), queratinócito humano (HaCat)] cedidas pelo Instituto Nacional do Cancer (NCI/EUA, Frederick MA). Todas as linhagens foram cultivadas em RPMI 1640 com 5% de soro fetal bovino inativado (SFB) e 1% de penicilina/estreptomicina (1000 U mL-1 : 1000 µg mL-1), em atmosfera de 5% de CO2, a 37 ºC e ambiente úmido. Teste de atividade antiproliferativa em cultura de células tumorais humanas Células tumorais humanas (100 µL suspensão celular compartimento-1, densidade de inoculação entre 3×104 e 6,5×104 cel mL-1) foram expostas a diferentes concentrações das amostras em DMSO/RPMI/SFB 5% (0.25, 2.5, 25 e 250 µg mL-1) e incubadas a 37 ºC, 5% de CO2, em ambiente úmido, por 48 h. A concentração final de DMSO não afetou a viabilidade celular. No momento da adição das amostras (T0) e após o período de exposição (T1), as células foram fixadas com ácido tricloroacético 50% e a proliferação celular foi determinada através da quantificação espectrofotométrica (540 nm) do conteúdo proteico celular usando-se o teste de sulforrodamina B.13 A partir da curva de proliferação celular em função da concentração de amostra, calculou-se a concentração efetiva GI50 (concentração necessária para inibir 50% da proliferação celular) através de regressão não linear usando o software ORIGIN 7.5 (OriginLab Corporation). Atividade antiproliferativa em formas epimastigotas e tripomastigotas de Trypanosoma cruzi Ensaios de toxicidade celular Os ensaios de toxicidade celular foram realizados com a linhagem LLCMK2 (células epiteliais de rim de Macaca mulata). As células foram mantidas em meio DMEM, suplementado com 10% de soro fetal bovino (SFB) e L-glutamina 2 mM, 50 mg mL-1 de gentamicina e tamponado com bicarbonato de sódio até pH 7,6, de acordo com metodologia da American Type Culture Collection (ATCC®). Em seguida, a cultura foi incubada em garrafas plásticas a 37 ºC e 5% de CO2, em estufa umidificada, para obtenção das células para ensaios. Para avaliar o efeito citotóxico sobre a linhagem LLCMK2 foi empregado o método de redução do MTT.14 A concentração dos compostos com capacidade de reduzir em 50% a densidade óptica das células tratadas, em comparação com o controle, foi determinada pelo cálculo da concentração citotóxica para 50% das células (CC50). Foram realizados três experimentos independentes em duplicata para cada ensaio. Atividade antiproliferativa in vitro em formas epimastigotas de T. cruzi Para o ensaio antiproliferativo, ressuspenderam-se em meio LIT suplementado com 10% de SFB as formas epimastigotas de T. cruzi, em fase exponencial de crescimento (96 h), o que resultou na concentração de 1,0 × 106 parasitos mL-1. Em placas de 24 poços, foi adicionado, em cada compartimento, 900 µL da suspensão descrita, bem como 100 µL de meio LIT, na presença ou não de diferentes concentrações dos compostos a serem testados. As placas foram incubadas a 28 ºC por 96 h e, em seguida, avaliou-se o crescimento celular através da contagem dos parasitos em câmara de Neubauer observada em microscópio óptico. Para mensurar o efeito antiproliferativo, foi tomado o percentual de inibição de crescimento dos parasitos em relação ao controle em 50% (IC50). A substância de referência utilizada como controle foi o Benzonidazol®. Foram realizados três experimentos independentes em duplicata para cada ensaio. Viabilidade in vitro em formas tripomastigotas de T. cruzi O sobrenadante da cultura de células LLMCK2 no pico parasistêmico (sexto dia de infecção) foi centrifugado para obtenção das formas tripomastigotas de T. cruzi. A seguir, as mesmas foram ressuspensas