
 |
 |
 |
 |
 |
Artigo
| Theoretical investigations of the bulk modulus in the tetra-cubic transition of PbTiO3 material |
|
Renan A. P. Ribeiro; Sergio R. de Lázaro*
Departamento de Química, Universidade Estadual de Ponta Grossa, 84030-900 Ponta Grossa - PR, Brasil Recebido em 05/12/2013 * e-mail: srlazaro@uepg.br Resulting from ion displacement in a solid under pressure, piezoelectricity is an electrical polarization that can be observed in perovskite-type electronic ceramics, such as PbTiO3, which present cubic and tetragonal symmetries at different pressures. The transition between these crystalline phases is determined theoretically through the bulk modulus from the relationship between material energy and volume. However, the change in the material molecular structure is responsible for the piezoelectric effect. In this study, density functional theory calculations using the Becke 3-Parameter-Lee-Yang-Parr hybrid functional were employed to investigate the structure and properties associated with the transition state of the tetragonal-cubic phase change in PbTiO3 material. INTRODUCTION Quantum mechanical simulations of material molecular structures play an important role in explaining material properties at the molecular level. Ferroelectricity, piezoelectricity, pyroelectricity, and other properties depend on the molecular structure of the materials because electronic density changes according to composition, atomic organization, and chemical bond. Actually, several materials may exhibit similar properties but different structures. Therefore, organic, inorganic, and biological structures can display the same properties. For instance, organic and inorganic materials can present spontaneous polarization properties; organic, inorganic, and biological molecular structures may show optical properties. In particular, oxides called perovskites, which mainly consist of titanate materials,1,2 exhibit ferro-, piezo-, opto-, and pyroelectricity properties3-6 that have been extensively applied to electrical systems such as memories, gas sensors, and transducers.7-9 Ferro- and piezoelectric properties rely on material composition and arrangement. For instance, the ferro- and piezoelectricity of PbTiO3 (PT) in the tetragonal phase are macroscopically characterized by determining the d33 values in the [111], [011], and [001] directions, because these properties stem from linear combinations of microscopic vectors in these directions. Generally, domain-engineered Relaxor-PT single crystals present extremely large piezoelectric coefficients for the longitudinal vibration mode while crystals with single domain states display very low longitudinal properties and ultrahigh shear.10 PT single crystals have shown a high electromechanical coupling constant and a large spontaneous polarization.11-13 Kushida and Takeuchi reported the formation of PT thin films presenting electromechanical coupling constants reaching 0.8 on patterned Pt electrode films embedded in SrTiO3 single crystals by seeded lateral overgrowth. These constants resulted from the orientation of the electrode-supported PT thin film on the c-axis, showing that piezo- and pyroelectricity depended on crystallographic axis orientation in this material.11 PT exhibits tetragonal and cubic phases, of which the tetragonal phase forms a more stable structure under ambient conditions. Tetragonal PT presents ferroelectric properties, a P4 mm spatial group with experimental lattice parameters a and c of 3.904 (a = b) and 4.152 Å (c/a = 1.064), respectively, and an experimental band gap of approximately 3.4 eV. Cubic PT is a dielectric material displaying a Pm3 m spatial group, an experimental lattice parameter a of 3.865 Å (a = b = c), and an experimental band gap of 3.6 eV.14-19 The tetragonal-cubic phase transition relies on temperature and pressure. The ferroelectric-paraelectric phase transition of PT occurred at a Curie temperature Tc of 766 K.20 The bulk modulus is defined as the pressure required for triggering phase transition. The experimental bulk modulus amounts to approximately 104 and 144 GPa for tetragonal and cubic PT phases, respectively.19 Under pressure, crystal structure and physical properties can be effectively changed by variations in the atom positions in the unit cell and the electron density.21 In PT, tetragonality and ferroelectricity are suppressed with increasing pressure at ambient temperature to produce a paraelectric cubic phase above 12 GPa.22 In recent years, numerous