
 |
 |
 |
 |
 |
Artigo
|
|
| Desenvolvimento de procedimento de extração para análise de sedativos e β-bloqueadores em rim suíno Development of extraction procedure for analysis of sedatives and β-blockers in swine kidney |
|
Lenise G. de OliveiraI,*; Marcia H. S. KurzI; Fábio F. GonçalvesI; Fabiano BarretoII; Gabriel RübensamII; Rodrigo HoffII
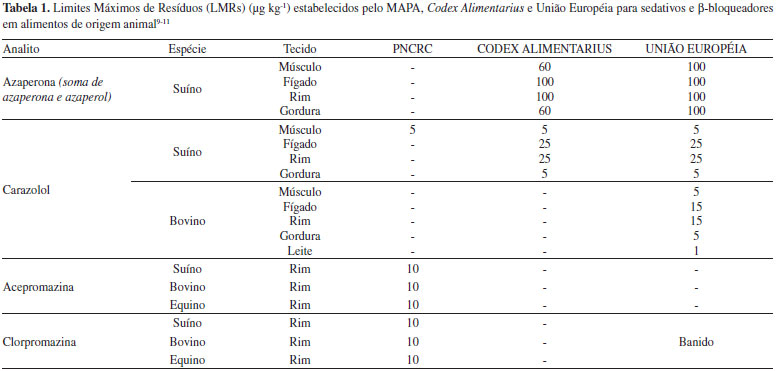
IEscola de Química e Alimentos, Universidade Federal do Rio Grande, 95500-000 Santo Antônio da Patrulha - RS, Brasil Recebido em 13/02/2014 *e-mail: leniseoliveira@furg.com A procedure was developed for determination of 5 sedatives and 14 β-blockers in swine kidney and subsequent analysis by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS). Three different procedures for extraction were tested, evaluated through recovery studies. The procedure using acetonitrile for extraction and cleanup with freezing at low temperature and dispersive solid phase extraction using 500 mg celiter 545 before the concentration step presented the better results. The dried samples were redissolved with methanol and analyzed using a LC-MS/MS system with electrospray ionization (ESI) operating in positive MRM mode. The recovery values for this procedure were in the 75-88% range. The robustness of the method was tested against small variations. The method was used to analyze carazolol, azaperone and azaperol in collaborative assay, obtaining results close to designed value. INTRODUÇAO O Brasil é destaque mundial na produçao de alimentos e, neste contexto, o setor de carnes vem crescendo gradativamente, impulsionado tanto pelo mercado interno quanto externo. Para suprir a demanda de mercado, o produtor pecuário nacional tem cada vez mais lançado mao de estratégias de manejo que primam pelo aumento do sucesso da cadeia produtiva de carne, pela melhoria do produto final e pela manutençao da saúde e bem-estar animal, conforme estabelecido nas Boas Práticas de Manejo (BPM).1 O emprego de medicamentos veterinários tem se configurado como uma prática-chave ao longo das diferentes etapas de produçao animal, pois reduz a incidência de doenças e melhora a qualidade de vida dos animais. No Brasil, as drogas veterinárias de maior emprego sao as antiparasitárias e as antimicrobianas2 devido, principalmente, ao regime de manejo (predominantemente extensivo) e às condiçoes climáticas (temperatura e umidade elevada) dos locais de produçao. No entanto, outras classes de drogas sao usadas com fins mais específicos. Este é o caso dos sedativos e β-bloqueadores. Tais compostos têm sido usados principalmente em suínos com o objetivo primário de reduzir a atividade excessiva e ansiedade dos animais durante seu transporte, além de possibilitar o aumento da taxa de conversao alimentar e reduzir o aparecimento de carnes do tipo PSE, - Pale, Soft e Exudative, que significa pálida, mole e exsudativa.3,4 Os suínos sao animais extremamente suscetíveis ao estresse durante o transporte podendo vir a óbito em decorrência de complicaçoes cardíacas.5 Assim, tanto os sedativos quanto os β-bloqueadores têm contribuído com a reduçao de perdas e a melhoria do bem-estar animal durante transporte. Entretanto, esses compostos apresentam a característica de acúmulo em tecidos, principalmente no rim e no fígado, e podem permanecer como resíduo nos alimentos caso a administraçao dos medicamentos seja feita poucas horas antes do abate.6,7 No que diz respeito aos resíduos de medicamentos veterinários em alimentos, diversos órgaos oficiais, nacionais e internacionais, determinam, por meio de suas normativas, os Limites Máximos de Resíduos (LMR) que podem estar presentes nos alimentos, considerando a ingestao diária aceitável do composto (IDA), protegendo a saúde do consumidor e garantindo a segurança do alimento oferecido para consumo humano através de programas de monitoramento.8 As normas referentes a LMRs para resíduos de medicamentos veterinários do Mercado Comum do Sul (MERCOSUL) e Food and Drug Administration (FDA) nao apresentam valores estabelecidos para sedativos e β-bloqueadores. Já o Programa Nacional de Controle de Resíduos e Contaminantes em Produtos de Origem Animal (PNCRC), do Ministério da Agricultura, Pecuária e Abastecimento (MAPA), o Codex Alimentarius e a Comunidade Europeia, por meio das suas normativas,9-11 apresentam limites para alguns compostos destas classes. Estes limites estao descritos na Tabela 1.