em meio DMEM, chegando-se a uma concentração de 1,0 × 107 parasitos mL-1. Cada compartimento das placas de 96 poços utilizadas para o ensaio recebeu, além da suspensão de protozoários, uma alíquota dos compostos a serem avaliados em diferentes concentrações, e também dos controles positivo (Benzonidazol®) e negativo (somente meio DMEM). Então, as placas foram incubadas por 24 h a 37 ºC e atmosfera de 5% de CO2 em estufa umidificada. A contagem dos parasitos foi realizada de acordo com o método de Pizzi-Brener.15 Dessa forma, foi possível quantificar o número de tripomastigotas em movimento, dado que foi utilizado para calcular a concentração de droga capaz de reduzir em 50% a viabilidade dos parasitos (EC50). Foram realizados três experimentos independentes em duplicata para cada ensaio. Atividade antiproliferativa em formas promastigotas e amastigotas axênica de Leishmania amazonensis Parasitos e macrófagos Promastigotas de Leishmania amazonensis (cepa WHOM/BR/75/JOSEFA) foram mantidas e cultivadas em meio Warren (infusão de cérebro e coração "Difco®" adicionado de hemina e ácido fólico), pH 7, suplementado com 10% de soro fetal bovino (SFB - Gibco) e incubados a 25 ºC. As amastigotas axênicas de L. amazonensis obtidas a partir de cultura de promastigotas foram cultivadas em meio Schneider (Sigma, St Louis, MO, USA), pH 4.5, suplementado com 20% de SFB e incubados a 32 ºC.16 Macrófagos de linhagem J774.A1 foram mantidos em meio RPMI 1640 (Roswell Park Memorial Institute medium - Sigma, St Louis, MO, USA), pH 7, suplementado com 10% de SFB e incubados a 37 ºC, em tensão de 5% de CO2. Ensaios antiproliferativos e de citotoxicidade in vitro A atividade antiproliferativa in vitro contra formas promastigotas e amastigotas axênicas de Leishmania amazonensis foi realizado como descrito por Britta et al.,17 enquanto que o ensaio citotóxico in vitro em macrófagos de linhagem J774.A1 foi descrito por Volpato et al.18
RESULTADOS E DISCUSSÃO O extrato bruto clorofórmico-metanólico de Maxillaria picta foi submetido a um fracionamento com hexano, CHCl3, AcOEt e n-BuOH em ordem de polaridade crescente, que resultou em suas respectivas frações e a fração H2O/MeOH que permaneceu após o particionamento. O extrato bruto e as cinco frações de M. picta foram avaliadas em onze linhagens de células tumorais evidenciando-se que a fração clorofórmica (MPC) foi a que apresentou atividade antiproliferativa mais promissora, com GI50 entre 6,9 e 26,5 mg mL-1 em dez linhagens de células. As demais frações ou apresentaram uma atividade mais fraca do que MPC ou foram inativas nas condições avaliadas. Vale a pena ressaltar ainda que, apesar de não ter apresentado uma atividade antiproliferativa média, a fração MPM apresentou uma ação citostática seletiva sobre a linhagem MCF-7 (mama, GI50 = 15,6 mg mL-1). Continuando a avaliação, MPC foi solubilizada com os solventes hexano, CHCl3, AcOEt e MeOH, resultando nas subfrações MPCH, MPCC, MPCA e MPCM, respectivamente, as quais também foram avaliadas quanto à atividade antiproliferativa em três linhagens tumorais humanas. Foi possível evidenciar que o fracionamento concentrou as substâncias com atividade antiproliferativa nas subfrações com CHCl3 (MPCC, média GI50 = 9,8 mg mL-1) e com AcOEt (MPCA, média GI50 = 7,2 mg mL-1), o que potencializou a ação citostática da fração MPC original. Por outro lado, as outras duas subfrações foram bem menos ativas do que a fração MPC original [MPCH (média GI50 = 61,5 mg mL-1) e MPCM (média GI50 = 51,2 mg mL-1)]. Todos esses resultados estão expostos na Tabela 1.