theoretical and experimental studies have been developed to investigate the phase transition of different perovskites.23,24 Supercell-based density functional theory (DFT) simulations have shown that the formation energies of possible vacancies in the band gap range of PT depend on oxygen partial pressures. These authors observed the influence of oxygen partial pressures on vacancy energetic states in this material.25 DFT simulations based on spin Hamiltonian models and Green's function techniques have been performed to investigate the influence of oxygen vacancy on the ferromagnetic properties of ferroelectric perovskite nanoparticles. This study demonstrated that the oxygen vacancy changes the spin electronic structure of the ferroelectric perovskite ABO3 nanoparticles without requiring additional magnetic impurities such as Co, Fe, and Ni. Polarization and magnetization depend on particle size, and an intermediate size interval is observed for multiferroic ferroelectric nanoparticles.26 An ab initio model based on DFT and a plane wave approximation has been proposed to study the influence of applied electric fields on piezoelectric response in perovskite superlattices formed by PT/SrTiO3 and BaTiO3/CaTiO3 interfaces. This model provided a relationship between electric field, displacement field, and PT composition that explains how the interface between these materials is influenced mainly in the [001] direction.27 The determination of timescales to study the phase transition with respect to symmetry and nanodomains in perovskites superlattices suggested piezoelectric field distortions with analysis pulses of approximately 300 ns.28 These previous reports described the PT phase transition along the [001] direction in the bulk and thin films. Theoretical studies on the nature of piezoelectricity and its governing parameters suggest that PT piezoelectricity rests on structural and electrical phase transitions involving an intermediate crystalline structure between tetragonal and cubic symmetries. However, this crystalline entity remains unclear mainly because it exhibits either a tetragonal or a cubic structure. Here we propose a new theoretical approach that provides insight into the crystalline symmetry in the piezoelectric phase transition of PT. This approach relies on the bulk modulus curve of PT and simulations of chemical transition states between tetragonal-cubic symmetries of this material at the same volume.
COMPUTATIONAL METHODOLOGY Molecular structure and calculations parameters The tetragonal PT phase was simulated in the P4 mm spatial group using experimental lattice parameters (a = b = 3.904 Å; c = 4.152 Å) and the internal coordinates (0, 0, 0) for Pb atoms, (0.5, 0.5, 0.49) for Ti atoms, as well as (0.5, 0.5, -0.112) and (0.5, 0.5, 0.388) for O atoms.15 The cubic PT phase was simulated in the Pm3 m spatial group using experimental lattice parameters (a = 3.865 Å) and the internal coordinates (0, 0, 0) for Pb atoms, (0.5, 0.5, 0.5) for Ti atoms, and (0.5, 0.5, 0) for O atoms. Bulk models (Figure 1) were constructed for tetragonal and cubic symmetries. All simulations were performed based on DFT using Becke's exchange functional29,30 corrected by the Lee-Yang-Parr31 correlation (B3LYP) and periodic boundary conditions implemented in the CRYSTAL09 code.32,33 Basis set combinations used in this work were selected through 56 combination tests and consisted of 86411-d31 for Ti atoms,34 [DB]-31G for Pb atoms,35 and 6-31G(p) for O atoms.36 [DB] represents the Duran-Barthelar pseudopotential.37 All models were simulated in the singlet electronic state. Tetragonal and cubic transition states were determined using the frequency package implemented in CRYSTAL09, and the lattice point group was used to reduce the maximum number of self-consistent field (SCF) and gradient calculations. At each point, the residual symmetry was exploited for the SCF calculation so that second derivative calculations were only performed on irreducible atoms. The full Hessian matrix was then generated by applying the point group symmetry to the irreducible part, and the corresponding mass-weighted matrix was diagonalized to obtain eigenvalues, which were transformed into frequencies (cm-1). The resulting eigenvectors were converted into normal modes. Lattice parameters and unit cell volumes were optimized in terms of system total energy using mono- and bielectronic integrals truncated to 10-8, a 4 × 4 Monkhorst-Gilat net, and a Phenon quadri-core AMD 64-bit processor.