O monitoramento destes resíduos só pode ser realizado se métodos analíticos que garantam resultados seguros estejam disponíveis. Faz-se necessário, portanto, que os procedimentos analíticos desenvolvidos sejam rápidos, fáceis e de baixo custo, viabilizando o monitoramento em larga escala. Os primeiros métodos de extraçao descritos para análise de sedativos e β-bloqueadores utilizavam detectores do tipo ultravioleta (UV) e fluorescência (FL) associados à cromatografia, e necessitavam de grande quantidade de amostra e reagentes, envolvendo inúmeras etapas complexas e laboriosas.12-18 No final dos anos 90, a utilizaçao de cromatografia líquida acoplada à espectrometria de massas sequencial (LC-MS/MS) ganhou espaço, permitindo a utilizaçao de menor quantidade de amostra e solventes, além de possibilitar a análise de maior número de compostos e identificaçao inequívoca destes, mesmo em baixas concentraçoes.5,19-27 Nos últimos anos, os métodos publicados têm apresentado muitas etapas de extraçao em comum. Basicamente, os procedimentos consistem na extraçao por solvente orgânico (geralmente, acetonitrila), separaçao extrato/matriz por centrifugaçao, seguida de evaporaçao sob fluxo de N2 a temperatura controlada. Após retomada da amostra com solvente, o extrato é submetido à etapa de purificaçao por meio da técnica de Extraçao em Fase Sólida (SPE), utilizando diferentes sorventes como sílica, C18 e NH2. A determinaçao de 19 β-bloqueadores e 11 sedativos em tecido animal (rim, fígado e músculo suíno e músculo bovino) foi realizada usando cromatografia líquida de alta eficiência (CLAE) acoplado à espectrometria de massas sequencial. As amostras foram extraídas com acetonitrila sob sonicaçao por 20 min a 40 ºC. O sobrenadante foi centrifugado e o resíduo reextraído. Os sobrenadantes combinados foram concentrados em evaporador rotatório e redissolvidos para purificaçao. Cartuchos de fase reversa (Oasis HLB e C18) e de fase normal (sílica e NH2) foram testados. A purificaçao usando cartuchos NH2 apresentou melhores recuperaçoes e menor efeito matriz.25 Recentemente, foi desenvolvido um método para determinaçao de 25 β2-agonistas e 23 β-bloqueadores em alimentos de origem animal (carne, fígado e rim de porco e amostras de frango). O método consistiu de hidrólise e extraçao com 10 mL de ácido tricloroacético 5% seguido de sonicaçao por 30 min. O meio fortemente ácido também proporcionou a desnaturaçao e precipitaçao de proteínas. A purificaçao consistiu na utilizaçao de cartuchos de SPE mistos (C8 ou C18 combinados com trocador catiônico forte) (MCX) seguido de um passo de purificaçao adicional com metanol e análise por CLAE acoplado a espectrometria de massas ion trap linear.27 Este trabalho consistiu no desenvolvimento de procedimento de extraçao adequado para determinaçao simultânea de resíduos de 5 sedativos (acepromazina, azaperol, azaperona, clorpromazina, xilazina) e 14 β-bloqueadores (acebutolol, atenolol, betaxolol, bisoprolol, carazolol, carvedilol, labtalol, metoprolol, nadolol, nebivolol, penbutolol, pindolol, propanolol, sotalol) em rim suíno e posterior análise por LC-MS/MS. O trabalho foi realizado em parceria com o Laboratório Nacional Agropecuário (LANAGRO/RS), nas instalaçoes do Laboratório de Resíduos de Pesticidas e Medicamentos Veterinários (RPM), que juntamente com demais laboratórios da rede do MAPA, integram a área técnica do PNCRC.
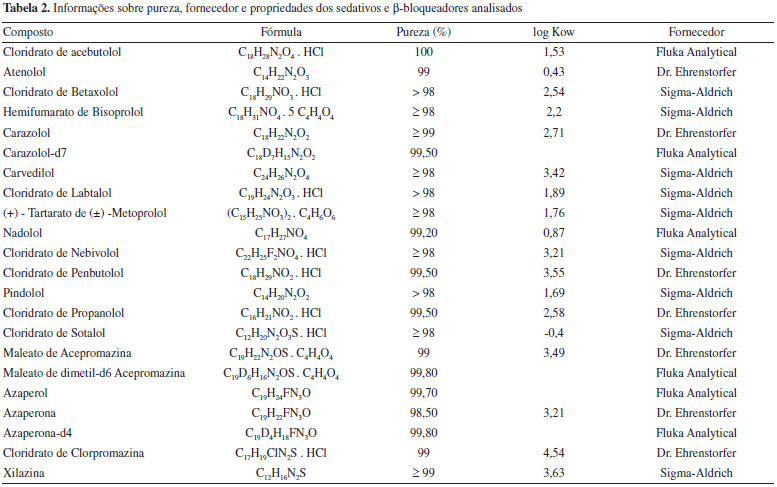
PARTE EXPERIMENTAL Reagentes e solventes Os padroes analíticos dos compostos foram adquiridos de 3 diferentes fornecedores: Dr. Ehrenstorfer GmH (Alemanha), Sigma-Aldrich (EUA) e Fluka Analytical (EUA). Além dos compostos que fazem parte do estudo, também foram utilizados 3 padroes deuterados como padroes internos (acepromazina-d6, azaperona-d4 e carazolol-d7). Os dados referentes a pureza, coeficiente de partiçao octanol-água (log Kow) e fornecedor dos padroes utilizados neste trabalho estao descritos na Tabela 2.
Os solventes utilizados foram: acetonitrila (ACN) grau HPLC (Sulpra Solv, Alemanha), metanol (MeOH) grau HPLC (Tedia Brasil, Brasil) e água ultrapurificada com ultrapurificador de água Master System (Gehaka, Brasil). Sorvente celite® 545 P.A. foi adquirido da empresa Impex (Brasil), cloreto de sódio 99% P.A. da empresa Dinamica (Brasil) e gás nitrogênio 99,996% da empresa IBG (Brasil).