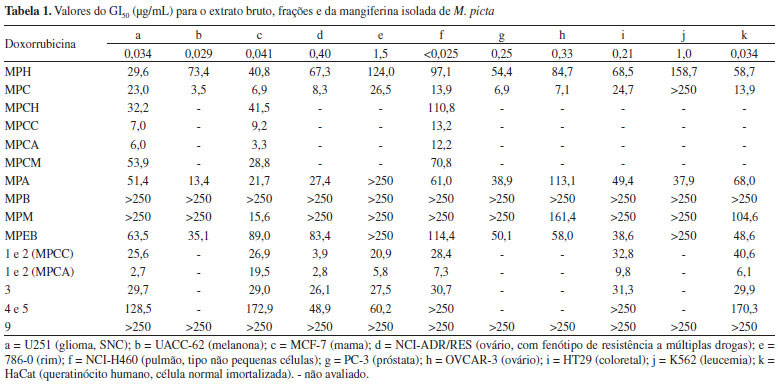
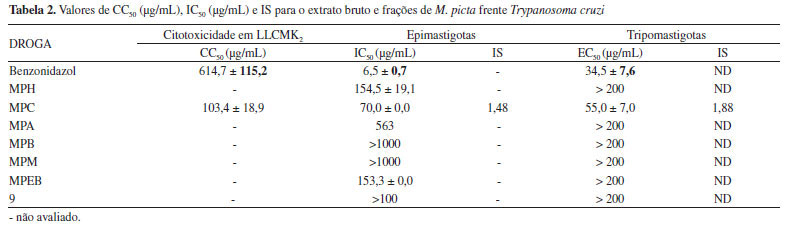
O extrato bruto e frações de M. picta também foram avaliados quanto à atividade antiproliferativa contra formas epimastigotas e tripomastigotas de T. cruzi. A fração MPC apresentou atividade antiproliferativa sobre as formas epimastigotas (IC50 70 µg mL-1) bem como afetou de modo expressivo a viabilidade das formas tripomastigotas (EC50 55 µg mL-1). Por manifestar ação relevante sobre esta última forma, a fração clorofórmica foi, então, submetida ao ensaio de citotoxicidade em células LLCMK2, apresentando uma CC50 de 103,35 µg mL-1, conforme observado na Tabela 2.
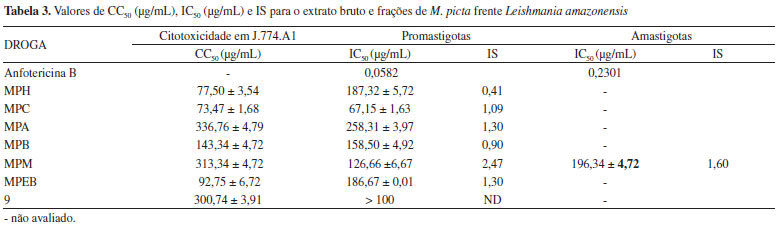
A avaliação da atividade antiproliferativa do extrato bruto, frações e substância precipitada durante a preparação dos extratos (mangiferina), contra formas promastigotas de L. amazonensis, mostrou que a fração MPC apresentou a melhor atividade sobre as formas evolutivas do parasito, com um IC50 de 67,15 µg mL-1. Já a fração H2O/MeOH apresentou moderada atividade sobre promastigotas (IC50 126,66 µg mL-1). No entanto, os valores de CC50 relativos a algumas frações suplantaram os IC50 das mesmas, sugerindo que a atividade está diretamente relacionada à citotoxicidade. As frações MPC, MPM, MPA apresentaram índice de seletividade (CC50/IC50) maior que 1, denotando que essas frações em particular são mais ativas contra os protozoários e menos ativas contra as células de mamíferos. Dentre essas três frações, a MPM foi a que apresentou o melhor índice de seletividade sendo, portanto, submetida ao ensaio antiproliferativo contra formas amastigotas axênicas de L. amazonensis, que resultou em uma IC50 de 196,34 µg mL-1 (Tabela 3).