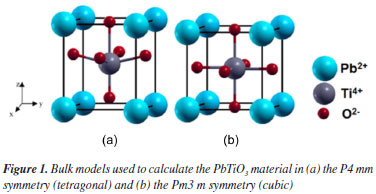 Figure 1. Bulk models used to calculate the PbTiO3 material in (a) the P4 mm symmetry (tetragonal) and (b) the Pm3 m symmetry (cubic)
The bulk modulus is the applied pressure necessary to decrease the solid volume, i.e., to cause spatial deformation in the unit cell, and thus, provides a measure of solid tenacity with respect to this pressure. To estimate this property, an equation of state (EOS) relating energy (E) and volume (V) for the different symmetries of the solid was required. Fitting this curve with a third-order polynomial gave the bulk modulus (B0) and its first derivative (B0'). These data were subsequently implemented in the Birch-Murnaghan state equation to calculate the applied pressure required for the phase transition.38
RESULTS AND DISCUSSIONS PT is well-known for its piezoelectric properties.39,40 Ferroelectric materials typically exhibit a spontaneous polarization in a direction than can be switched under an external electric field or stress. Piezoelectric PT exists as a noncentrosymmetric tetragonal crystal structure presenting a spontaneous polarization in the (001) direction because of the spatial displacement of the oxygen and titanium atoms with respect to the symmetry center position.41-43 In contrast, cubic PT does not exhibit spontaneous polarization and, therefore, no piezoelectric properties. However, a tetragonal-cubic phase transition is observed when pressure is applied to tetragonal PT. This pressure changes the molecular structure of PT, increasing the electronic energy and modifying its molecular energy state. From this perspective, piezoelectric phenomena are tetra-cubic phase transitions that can be investigated as chemical reactions in which the tetragonal phase is the reagent, the cubic phase is the product, and the activation energy is transferred through applied pressure. Possible molecular structures for the transition states of this chemical reaction may display a tetragonal or a cubic arrangement. Electronic structure Table 1 shows experimental and theoretical results for the tetragonal and cubic symmetries of PT. These data indicated that lattice parameters obtained in this study are in close agreement with experimental data and other theoretical studies.
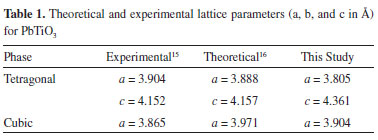
Brillouin zones for tetragonal and cubic PT are shown in Figure 2. Band structures provide possible electronic excitation and decay pathways between valence and conduction bands. These pathways usually define direct and indirect band gaps, which occur between identical or different k symmetry points of the Brillouin zone, respectively.44 In the tetragonal band structure (Figure 2a), the smallest band gap was an indirect band gap of 3.613 eV localized between X and Γ symmetry points, consistent with the experimental band gap for tetragonal PT (3.60 eV).15 For the cubic band structure (Figure 2b), the smallest band gap corresponded to a direct band gap of 3.045 eV localized between X and X symmetry points. The experimental value for the band gap of cubic PT amounted to approximately 3.40 eV.45
 Figure 2. Band structures of (a) tetragonal and (b) cubic PbTiO3 phases. High symmetry points in the Brillouin Zone are described as Γ (0,0,0); X (0.5,0,0); M (0.5,0.5,0); Z (0,0,0.5); R (0.5,0,0.5); A (0.5,0.5,0.5); and Γ (0,0,0); X (0.5,0,0); M (0.5,0.5,0); R (0.5,0,0.5) for tetragonal and cubic phases, respectively
Changes in band gap stem from the influence of molecular organization on material electronic structure. Here PT showed a Fermi energy, or maximum valence band, of -3.324 eV in the tetragonal symmetry (Figure 2a) and -2.913 eV in the cubic symmetry (Figure 2b). However, the conduction band minima displayed the same energy for both phases, suggesting that band gap modifications mainly resulted from the redistribution of the electronic density in PT. A transition state displaying an indirect band gap of 4.28 eV was localized between X and Γ symmetry points. The Fermi energy for this phase equaled -3.147 eV; therefore, the PT transition state exhibited the same band gap orientation (indirect) between the same symmetry points as tetragonal PT. However, its band gap was considerably larger than those for cubic and tetragonal phases, and its Fermi energy was localized between the levels obtained for these phases. These results indicate that the PT transition state presents the same electronic features as tetragonal PT but its conduction band is significantly different. Bulk modulus and chemical transition state Table 2 shows experimental and theoretical results for the bulk modulus of PT. The data indicated that the theoretical bulk modulus obtained for the cubic phase was closer to experimental result than previous theoretical studies.16 However, theoretical results for the tetragonal phase deviated from other theoretical studies to a greater extent.16 These new theoretical results were obtained using a basis set combination, which previously showed that different basis set combinations provided different bulk modulus calculations.