EQUIPAMENTO O sistema LC-MS/MS utilizado consistiu em um cromatógrafo Agilent série 1260 (Agilent, EUA), acoplado a um espectrômetro de massas QTrap 5500 (ABSciex, EUA), equipado com uma fonte de ionizaçao por eletronebulizaçao (ESI) operada no modo positivo. A separaçao cromatográfica foi realizada em uma coluna Poroshell 120 EC-C18 (3,0 x 50 mm, 2,7 µm) provida pela Agilent. Os dados foram adquiridos e tratados utilizando o sistema de aquisiçao Analyst 1.6.2 (ABSciex). Preparo das soluçoes analíticas As soluçoes estoque de 1000 mg L-1 foram preparadas individualmente em metanol e armazenadas em frascos de polipropileno revestidos com papel alumínio para evitar degradaçao, e armazenados em freezer a -20 ºC. A soluçao intermediária foi preparada a partir da diluiçao de cada soluçao estoque com metanol, de forma a obter uma soluçao com todos os analitos na concentraçao de 10 mg L-1. A soluçao dos padroes internos (10 mg L-1) foi preparada de forma similar, exceto para carazolol e carazolol-d7 cujas concentraçoes finais foram 5 mg L-1. A partir das soluçoes intermediárias, foram preparadas soluçoes de trabalho ou soluçoes de fortificaçao com concentraçoes de 1 mg L-1, exceto para carazolol e carazolol-d7 (0,5 mg L-1), sempre em metanol. Estudo de estabilidade A estabilidade foi verificada a partir da análise dos compostos em soluçao e extrato. Para a determinaçao da estabilidade em soluçao, foram analisadas as soluçoes de fortificaçao (1 mg L-1) preparadas semanalmente, de forma a obter tempo de armazenamento de aproximadamente 5 semanas. Três alíquotas de cada soluçao foram diluídas para obtençao de soluçao com concentraçao correspondente a 20 µg L-1 e injetadas para obtençao de média. O cálculo da concentraçao do analito em cada soluçao analisada foi realizado por comparaçao com a soluçao de mesma concentraçao teórica recém diluída, que foi considerada 100%, conforme Equaçao 1.
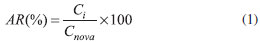
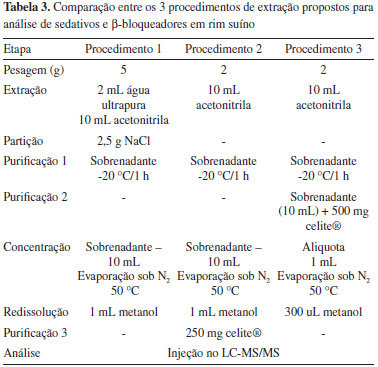
na qual AR corresponde a variaçao do analito remanescente, Ci é a concentraçao média da soluçao e Cnova é a concentraçao média da soluçao nova. Para determinaçao da estabilidade do analito em extrato, foi realizado fortificaçao em extrato obtido de amostra branca, sendo analisado no dia do preparo e 24 h após o preparo. Durante este período, elas permaneceram a uma temperatura de 20 ºC em frasco de polipropileno. Foi utilizado o mesmo cálculo descrito anteriormente, onde Cnova será a concentraçao obtida na análise realizada no dia do preparo. Amostras As amostras brancas de rim suíno foram obtidas junto a estabelecimentos sob Serviço de Inspeçao Federal (SIF) do MAPA. Foram retiradas pequenas porçoes da parte externa do órgao, já isento de cápsula fibrosa, e processadas até homogeneizaçao com auxílio de processador de amostras. A amostra foi acondicionada e mantida em freezer a -20 ºC, em recipiente plástico, para posterior utilizaçao. Otimizaçao do método por LC-MS/MS Condiçoes cromatográficas A escolha da fase móvel foi realizada inicialmente através de revisao bibliográfica. Foi utilizado (A) soluçao aquosa acidificada com 0,1% de ácido fórmico (v/v) e (B) metanol acidificado com 0,1% de ácido fórmico (v/v).25 Também foi testada a seguinte composiçao: (A) soluçao aquosa acidificada com 0,1% de ácido fórmico (v/v) e (B) acetonitrila acidificada com 0,1% de ácido fórmico (v/v). Sistema de detecçao Para otimizaçao dos parâmetros dependentes do composto, inicialmente foi realizado infusao dos compostos em solvente na concentraçao de aproximadamente 200 µg L-1 com o objetivo de selecionar no mínimo 2 fragmentos (íons produto) de maior intensidade ou seletividade para cada analito, sendo o mais intenso utilizado como íon quantificador e o segundo como qualificador. Para otimizaçao dos parâmetros dependentes da fonte de ionizaçao foram realizados os experimentos de injeçao em fluxo (Flow Injection Analysis, FIA) visando obter as melhores condiçoes para o conjunto de compostos. Desenvolvimento e otimizaçao dos procedimentos de extraçao Três métodos de extraçao foram propostos com o objetivo de verificar qual apresentaria melhor recuperaçao, além de produzir extratos com menor concentraçao possível de coextrativos para análise de sedativos e β-bloqueadores em rim suíno por LC-MS/MS. Procedimento 1 O procedimento 1 consistiu na pesagem de 5 g de amostras brancas em tubos de polipropileno com capacidade para 50 mL e fortificadas na concentraçao de 10 µg kg-1 (apenas A1, A2 e A3). Após homogeneizaçao em vórtex por 20 s, foi realizada adiçao de 2 mL de água ultrapura e 10 mL de acetonitrila. As amostras foram trituradas e homogeneizadas em ultra-turrax e, em seguida, agitadas em mesa agitadora a 180 rpm por 20 min. Após agitaçao foi adicionado 2,5 g de NaCl, seguido de nova agitaçao por 5 min nas mesmas condiçoes anteriores. As amostras foram centrifugadas a 5670 g por 5 min a 5 ºC, o sobrenadante foi transferido para tubo de polipropileno de 15 mL e acondicionado em freezer a -20 ºC por 1 h. Posteriormente, a amostra foi centrifugada a 5670 g e 0 ºC por 20 min, o sobrenadante foi transferido para tubo de polipropileno de 50 mL e evaporado até secura sob fluxo de N2 a 50 ºC. As amostras secas foram fortificadas na concentraçao de 10 µg kg-1 (apenas B1, B2 e B3) e retomadas com metanol obtendo-se volume de 1 mL, agitadas em vórtex por 20 s e transferidas para tubos eppendorf para centrifugaçao a 16200 g por 10 min a 0 ºC. Após centrifugaçao, as amostras foram transferidas para frasco com capacidade para 2 mL e analisadas por LC-MS/MS. Procedimento 2 O procedimento 2 consistiu na pesagem de 2 g de amostras brancas em tubos de polipropileno com capacidade para 50 mL e fortificadas na concentraçao de 10 µg kg-1 (apenas A1, A2 e A3). Após homogeneizaçao em vórtex por 20 s, foi adicionado 10 mL de acetonitrila. Com auxílio de ultra-turrax as amostras foram trituradas e homogeneizadas. Em seguida, foram colocadas em mesa agitadora por 20 min a 