Considerando a ação significativa nas formas tripomastigotas e epimastigotas de T. cruzi e formas promastigotas de L. amazonensis da fração MPC, bem como os bons resultados da atividade antiproliferativa em células tumorais das subfração MPCA, esta subfração foi selecionada para dar início ao isolamento dos constituintes químicos de M. picta. A filtração em Sephadex LH-20 dessa subfração resultou na identificação da mistura dos estilbenos foiumbeno B (1) e foiumbeno C (2), na proporção 6:1, e também do ácido florético (3).5,19 A mistura de estilbenos também foi isolada da subfração MPCC na proporção 3:2. Adicionalmente, outros nove compostos foram obtidos e identificados de outras frações, tais como os triterpenos eburicol (4) e 24-metilenocicloartenol (5),20,21 dos compostos esteroidais sitosterol (6), estigmasterol (7) e campesterol (8),22 a xantona mangiferina (9),23 o esteroide glicosilado daucosterol (10)24 e os ácidos 4-O-cafeoilchiquímico (11),25 5-O-cafeoilchiquímico (12) e 5-O-cafeoilquínico (13).26,27 As estruturas das substâncias isoladas foram elucidadas com base na análise dos dados espectroscópicos (RMN 1H e 13C, DEPT, COSY, HMQC, HMBC e NOESY), por comparação com os dados disponíveis na literatura. A mistura das substâncias 1 e 2 foi isolada como um óleo avermelhado. No cromatograma obtido na análise por CG-EM (70 eV) para a amostra, observou-se a existência de duas substâncias, com tempos de retenção de 16,82 min e 17,96 min. A substância 1, com o tempo de retenção 17,96 min, apresentou um espectro de massas com o pico do íon molecular em m/z 302. O espectro de RMN de 1H mostrou dupletos em δH 6,90 (H-α') e δH 7,24 (H-α) ambas com J = 16,5 Hz, indicando uma configuração trans para esses hidrogênios e dupletos em δH 6,63 (J = 8,7 Hz, H-5') e em δH 7,24 (J = 8,7 Hz, H-6'), indicando a presença de um anel aromático 1, 2, 3, 4 substituído. O tripleto em δH 6,26 (J = 1,8 Hz), juntamente com os multipletos em δH 6,54 e δH 6,57, sugerem a presença de um anel aromático 1, 3 e 5 substituído. Também foram observados sinais referentes de hidrogênios oximetílicos em δH 3,87, δH 3,86 e δH 3,78. O espectro de RMN de 13C apresentou sinais em δC 128,3 (C-α') e δC 124,0 (C-α), correspondentes à ligação dupla e sinais em δC 61,6, δC 61,1 e δC 55,6, correspondendo aos carbonos oximetílicos. Os demais sinais foram atribuídos aos carbonos dos anéis aromáticos. Através do mapa de contornos HMBC foi estabelecida a posição dos grupos oximetílicos, onde os hidrogênios localizados em δH 3,78 apresentaram correlação com o carbono C-5 em δC 162,5 além das correlações do sinal em δH 3,87 com o carbono C-2', em δC 153,0 e em δH 3,86 com C-3', em δC 142,2. Os dados espectroscópicos de 1 foram concordantes com os dados da literatura para o trans-3,4'dihidróxi-2',3',5-trimetóxiestilbeno ou foiumbeno B.5 A substância 2, com o tempo de retenção de 16,82 min., apresentou um espectro de massas com o pico do íon molecular em m/z 272. A análise do espectro de RMN de 1H para a substância 2 também apresentou sinais característicos de uma configuração trans, em δH 7,04 (H-α') e δH 7,33 (H-α), além de sinais indicando a existência de um segundo anel aromático 1, 3 e 5 substituído. Também foram observados duplos dupletos em δH 7,12 (J = 1,2 e 8,1 Hz) e em δH 6,77 (J = 1,2 e 8,1 Hz), além de um tripleto em δH 6,93 (J = 8,1 Hz), referente aos H-4', H-6' e H-5', respectivamente, indicando a presença de um anel aromático 1, 2 e 3 substituído. Os sinais em δH 3,80 e δH 3,78 foram atribuídos aos hidrogênios oximetílicos. O espectro de RMN de 13C mostrou, entre outros, sinais em δC 61,4 e δC 55,7 correspondendo aos carbonos oximetílicos. A posição dos grupos oximetílicos foi confirmada no mapa de contornos HMBC, onde observou-se a correlação dos hidrogênios oximetilênicos localizados em δH 3,78 com o carbono C-5 em δC 162,6 e entre os hidrogênios em δH 3,80 com o C-2', em δC 147,0. Os dados espectroscópicos de 2 foram comparados com os dados existentes na literatura para o trans-3,3'dihidróxi-2',5-dimetóxiestilbeno ou foiumbeno C, sendo concordantes.5