In piezoelectricity, a solid phase transition occurs under applied pressure to produce an electric signal. From the chemical perspective, this electrical phenomenon can be interpreted as a phase transition between two specific phases connected by a transition state. Because tetragonal PT is the reagent and cubic PT is the product, this transition state is observed when pressure is applied on the solid. Interestingly, this suggests that a tetragonal-cubic transition point can be identified and related to a chemical transition state point. Here the theoretical EOS curves (Figure 3a) showed that the tetragonal minimum energy was localized in a volume range larger than that of the cubic minimum energy. Moreover, the intersections between the third-order polynomials applied to the EOS curves (Figure 3a) provided identical unit cell volumes of 58.690 Å3 for tetragonal and cubic phases. Therefore, the transition state for this piezoelectric system was observed for this volume. The PT structure changes from tetragonal to cubic in the piezoelectric process.
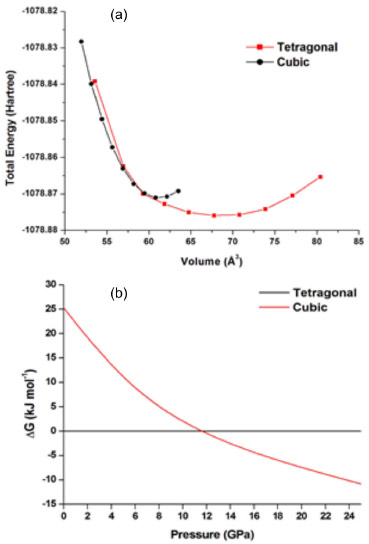 Figure 3. (a) Theoretical EOS curves for cubic and tetragonal phases. (b) Phase diagram for PbTiO3
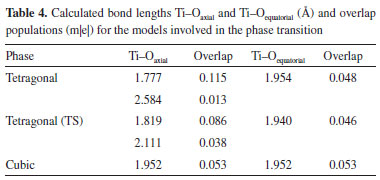
Figure 3b shows the phase diagram obtained from the theoretical EOS curves for cubic and tetragonal PT. The calculations indicate that the cubic PT becomes more stable than its tetragonal counterpart at pressures surpassing 11.50 GPa, in excellent agreement with experimental results (12 GPa).22 Frequency calculations were conducted for both symmetries at a volume of 58.690 Å3 to identify the molecular structure of the transition state between tetragonal and cubic phases. These calculations showed that only the tetragonal symmetry exhibited a single imaginary frequency (ν = -187.7122 cm-1) corresponding to a transition state (Table 3). This frequency was attributed to the stretching vibrational modes of Ti-O bonds in axial and equatorial planes. Furthermore, a comparison between the total energies of tetragonal and cubic phases (Table 3) showed that cubic PT displayed a higher total energy than tetragonal PT for the same volume. This demonstrates that the tetragonal transition state is more stable than the cubic minimum structure. Thermodynamic approximations allow the piezoelectric process to be considered as a reversible phase transition. These approximations, combined with these vibrational results, suggest that the transition state exhibiting only one negative vibrational mode must be fully relaxed to a minimum energy structure with the same unit cell volume (58.690 Å3). This reversible process is proposed as an automatic rearrangement of the internal atomic positions upon applied pressure on the lattice parameters of PT. The relaxed lattice parameters are shown in Table 3.

The redistribution of the electronic density is responsible for the piezoelectric properties of PT, resulting in loss of spontaneous polarization in the cubic phase. To investigate this loss, electronic density maps were analyzed for tetragonal and cubic symmetries (Figure 4). Electronic density maps were obtained from wave function and electronic density matrix isolines that describe the density in an area.46 For better visualization, the unit cell was expanded in the z-direction. These electronic density maps were described along the Ti-O bond direction of the material, which corresponds more specifically to the (111) and (010) Miller indices for cubic and tetragonal symmetries (Figure 4).