180 rpm. Após agitaçao, as amostras foram centrifugadas a 5670 g por 5 min a 5 ºC. Os sobrenadantes foram transferidos para tubo de polipropileno de 15 mL e acondicionados em freezer a -20 ºC por uma hora. Posteriormente, a amostra foi centrifugada a 5670 g por 20 min a 0 ºC. O sobrenadante foi transferido para tubo de polipropileno de 50 mL e evaporado até secura sob fluxo de N2 a 50 ºC. As amostras secas foram fortificadas na concentraçao de 10 µg kg-1 (apenas B1, B2 e B3) e retomadas com metanol obtendo-se volume de 1 mL, agitadas em vórtex por 20 s, transferidas para tubos eppendorf contendo 250 mg de celite® 545 e novamente homogeneizadas em vórtex por 20 s. Após homogeneizaçao, as amostras em tubos eppendorf foram centrifugadas a 16200 g por 20 min a 0 ºC e transferidas para frasco com capacidade para 2 mL para posterior análise por LC-MS/MS. Procedimento 3 Este procedimento consistiu na pesagem de 2 g de amostras brancas, previamente homogeneizadas, em tubos de polipropileno com capacidade para 50 mL e fortificadas na concentraçao de 10 µg kg-1 (apenas A1, A2 e A3), seguido de adiçao de 10 mL de acetonitrila. Com auxílio de ultra-turrax as amostras foram trituradas e homogeneizadas. Em seguida, foram colocadas em mesa agitadora e agitadas por 20 min a 180 rpm. Após agitaçao, as amostras foram centrifugadas a 5670 g por 5 min a 5 ºC, o sobrenadante foi transferido para tubo de polipropileno de 15 mL e acondicionado em freezer a -20 ºC por uma hora. Posteriormente, a amostra foi centrifugada a 5670 g por 20 min a 0 ºC e o sobrenadante foi transferido para novo tubo de polipropileno de 15 mL contendo 500 mg de celite® 545. As amostras foram agitadas em vórtex por 20 s e novamente submetidas a centrifugaçao a 5670 g por 20 min a 0 ºC. Uma alíquota de 1 mL foi transferida para tubo de polipropileno de 50 mL e evaporada a 50 ºC, sob fluxo de N2 até secura. As amostras foram fortificadas na concentraçao de 10 µg kg-1 (apenas B1, B2 e B3), e retomadas em metanol obtendo-se volume de 300 µL, agitadas em vórtex por 20 s e transferidas para frasco com insert fixo com capacidade para 300 µL para posterior análise por LC-MS/MS. A Tabela 3 apresenta uma comparaçao entre os 3 procedimentos propostos, salientando-se as principais diferenças entre eles.
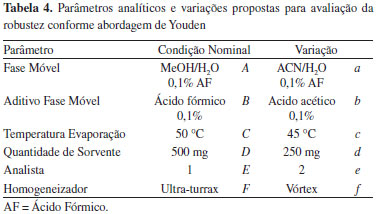
Avaliaçao dos procedimentos de extraçao Recuperaçao A avaliaçao da eficiência dos procedimentos empregados (procedimentos 1, 2 e 3) consistiu na análise das recuperaçoes obtidas para cada composto e também análise das amostras brancas obtidas, de forma a verificar presença de interferentes que prejudicariam a quantificaçao dos compostos. Para isso, foi utilizado amostras brancas fortificadas na concentraçao desejada (10 µg kg-1) antes da extraçao (denominadas A1, A2 e A3), e extratos de amostras brancas fortificadas no final do processo de extraçao (denominadas B1, B2 e B3), além da análise de amostra branca. Robustez O estudo da robustez foi realizado utilizando a abordagem de Youden, que consiste em uma análise multivariada de variáveis, as quais quando testadas nas condiçoes normais sao identificadas por letras maiúsculas e quando alteradas, por letra minúscula. O desvio padrao obtido foi utilizado para análise dos testes realizados.28 Esta abordagem foi utilizada para avaliar a variaçao produzida pelos seguintes fatores: tipo de fase móvel, aditivo utilizado na fase móvel, temperatura de evaporaçao, analista, forma de homogeneizaçao das amostras e quantidade de sorvente em amostras brancas fortificadas na concentraçao de 10 µg kg-1 utilizando procedimento selecionado para análise de sedativos e β-bloqueadores em rim suíno, conforme Tabela 4.
Aplicabilidade do procedimento A aplicabilidade do procedimento foi testada através de análise de amostras de rim suíno enviadas pela Canadian Food Inspection Agency para determinaçao de carazolol, azaperona e azaperol, em ensaio colaborativo. Cinco amostras foram recebidas em embalagem devidamente identificada e lacrada, sendo que uma delas consistia em branco de rim suíno, portanto isenta dos analitos, para construçao de curva de calibraçao. A estimativa da reprodutibilidade dos procedimentos utilizados pelos diferentes laboratórios é dada pela avaliaçao do Z-Score, que considera os resultados enviados por cada um dos laboratórios e os desvios padrao obtidos no ensaio colaborativo, conforme Equaçao 2.

na qual x corresponde ao resultado de cada participante, X à concentraçao atribuída e σp ao desvio padrao obtido no ensaio colaborativo. Valores de Z-Scores inferiores a 2 sao considerados satisfatórios, entre 2 e 3 questionáveis e superiores a 3 insatisfatórios.
RESULTADOS E DISCUSSAO Otimizaçao do método por LC-MS/MS Condiçoes cromatográficas Dois tipos de fases móveis foram testadas durante o processo de otimizaçao do método. Inicialmente foi utilizada fase móvel que consistia em (A) soluçao aquosa acidificada com 0,1% ácido fórmico (v/v) e (B) acetonitrila 0,1% ácido fórmico (v/v). Esta fase móvel foi utilizada com o objetivo de garantir limpeza eficiente da coluna, assegurando sua vida útil. Os resultados obtidos nas análises utilizando esta fase móvel foram satisfatórios e reprodutíveis, porém, com resoluçao insatisfatória para os picos do atenolol e sotalol, além de tempo de eluiçao de aproximadamente 1 minuto para a maioria dos compostos. A segunda fase móvel utilizada consistiu em (A) soluçao aquosa acidificada com 0,1% ácido fórmico (v/v) e (B) metanol 0,1% ácido fórmico (v/v). Os resultados obtidos foram satisfatórios, reprodutíveis, com picos bem resolvidos para atenolol e sotalol, resultando em tempo de eluiçao dos analitos de aproximadamente 3 minutos (Figura 1). A utilizaçao desta fase móvel apresenta como única desvantagem eluiçao de maior quantidade de coextrativos, porém, nenhum destes coelui com os analitos. O aumento do tempo de retençao de compostos coextraídos é compatível com o comportamento de aumento de tempo de retençao dos analitos.