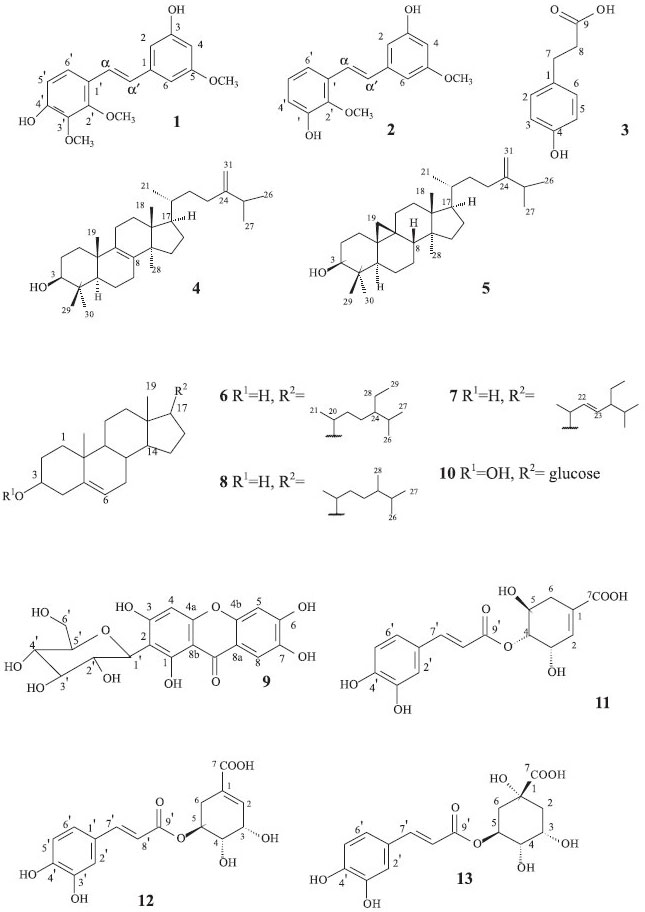 Figura 1. Estruturas dos metabólitos secundários isolados de M. picta
No estudo realizado por Wang et al,28 testou-se a atividade inibitória dos estilbenos isolados a partir da orquídea Pholidota yunnanensis, frente a células de carcinoma hepatocelular (HepG2) e células de hepatoma humano (FHCC-98). Nesse estudo, o Foiumbeno C apresentou uma pequena atividade inibitória sobre a linhagem HepG2, mas não inibiu o crescimento da linhagem FHCC-98, enquanto que o Foiumbeno B mostrou-se capaz de inibir fortemente essas duas linhagens. A presença destas substâncias na subfração CHCl3-AcOEt obtida a partir de M. picta permite compreender, portanto, sua grande atividade citostática frente a diversas linhagens de células tumorais humanas testadas no presente trabalho. A substância 3 foi isolada como um sólido avermelhado solúvel em AcOEt e seu espectro de massas de baixa resolução apresentou o pico do íon molecular em m/z 166. O espectro de RMN 1H mostrou dupletos em δH 7,05 (J = 7,8 Hz, H-2 e H-6) e δH 6,73 (J = 8,4 Hz, H-3 e H-5), sugerindo a presença de um anel aromático p-substituído. Também foram observados tripletos em δH 2,85 (J = 7,8 Hz) e δH 2,56 (J = 7,8 Hz), correspondendo aos hidrogênios H-7 e H-8, respectivamente. No espectro de RMN 13C foi observado um sinal de carbono carbonílico C-9 em δC 177,8. Os dados espectroscópicos de RMN de 1H e 13C foram comparados com os da literatura e se mostraram concordantes para o ácido 3-(4-hidroxifenil)-propiônico ou ácido florético.15 A mistura das substâncias 4 (majoritária) e 5 foi isolada na forma de um sólido amarelado amorfo solúvel em CHCl3. O cromatograma obtido na análise por CG-EM para a amostra mostrou a presença de duas substâncias, com tempos de retenção de 24,86 min e 25,94 min, e o espectro de massas apresentou o pico do íon molecular em m/z 440 nesses dois tempos de retenção. O espectro de RMN 1H para essas substâncias mostrou um singleto em δH 4,71 e um dupleto em δH 4,66 com constante de acoplamento de 1,2 Hz característico de acoplamento geminal (H-31). Também foram observados dupletos em δH 0,92 (J = 6,3 Hz), δH 1,03 (J = 6,6 Hz) e 1,02 (J = 7,2 Hz) referentes aos grupos metílicos C-21, C-26 e C-27. Nos espectros de RMN de 13C e DEPT foram observados sinais na região de carbonos olefínicos em δC 157,1 (C-24) e δC 106,1 (C-28), para as duas substâncias, além de um sinal adicional em δC 134,6 (C-8 e C-9) para 4. Em δC 79,2 e δC 79,0 foram observados os sinais correspondentes ao carbono carbinólico (C-3) das substâncias 4 e 5, respectivamente. Os dados espectroscópicos para as substâncias 4 e 5 foram condizentes com os da literatura para o triterpeno eburicol (24-metileno-24,25-dihidrolanosterol) e para o triterpeno cicloartânico 24-metilenocicloartenol.20,21 A mistura de substâncias 6, 7 e 8 foi isolada sob a forma de um sólido branco solúvel em clorofórmio e caracterizada como sendo os esteroides sitosterol, estigmasterol e campesterol por conta dos dados de RMN serem concordantes com os da literatura.22 Através da comparação