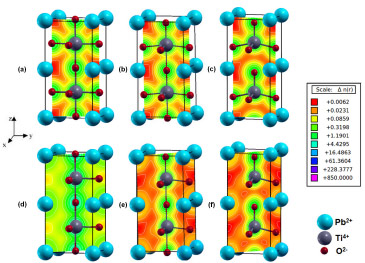 Figure 4. Charge density maps along the (010) direction for (a) cubic, (b) tetra-TS, (c) tetragonal geometries. Charge density maps along the (111) direction for (d) cubic, (e) tetra-TS, and (f) tetragonal geometries
Electronic density isolines around titanium and oxygen atoms (Figure 4f) showed the formation of spontaneous dipole moments between these atoms, consistent with spontaneous polarization in the (111) direction. A spontaneous quadrupole moment was also observed between one titanium atom and three oxygen atoms along the (010) direction of the tetragonal symmetry (Figure 4c). These results show that the piezoelectric phenomenon results from more than one type of spontaneous polarizations in different crystalline directions.41 For the transition state model (Figures 4b and 4e), a connection was determined between the Ti-O bonds along the (111) and (010) directions, suggesting a charge transfer from the tetragonal geometry. Charge density maps (Figure 4) allowed charges to be attributed to spontaneous polarization elements (electric moments). These charges amounted to 23.1, 85.9, and 319.8 m|e| for tetragonal, transition state, and cubic models, respectively. Therefore, a charge corridor formed between Ti and O atoms (Figures 4a and 4d) under pressure during the PT phase transition, leading to a charge increase in the cubic phase. On the other hand, the transformation into cubic PT caused a loss of the spontaneous polarization (Figures 4a and 4d) along (111) and (010) directions. In this phase, polar moments were replaced by chemical bonds between titanium and oxygen atoms, causing the piezoelectric properties to disappear. Table 4 shows the calculated overlap population and bond lengths (Ti-O) for the tetragonal, transition state, and cubic models. The tetragonal phase (Figure 1a) exhibited four equatorial Ti-O bonds of equal length and two axial Ti-O bonds of different lengths because of the displacement of the Ti atom with respect to the central position of the unit cell (Table 4). After the transition, the PT cubic phase displayed equal Ti-O bond lengths because the Ti atom occupies the central position of the unit cell, consistent with the influence of the external pressure on the PT crystal structure. The most significant changes in overlap population occurred in the Ti-O axial bonds of the cubic phase. Specifically, changes in electronic density after the phase transition resulted in the loss of the overlap population and, consequently, loss of the spontaneous polarization (Figures 4a and 4d). Interestingly, intermediate deformations between tetragonal and transition state models modified the PT crystalline structure and these structures were also electrically charged.41
A potential energy surface was plotted based on the Gibbs free energy (ΔG) for the tetragonal-cubic transition (Figure 5) using only a tetragonal symmetry to represent the transition state (TS) (Table 3). The calculated Gibbs activation energy barrier (ΔG≠tetra-TS) amounted to 19.5 kJ.mol-1, consistent with a nonspontaneous process. In the TS-cubic transition step of this chemical pathway, the calculated ΔG≠TS-Cubic equaled -0.90 kJ.mol-1, indicative of a spontaneous process. In this second step, the energy approximates the energy available in the environment. The estimated electrical potential charged in one PT unit cell amounted to 86 mV/unit cell using Gibbs standard free energy (ΔGθ) for the overall process between cubic and tetragonal phases. Therefore, approximately 12 unit cells of PT material oriented along the z-direction under pressure are required to obtain an electrical potential of 1 V.