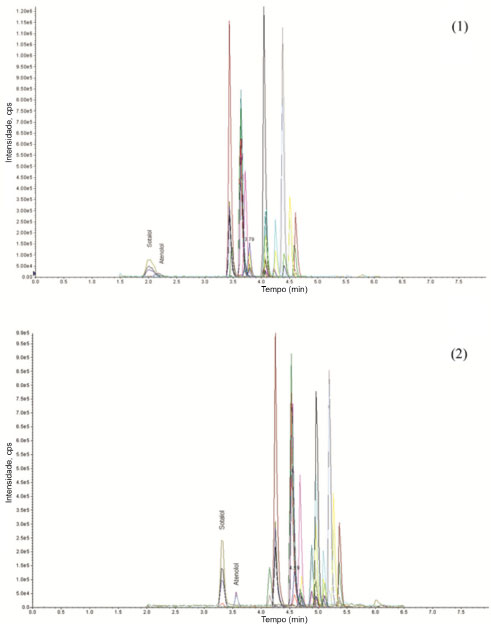 Figura 1. Cromatogramas dos compostos em solvente com concentraçao correspondente a 20 µg L-1, utilizando (1) fase móvel composta por (A) soluçao aquosa acidificada com 0,1% ácido fórmico (v/v) e (B) acetonitrila acidificada com 0,1% de ácido fórmico (v/v); e (2) fase móvel composta por (A) soluçao aquosa acidificada com 0,1% ácido fórmico (v/v) e (B) metanol acidificado com 0,1% de ácido fórmico (v/v)
O tempo necessário para análise cromatográfica de todos os compostos por LC-MS/MS foi de 8 minutos, seguido de 2 minutos de equilíbrio, resultando em um tempo total de 10 minutos para cada injeçao de amostra. As condiçoes cromatográficas utilizadas para análise de resíduos de sedativos e β-bloqueadores em rim suíno por LC-MS/MS estao descritas na Tabela 5.

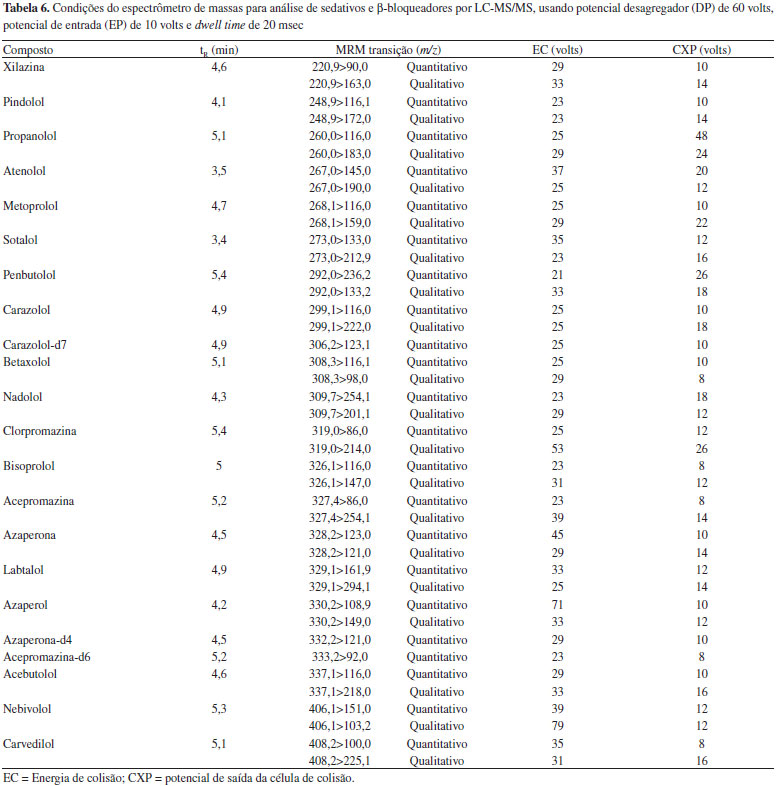
Sistema de detecçao Os parâmetros definidos para análise de sedativos e β-bloqueadores foram obtidos na etapa de infusao dos analitos e otimizaçao usando FIA, os quais sao otimizados automaticamente pelo software Analyst. Os parâmetros da fonte de ionizaçao por eletronebulizaçao no modo positivo que foram usados neste método sao gás de colisao high, voltagem do Spray eletrônico de 5500 volts, temperatura de 700 ºC, gás de nebulizaçao e gás auxiliar de 55 psi, e gás de cortina de 30 volts. Os parâmetros do espectrômetro de massas e tempos de retençao dos compostos estao decritos na Tabela 6.