dos dados de RMN da substância 9 com os constantes na literatura para a xantona mangiferina,23 concluiu-se que trata-se deste composto, na forma de um sólido amorfo amarelado solúvel em DMSO. Estudos recentes mostraram que a mangiferina apresenta grande atividade antihiperlipidêmica, antidiabética e antiaterogênica em ratos com diabetes mellitus,29,30 além de ser eficaz na prevenção de câncer intestinal neste tipo de animal.31 A molécula também mostrou bons resultados nos ensaios relacionados à atividade inibitória sobre a replicação do HIV-1 e antioxidante,32,33 além de outras ações farmacológicas como antihelmíntica,34 radioprotetora e imunomodulatória.35,36 Tais relatos mostram que a mangiferina é uma substância promissora para o desenvolvimento de novas terapias no combate a diversas doenças. O composto 10 foi obtido com um sólido branco e identificado como sendo o esteroide glicosilado dauscosterol de acordo com a comparação dos dados espectroscópicos de RMN com a literatura.24 Os compostos 11 e 12 foram obtidos como um sólido marrom e solúvel em MeOH. Foram observados, através do espectro de RMN de 1H, sinais característicos de uma unidade cafeoíla devido aos sinais correspondentes a aromático 1,3,4-trissubstituído, além de dupletos cuja constante de acoplamento indica uma configuração trans para esses hidrogênios. Também foram observados sinais na região de δH 2,30 a δH 2,90 típicos de hidrogênios metínicos do grupo chiquímico. O espectro de RMN de 13C apresentou sinais referentes aos carbonos metínicos C-2', C-5' e C-6' e sinais característicos de carbonila de éster α,β-insaturado de grupo cafeoíla. Também foram observados sinais de carbonos metilênicos em δC 30,4 e δC 32,2 referentes ao C-6, sinais de carbonos metínicos em δC 135,6 e δC 138,2, correspondentes ao C-2, além de sinais de carbonos carbonílicos (C-7) em δC 168,8 e δC 169,8 para as substâncias 11 e 12, respectivamente. A conectividade do grupo cafeoila com o grupo chiquímico para as substâncias foi confirmada através dos deslocamentos químicos dos hidrogênios em relação aos dados da literatura e foram concordantes para o 4-O-ácido cafeoilchiquímico e 5-O-ácido cafeoilchiquímico.21,22 O composto 13 foi isolado como um sólido marrom solúvel em MeOH. Os dados de RMN da substância foram concordantes aos da literatura para o ácido 5-O-cafeoilquínico.23 A mistura de triterpenos 4 e 5 foi submetida ao ensaio de atividade antiproliferativa in vitro apresentando uma moderada atividade sobre as linhagens NCI-ADR/RES (ovário, com fenótipo de resistência a múltiplas drogas, GI50 = 48,9 µg mL-1) e 786-0 (rim, GI50 = 60,2 µg mL-1), enquanto que o ácido florético (3) apresentou uma ação citostática significativa em todas as linhagens testadas, com GI50 entre 26,1 e 31,3 µg mL-1. A mistura dos estilbenos 1 e 2 proveniente da fração MPCC na proporção 6:1, apresentou boa seletividade sobre a linhagem NCI-ADR/RES (GI50 = 3,9 µg mL-1), enquanto que as demais linhagens obtiveram um GI50 entre 20,9 e 40,6 µg mL-1. No entanto, a mesma mistura isolada na fração MPCA em proporção 3:2, apresentou promissora atividade antiproliferativa, inibindo fortemente o crescimento de todas as linhagens testadas, com valores de GI50 entre 2,7 e 19,5 µg mL-1 (média GI50 = 7,7 µg mL-1). Através desses resultados, pode-se concluir que foiumbeno B (2) é, ao menos em parte, responsável pela atividade observada no extrato bruto clorofórmico-metanólico e na fração clorofórmica. A mangiferina (9), avaliada nas onze linhagens celulares tumorais humanas, mostrou-se inativa nas condições empregadas.