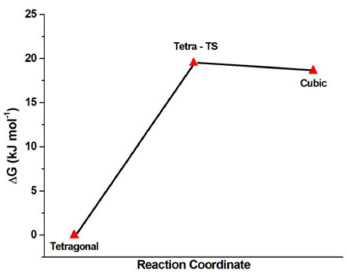 Figure 5. Potential energy surface (PES) for the tetragonal-cubic transition using tetragonal phase as a reference state
Another way to demonstrate this chemical process relies on the Kröger-Vink defect notation for crystalline structures.47 In this notation, the sign "x" represents the formation of a neutral cluster while the sign "'" denotes the accumulation of negative charge in the corresponding cluster. Researchers have used this notation to investigate optical and structural properties in electronic ceramics based on modifications of molecular clusters such as TiO6,48,49 WO4,50 and ZrO6.51 Therefore, the defect equation sequence proposed for this chemical process represents the transformation of the [TiO5.Vo]X cluster in the tetragonal phase into the [TiO6]' cluster in the cubic phase. These molecular clusters were chosen based on calculated bond lengths (Table 3).
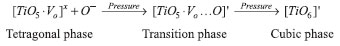
This sequence agrees with the charge density maps (Figure 4), which showed the oxygen and vacancy charge densities for the [TiO5.Vo]X cluster in the tetragonal phase (Figures 4c and 4f), the [TiO5.Vo...O]' cluster in TS (Figures 4b and 4e), and the [TiO6]' cluster in the cubic phase (Figures 4a and 4d).
CONCLUSIONS The theoretical DFT/B3LYP calculations of PT molecular structures in the tetragonal and cubic phases agreed well with previous structural and electronic results. Charge density maps along the crystallographic directions provided a visualization of spontaneous polarization, which resulted from the spontaneous formation of electric dipole moments in the tetragonal phase, qualitatively confirming the piezoelectric property. Bulk modulus results for the tetragonal phase showed a good agreement with other theoretical results. This theoretical analysis was essential for determining the volume of the transition state unit cell (58.690 Å3) under a pressure of 11.50 GPa, leading to a potential chemical pathway for the piezoelectric process. This proposed pathway exhibited a Gibbs activation energy of 19.5 kJ.mol-1, and the calculated electric potential charged on the unit cell amounted to 86 mV/unit cell. The modifications of Ti-O bond lengths along various directions were observed to be the most significant along the z-direction; this is experimentally characterized by the spontaneous polarization of PT.
ACKNOWLEDGEMENT The authors thank UEPG, CAPES, CNPq, and the Araucaria Foundation for financial support.
REFERENCES 1. Wang, J.; Pang, X.; Akinc, M.; Lin, Z.; J. Mater. Chem. 2010, 20, 5945. 2. Qi, T.; Grinberg, I.; Rappe, A. M.; Phys. Rev. B: Condens. Matter Mater. Phys. 2010, 82, 134113. 3. Chen, Z.-X.; Chen, Y.; Jiang, Y.-S.; J. Phys. Chem. B 2002, 106, 9986. 4. Damjanovic, D.; Brem, F.; Setter, N.; Appl. Phys. Lett. 2002, 80, 652. 5. Lemanov, V. V.; Sotnikov, A. V.; Smirnova, E. P.; Weihnacht, M.; Kunze, R.; Solid State Commun. 1999, 110, 611. 6. Tuzun, R. E.; Noid, D. W.; Sumpter, B. G.; J. Comput. Chem. 2000, 21, 553. 