No decorrer da otimizaçao dos procedimentos de extraçao, alguns analitos apresentaram relaçao sinal/ruído baixos, já que estavam em matriz e nao mais em solvente. Para estes compostos foi realizada nova otimizaçao, desta vez manual, para garantir fragmentaçao eficiente do íon precursor e seleçao de fragmentos que apresentassem maior seletividade. Estudo de estabilidade No estudo de estabilidade, admite-se variaçao de até 15%29 nos resultados obtidos, sendo que para valores acima ou abaixo considera-se que a substância é instável a partir do período avaliado. A estabilidade das soluçoes de fortificaçao preparadas para análise de sedativos e β-bloqueadores, armazenadas a -20 ºC, no escuro, foram monitoradas durante cinco semanas e foi possível observar que todos os compostos permaneceram estáveis neste período. Os resultados de concentraçao obtidos variaram entre 4 e 8%. No estudo de estabilidade dos compostos em extrato, as amostras foram analisadas no dia do preparo e após 24 h. A variaçao dos resultados obtidos ficou entre 2 e 14%, exceto para carvedilol em que o resultado variou cerca de 27%. Assim, considera-se que os analitos sao estáveis tanto em soluçao quanto no extrato no período avaliado, com exceçao do composto carvedilol em extrato na temperatura de 20 ºC. Otimizaçao do procedimento de extraçao A etapa de extraçao dos compostos é uma das mais relevantes, devido à complexidade da matriz e os baixos níveis de concentraçao desejados. A matriz, rim suíno, é constituída basicamente por cerca de 80% de água, 16% de proteínas, 3% de gorduras totais, e 1% de vitaminas e minerais.30 Além disso, conforme mostrado na Tabela 2, o coeficiente de partiçao octanol água dos analitos varia de 0 a 4.31 A utilizaçao de acetonitrila é uma opçao interessante, pois permite a extraçao de compostos com faixa de polaridade mais ampla, quando comparado a outros solventes como acetato de etila, acetona, metanol, água, entre outros. A adiçao de sal, além de possibilitar a retirada de água do sistema, tem efeito salting out, transferindo os analitos da fase aquosa para fase orgânica.32 Para a retirada de proteínas e gorduras da amostra, um procedimento bastante comum como alternativa de purificaçao é o congelamento do extrato da amostra à baixa temperatura (-20 ºC), pois auxilia na precipitaçao destes interferentes, os quais podem ser posteriormente removidos do extrato por centrifugaçao.33 Outra prática, que substituiria a SPE, corresponde à Dispersao em Fase Sólida, d-SPE, que consiste na utilizaçao de sorvente que atua como filtro na retirada de coextrativos. A utilizaçao desta técnica é uma alternativa simples e rápida, em que a adiçao do sorvente escolhido é realizada diretamente no extrato, seguido de agitaçao para homogeneizaçao da amostra e centrifugaçao para separaçao extrato/sorvente.34,35 Quando se deseja determinar concentraçoes muito baixas, uma etapa de pré-concentraçao do extrato torna-se necessária. Uma opçao interessante e viável, em substituiçao à SPE, seria a evaporaçao à temperatura controlada, sob fluxo de nitrogênio. Recuperaçao Tendo em vista as características da matriz e dos compostos, os procedimentos foram propostos com o intuito de garantir a máxima extraçao dos analitos e limpeza eficiente do extrato, para posterior análise por LC-MS/MS. A Tabela 7 apresenta os resultados de recuperaçao obtidos para os 3 procedimentos propostos.
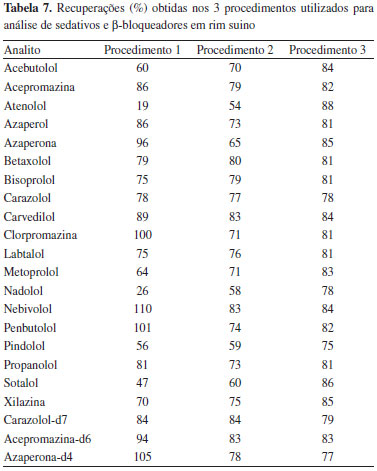
O procedimento 1 proporcionou recuperaçoes baixas para atenolol, nadolol e sotalol (19%, 26% e 47%, respectivamente). Estes compostos apresentam log Kow muito próximo de zero, portanto, é provável que a etapa de partiçao tenha sido ineficiente e os compostos tenham sido removidos do extrato juntamente com a água. Para os demais analitos, o procedimento apresentou recuperaçoes que variaram entre 56 e 110%. A utilizaçao de menor quantidade de amostra no procedimento 2 permitiu a quantificaçao de todos os analitos, porém apresentando como grande vantagem a obtençao de extrato mais límpido. Neste procedimento foi possível observar uma melhora significativa nas recuperaçoes obtidas para atenolol, nadolol e sotalol, que foram de 54%, 58% e 60% respectivamente, provavelmente devido à retirada da etapa de partiçao. Com isso, o extrato foi evaporado contendo a água oriunda da matriz. Cabe ressaltar que este procedimento se tornou bastante oneroso devido ao tempo gasto para evaporaçao do extrato obtido (cerca de 11 mL), que permaneceu sob fluxo de nitrogênio, a 50 ºC, por cerca de 3 a 4 horas. Para os demais compostos, as recuperaçoes obtidas ficaram entre 65 e 84%. O procedimento 3 consistiu no procedimento 2 otimizado, com o objetivo principal de reduzir o tempo de análise (pela diminuiçao no tempo de evaporaçao), além de diluir a amostra, reduzindo a quantidade de coextrativos a serem inseridos no sistema cromatográfico. Para isso, a etapa de purificaçao utilizando celite® 545 foi antecipada, sendo realizada logo após a precipitaçao de proteínas à baixa temperatura. Uma alíquota de 1 mL do extrato foi retirada para evaporaçao sob fluxo de N2 a 50 ºC. Esta otimizaçao reduziu significativamente o tempo de evaporaçao para cerca de 20 min. As recuperaçoes obtidas neste procedimento foram satisfatórias para todos os compostos, proporcionando valores de recuperaçao entre 75 e 88%. Portanto, o procedimento 3 descrito foi escolhido para análise de sedativos e β-bloqueadores em rim suíno devido suas vantagens de custo, praticidade e valores de recuperaçoes satisfatórios para todos os compostos. Procedimento de extraçao para análise de