CONCLUSÃO A fração MPC de M. picta apresentou atividade citostática contra diversas linhagens de células cancerosas humanas e uma efetiva atividade sobre a viabilidade das diferentes formas dos parasitos T. cruzi e L. amazonensis. O estudo da fração MPC da espécie vegetal M. picta resultou no isolamento dos compostos esteroidais campesterol, sitosterol e estigmasterol, dos triterpenos eburicol e 24-metilenocicloartenol, além do ácido florético e da mistura de estilbenos foiumbeno B e foiumbeno C. A mistura contendo uma maior proporção do estilbeno foiumbeno B foi a que apresentou o resultado mais promissor, concluindo-se que a substância é a principal responsável pela atividade observada no extrato bruto, frações e subfrações da espécie vegetal. Adicionalmente foram isolados o 4-O-cafeoilchiquímico, 5-O-cafeoilchiquímico, 5-O-cafeoilquínico, o esteroide glicosilado daucosterol e a xantona mangiferina. Todos esses compostos são inéditos para o gênero Maxillaria.
MATERIAL SUPLEMENTAR Os espectros de RMN unidimensionais das substâncias 1 e 2, bem como os dados espectroscópicos das substâncias 1, 2, 3, 4, 11 e 12 encontram-se disponíveis na forma de arquivo PDF, disponível em http://quimicanova.sbq.org.br, com acesso livre.
AGRADECIMENTOS Os autores agradecem à Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), pelo suporte financeiro.
REFERÊNCIAS BIBLIOGRÁFICAS 1. Cechinel Filho, V.; Quim. Nova 2000, 23, 680. 2. Wang, H.; Zhao, T.; Che, C. T.; J. Nat. Prod. 1985, 48, 796. 3. Hwang, J. S.; Lee, S. A.; Hong, S. S.; Han, X. H.; Lee, C.; Kang, S. J.; Lee, D.; Kim, Y.; Hong, J. T.; Lee, M. K.; Hwang.; Bioorg. Med. Chem. Lett. 2010, 20, 3785. 4. Pomini, A. M.; Santin, S. M. O.; Silva, C. C.; Faria, T. J.; Faria, R. T.; Ruiz, A. L. T. G.; Carvalho, J. E. C.; Dela Porte, L. F.; Planta Med. 2012, 78, 1169. 5. Guo, X. Y.; Wang, J.; Wang, N. L.; Kitanaka, S.; Liu, H. W.; Yao, X. S.; Biol. Pharm. Bull. 2006, 54, 21. 6. Sinha, A. K.; Verma, S. C.; Sharma, U. K.; J. Sep. Sci. 2007, 30, 15. 7. Gutiérrez, R. P. M.; J. Med. Plants Res. 2010, 4, 592. 8. Estrada, S.; López-Guerrero, J. J.; Villalobos-Molina, R.; Mata, R.; Fitoterapia 2004, 75, 690. 9. Déciga-Campos, M.; Rivero-Cruz, I.; Arriaga-Alba, M.; Castañeda-Corral, G.; Angeles-López G. E.; Navarrete, A.; Mata, R.; J. Ethnopharmacol. 2007, 110, 334. 10. Singer, R. B.; Koehler, S.; Lankesteriana 2003, 7, 57. 11. Singer, R. B.; Lankesteriana 2003, 7, 111. 12. Flach, A.; Tese de doutorado, Universidade Estadual de Campinas, Brasil, 2005. 13. Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A.; Gray-Goodrich, M.; Campbell, H.; Mayo, J.; Boyd, M.; J. Natl. Cancer Inst. 1991, 83, 757. 