sedativos e β-bloqueadores O procedimento 3 selecionado consistiu em extraçao simples com solvente orgânico, seguido de etapas de purificaçao à baixa temperatura e d-SPE, além de etapa rápida de concentraçao da amostra sob fluxo de N2 e, por fim, retomada da amostra para posterior análise por LC-MS/MS. O tempo total do procedimento de extraçao foi de no máximo 5 horas para cerca de 50 amostras, o que corresponde a aproximadamente 6 minutos por amostra. A utilizaçao de acetonitrila na etapa de extraçao permitiu recuperaçoes satisfatórias para todos os compostos, independente de sua polaridade, além de reduzir a extraçao de coextrativos presentes no rim suíno, principalmente gorduras, quando comparado ao uso de solventes como metanol. O uso de homogenizador mecânico aumenta significativamente a superfície de contato solvente/matriz, permitindo melhor extraçao dos compostos. O uso de mesa agitadora por 20 minutos teve como principal objetivo favorecer a precipitaçao de proteínas, enquanto a posiçao dos tubos no sentido horizontal favorece a agitaçao. A última etapa, de centrifugaçao, é necessária para separar o extrato da matriz para posterior etapa de purificaçao. Na etapa de purificaçao, dois procedimentos simples foram empregados. A precipitaçao de proteínas à baixa temperatura é uma alternativa de purificaçao bastante difundida. Após uma hora já foi possível observar a deposiçao de quantidade significativa de proteínas que ficaram retidas no fundo do tubo após centrifugaçao. A adiçao de celite® 545 como sorvente para d-SPE teve como objetivo retirar principalmente gordura, além de proteínas e fosfolipídios, que podem estar presentes nesta matriz e que sao coextrativos de difícil remoçao. A grande vantagem na utilizaçao deste sorvente está relacionado ao baixo custo se comparado a sorventes como C18 e PSA. A etapa de concentraçao consiste na utilizaçao de recurso simples de evaporaçao sob temperatura controlada e fluxo de N2. Este procedimento é bastante rápido e eficiente, e é necessário já que os níveis de concentraçao a serem alcançados neste procedimento sao significativamente baixos. Robustez O estudo da robustez tem por objetivo demonstrar que o procedimento em questao apresenta resultados confiáveis, mesmo quando sujeito a pequenas variaçoes como diferentes fabricantes de insumos, colunas cromatográficas, concentraçoes de fase móvel, entre outros.36 Uma alternativa sugerida pela 2002/657/CE37 seria a verificaçao da robustez por meio da abordagem de Youden.28 A Tabela 8 apresenta os resultados de desvio padrao relativo obtidos no estudo de robustez utilizando o procedimento 3.
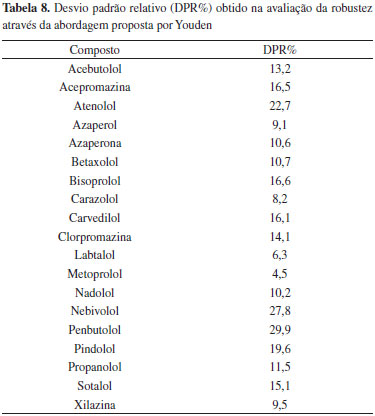
Considerou-se 30% como desvio aceitável no estudo de robustez devido a complexidade da matriz e o baixo valor de concentraçao (10 µg kg-1). Os desvios obtidos para a maioria dos compostos foram inferiores a 20%. Também foi possível observar por meio dos dados estatísticos que a quantidade de sorvente utilizado (d) corresponde ao fator que apresentou maior influência sobre os desvios obtidos para penbutolol e nebivolol, enquanto que para atenolol, o desvio padrao obtido é influenciado principalmente pela utilizaçao de menor temperatura de evaporaçao (c) e de vórtex (f) no processo de extraçao. Cabe salientar que foram propostas alteraçoes mais robustas que o recomendado (reduçao de 50% da quantidade de sorvente, por exemplo). Por este motivo, é possível afirmar que os fatores estudados nao afetaram significativamente o desempenho do procedimento, permitindo concluir que o mesmo foi robusto a estas alteraçoes. Aplicabilidade do procedimento A Tabela 9 apresenta os valores admitidos como reais das amostras, emitidos pela agência canadense, os valores emitidos pelos demais laboratórios participantes e os obtidos utilizando o procedimento desenvolvido neste trabalho com seu respectivo valor de Z-Score.
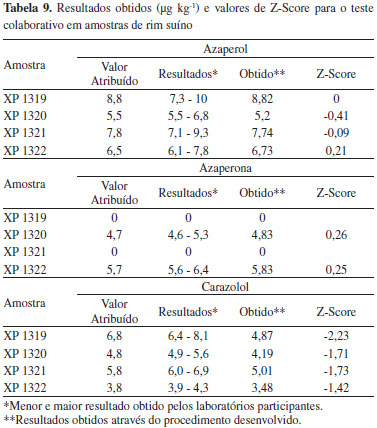
Analisando os resultados e os valores de Z-Scores, é possível concluir que o procedimento é adequado para análise de azaperona, azaperol e carazolol em rim suíno, apresentando valores muito próximos do esperado, sendo classificado como satisfatório para a maioria das amostras. Apenas a análise de carazolol na amostra XP 1319 apresentou valor de Z-Score na faixa considerada questionável, porém, devido ao tempo em que amostra permaneceu armazenada (cerca de 9 meses) até realizaçao da análise é possível considerar como satisfatório o resultado obtido. Este procedimento será validado conforme as normas do MAPA36 e incluído no PNCRC para monitoramento de sedativos e β-bloqueadores em amostras de rim suíno. Posteriormente, o escopo do método será ampliado para outras matrizes de origem animal.
CONCLUSAO Foi demonstrado o desenvolvimento de um procedimento para extraçao dos resíduos de 5 sedativos e 14 β-bloqueadores em rim suíno. O procedimento foi considerado adequado para a produçao de um extrato compatível com sistema de LC-MS/MS. O tempo do procedimento de extraçao foi de aproximadamente 6 minutos por amostra por analista. O procedimento também foi considerado robusto e passível de ser prontamente implementado em outras unidades laboratoriais. A utilizaçao de pequena quantidade de solvente e de sorvente estao de acordo com a necessidade de geraçao mínima de resíduos, atendendo, portanto, aos princípios da Química Verde. Dessa forma, o procedimento desenvolvido neste trabalho mostrou-se simples, rápido, prático, robusto e de baixo custo, sendo uma alternativa altamente viável aos procedimentos de extraçao que utilizam SPE como etapa de purificaçao.