14. Mossmann, T.; J. Immunol. Methods 1983, 65, 55. 15. Brener, Z.; Rev. Inst. Med. Trop. Sao Paulo 1962, 4, 386. 16. Ueda-Nakamura, T.; Attias, M.; Souza, W.; Parasitol. Res. 2001, 87, 89. 17. Brita, E. A.; Silva, A. P. B.; Ueda-Nakamura, T.; Dias-Filho, B. P.; Silva, C. C.; Sernaglia, R. L.; Nakamura, C. V.; PLoS One 2012, 7, 12. 18. Volpato, H.; Desoti, V. C.; Cogo, J.; Panice, M. R.; Sarragiotto, M. H.; Silva, S. O.; Ueda-Nakamura, T.; Nakamura, C. V.; Evid Based Complement Alternat Med (2013), doi:10.1155/2013/874367. 19. Owen, R. W.; Haubner, R.; Mier, W.; Giacosa, A.; Hull, W. E.; Spiegelhalder, B.; Bartsch, H.; Food Chem. Toxicol. 2003, 41, 703. 20. Shirane, N.; Murabayashi, A., Masuko, M.; Uomori, A.; Yoshimura, Y.; Seo, S.; Uchida, K.; Takeda, K.; Phytochemistry 1990, 29, 2513. 21. Lee, S.Y.; Choi, S. U.; Lee, J. H.; Lee D. U.; Lee K. R.; Arch. Pharmacal Res. 2010, 33, 515. 22. Goulart, M. O. F.; Sant'ana, A. E. G.; Lima, R. A.; Calvacante, S.; Carvalho, M. G.; Braz-Filho, R.; Quim. Nova 1993, 16, 95. 23. Kim, C. Y.; Ahn, M, J.; Kim, J.; J. Liq. Chromatogr. Relat. Technol. 2007, 29, 869. 24. Lendl, A.; Werner, I.; Glasl, S.; Kletter, C.; Mucaji, P.; Presser, A.; Reznicek, G.; Jurenitsch, J.; Taylor, D.; Phytochemistry 2005, 66, 2381. 25. Saito, T.; Yamane, H.; Murofushi, N.; Takahashi, T.; Phinney, B. O.; Biosci., Biotechnol., Biochem. 1997, 61, 1397. 26. Ivanova A.; Mikhova, B.; Kostova, I.; Evstatieva L.; Chem. Nat. Compd. 2010, 46, 294. 27. Satake, T.; Biol. Pharm. Bull. 2007, 30, 935. 28. Wang, G.; Guo, X.; Chen, H; Lin, T.; Xu, Y.; Chen, Q.; Liu, J.; Zeng, J.; Zhang, X. K.; Yao, X.; Bioorg. Med. Chem. Lett. 2012, 22, 2114. 29. Maruganandan, S.; Gupta, S.; Kataria, M.; Lal, J.; Gupta, P. K.; Toxicology 2002, 3, 165. 30. Maruganandan, S.; Gupta, S.; Kataria, M.; Lal, J.; Gupta, P. K.; J. Ethnopharmacol. 2005, 97, 497. 31. Yoshimi, N.; Matsunaga, K.; Katayama, M.; Yamada, Y.; Kuno, T.; Qiao, Z.; Hara, A.; Yamahara, J.; Mori, H.; Cancer Lett. 2001, 163, 163. 32. Wang, R. R.; Gao, Y. D.; Ma, C. H.; Zhang, X. J.; Huang, C. G.; Huang, J. F.; Zheng, Y. T.; Molecules 2011, 16, 4264. 33. Chen, Y. H.; Chang, F. R.; Lin, Y. J.; Hsieh, P. W.; Wub, M. J.; Wu, Y. C.. Food Chem. 2008, 2, 684. 34. García, D.; Escalante, M.; Delgado, R.; Ubeira, F. M.; Leiro, J.; Phytother. Res. 2003, 17, 1203. 35. Jagetia, G. C.; Baliga, M. S.; Phytomedicine 2005, 12, 209. 36. Leiro, J.; Arranz, J. A.; Yáñes, M.; Ubeira, F. M.; Sanmartín, M. L.; Orallo, F.; Int. J. Immunopharmacol. 2004, 4, 763. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access