AGRADECIMENTOS A Universidade Federal do Rio Grande e ao Programa de Pós-Graduaçao em Química Tecnológica e Ambiental por oportunizar o desenvolvimento deste trabalho em conjunto com o Laboratório Nacional Agropecuário- LANAGRO/RS ao qual agradeço pela disponibilidade de estrutura e equipamentos, além de esforço incansável de todos os seus colaboradores para a realizaçao deste trabalho.
REFERENCIAS 1. http://www.agricultura.gov.br, acessada em Junho 2014. 2. http://www.cpvs.com.br/cpvs, acessada em Junho 2014. 3. Cooper, J.; Delahaut, P.; Fodey, T. L.; Elliott, C. T.; The Analyst 2004, 129, 169. 4. Stolker, A. A. M.; Brinkman, U.A.Th.; J. Chromatogr. A 2005, 1067, 15. 5. Delahaut, P.; Brasseur, P. Y.; Dubois, M.; J. Chromatogr. A 2004, 1054, 373. 6. Botsoglou, N. A.; Fletouris, D. J.; Drug Residues in Foods: pharmacology, food safety and analysis, 1st ed., Marcel Dekker: New York, 2001. 7. Frenich, A. G.; Bolanos, P. P.; Aguilera, L. M. M.; Martínez, V. J. L.; Veterinary drugs and growth-promoting agent analyses, 1st ed., Nova Science Publishers: New York, 2010. 8. Paschoal, J. A. R.; Rath, S.; Airoldi, F. P. S.; Reyes, F. G. R.; Quim. Nova 2008, 31, 1190. 9. Brasil. Ministério da Agricultura, Pecuária e Abastecimento. Secretaria de Defesa Agropecuária. Instruçao Normativa Nº 17 de 29 de maio de 2013. Plano Nacional de Controle de Resíduos em produtos de origem animal. 2013. Diário Oficial da Uniao de 31 de maio de 2013. Brasília (Brasil). 10. Codex Alimentarius; Límites máximos de residuos para medicamentos veterinarios en los alimentos, 35a Sesión de la Comisón del Codex Alimentarius Commission, Comisión del Codex Alimentarius, 2012. 11. Regulamento EU 37/2010 da Comissao Reguladora, de 22/12/2009, relativo à substâncias farmacologicamente activas e respectiva classificaçao em relaçao à limites máximos de resíduos nos alimentos de origem animal, Official Journal of European Communities, 2010. 12. Engelsma, J. W.; Simons, J.; The Veterinary Quarterly 1985, 7, 73. 13. Rudolph, M.; Steinhart, H.; J. Chromatogr. 1987 392, 371. 14. Keukens, H. J.; Aerts, M. M. L.; J. Chromatogr. 1989 464, 149. 15. Van G. L. A.; Schwillens, P. L. W. J.; Olling, M.; Anal. Chim. Acta 1989, 225, 137. 16. Rose, M. D.; Shearer, G.; J. Chromatogr. 1992, 624, 471. 17. Quintana, M. C.; Blanco, M. H.; Lacal, J.; Fernández, L.; Talanta 2003, 59, 417. 18. Cerkenik-Flajs, V.; Anal. Chim. Acta 2007, 586, 374. 19. Delahaut, Ph.; Levaux, C.; Eloy, P.; Dubois, M.; Anal. Chim. Acta 2003, 483, 335. 20. Govaert, Y.; Batjoens, P.; Tsilikas, K.; Degroodt, J.M.; Srebrnik, S.; The Analyst 1998, 123, 2507. 21. Fluchard, D.; Kiebooms, S.; Dubois, M.; Delahaut, Ph.; J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2000, 744, 139. 22. Dupuis, C.; Gaulier, J.M.; Pélissier-Alicot, A. L.; Marquet, P.; Lachâtre, G.; J. Anal. Toxicol. 2004, 28, 674. 23. Mitrowska, K.; Posyniak, A.; Zmudzki, J.; Anal. Chim. Acta 2009, 637, 185. 24. Aoki, Y.; Hakamata, H.; Igarashi, Y.; Uchida, K.; Kobayashi, H.; Hirayama, N.; Kotani, A.; Kusu, F.; J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2009, 877, 166. 25. Zhang, J.; Shao, B.; Yin, J.; Wu, Y.; Duan, H.; J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2009, 877, 1915. 26. Ortelli, D.; Cognard E.; Jan, P.; Edder, P.; J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2009, 877, 2363. 27. Sai, F.; Hong, M.; Yufeng, Z.; Huijing, C.; Yongning, W.; J. Agric. Food Chem. 2012, 60, 1898. 28. Karageorgou, E.; Myridakis, A.; Stephanou, E. G.; Samanidou, V.; J. Sep. Sci. 2013, 36, 2020. 29. Agência Nacional de Vigilância Sanitária (ANVISA), Resoluçao RE nº 899, de 29 de maio de 2003, Guia para validaçao de métodos analíticos e bioanalíticos. 2003. Diário Oficial da Uniao de 02 de junho de 2003. Brasília (Brasil). 30. http://ndb.nal.usda.gov/ndb/foods/show/2633, acessada em Junho 2014. 31. http://www.chemicalize.org, acessada em Junho 2014. 32. Prestes, O. D.; Friggi, C. A.; Adaime, M. B.; Zanella, R.; Quim. Nova 2009, 32, 1620. 33. http://quechers.cvua-stuttgart.de/pdf/cleanup.pdf, acessada em Junho 2014. 34. Anastassiades, M.; Lehotay, S.; Stajnbaher, D.; Schenck, F. J.; J. AOAC Int. 2003, 86, 412. 35. Cabrera, L. C.; Martins, M. L.; Primel, E. G.; Prestes, O. D.; Adaime, M. B.; Zanella, R.; Scientia Chromatographica 2012, 4, 227. 36. Brasil, Ministério da Agricultura, Pecuária e Abastecimento; Manual de garantia da qualidade analítica, Ministério da Agricultura, Pecuária e Abastecimento, Secretaria de Defesa Agropecuária, Brasília, 2011. 37. Diretiva 2002/657/EC do Conselho, de 12/08/2002, que dá execuçao ao disposto na Diretiva 96/23/CE do Conselho relativamente ao desempenho de métodos analíticos e à interpretaçao de resultados, Official Journal of European Communities 2002. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access