7. Bennett, J.; Bell, A. J.; Stevenson, T. J.; Comyn, T. P.; Appl. Phys. Lett. 2013, 103, 152901 8. Kong, L.; Liu, G.; Zhang, S.; Liu, H.; J. Appl. Phys. (Melville, NY, U. S.) 2013, 114, 144106 9. Li, L.; Liu, X.; Zhang, Y.; Salvador, P. A.; Rohrer, G. S.; Int. J. Hydrogen Energy 2013, 38, 6948. 10. Zhang, S.; Li, F.; J. Appl. Phys. (Melville, NY, U. S.) 2012, 111, 031301 11. Kushida, K.; Takeuchi, H.; Appl. Phys. Lett. 1987, 50, 1800. 12. Kushida, K.; Takeuchi, H.; Ferroelectrics 1990, 108, 3. 13. Ghonge, S. G.; Goo, E.; Ramesh, R.; Appl. Phys. Lett. 1993, 62, 1742. 14. de Lazaro, S.; Longo, E.; Sambrano, J. R.; Beltrán, A.; Surf. Sci. 2004, 552, 149. 15. Hermet, P.; Veithen, M.; Ph, G.; J. Phys.: Condens. Matter 2009, 21, 215901. 16. Liu, Y.; Xu, G.; Song, C.; Ren, Z.; Han, G.; Zheng, Y.; Mat. Sci. Eng. A 2008, 472, 269. 17. Hosseini, S. M.; Movlarooy, T.; Kompany, A.; Phys. B (Amsterdam, Neth.) 2007, 391, 316. 18. Piskunov, S.; Heifets, E.; Eglitis, R. I.; Borstel, G.; Comput. Mater. Sci. 2004, 29, 165. 19. Li, Z.; Grimsditch, M.; Foster, C. M.; Chan, S. K.; J. Phys. Chem. Solids 1996, 57, 1433. 20. Sani, A.; Hanfland, M.; Levy, D.; J. Solid State Chem. 2002, 167, 446. 21. Zhu, J.; Zhang, J.; Xu, H.; Vogel, S. C.; Jin, C.; Frantti, J.; Zhao, Y.; Sci. Rep. 2014, 4, 3700. 22. Sanjurjo, J. A.; López-Cruz, E.; Burns, G.; Phys. Rev. B: Condens. Matter Mater. Phys. 1983, 28, 7260. 23. Young, S. E.; Guo, H. Z.; Ma, C.; Kessler, M. R.; Tan, X.; J. Therm. Anal. Calorim. 2014, 115, 587. 24. Blokhin, E.; Gryaznov, D.; Kotomin, E.; Evarestov, R.; Maier, J.; Integr. Ferroelectr. 2011, 123, 18. 25. Zhang, Z.; Wu, P.; Lu, L.; Shu, C.; Appl. Phys. Lett. 2006, 88, 142902 26. Bahoosh, S. G.; Wesselinowa, J. M.; J. Appl. Phys. (Melville, NY, U. S.) 2012, 112, 053907 27. Swartz, C. W.; Wu, X.; Phys. Rev. B: Condens. Matter Mater. Phys. 2012, 85, 054102. 28. Pice, C.; Ji Young, J.; Ho Nyung, L.; Eric, M. D.; Serge, M. N.; Paul, G. E.; New J. Phys. 2012, 14, 013034. 29. Becke, A. D.; Phys. Rev. A: At., Mol., Opt. Phys. 1988, 38, 3098. 30. Becke, A. D.; J. Chem. Phys. 1993, 98, 5648. 31. Lee, C.; Yang, W.; Parr, R. G.; Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785. 32. Dovesi, R.; Orlando, R.; Civalleri, B.; Roetti, C.; Saunders, V. R.; Zicovich-Wilson, C. M.; Z. Kristallogr. 2005, 220, 571. 33. Dovesi, R.; Saunders, V. R.; Roetti, C.; Orlando, R.; Zivocih-Wilson, C. M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N. M.; Bush, I. J.; D'Arco, P.; Llunell, M.; CRYSTAL09 User's Manual, University of Torino: Torino, 2009. 34. Corà, F.; Mol. Phys. 2005, 103, 2483. 35. Nizam, M.; Bouteiller, Y.; Silvi, B.; Pisani, C.; Causa, M.; Dovesi, R.; J. Phys. C: Solid State Phys. 1988, 21, 5351. 36. Corno, M.; Busco, C.; Civalleri, B.; Ugliengo, P.; Phys. Chem. Chem. Phys. 2006, 8, 2464. 37. Durand, P.; Barthelat, J.-C.; Theor. Chim. Acta 1975, 38, 283. 38. Chaplot, S. L.; Thermodynamic Properties of Solids, 1st ed., Wiley-Vch: Weinheim, 2010. 39. Chen, T.-Y.; Chu, S.-Y.; J. Eur. Ceram. Soc. 2003, 23, 2171. 40. Zhang, S.; Li, H.; Li, M.; Mater. Lett. 2008, 62, 2438. 41. Bhattacharya, K.; Ravichandran, G.; Acta Mater. 2003, 51, 5941. 42. Miura, K.; Funakubo, H. Em Advances in Ferroelectrics; Barranco, A. E., eds.; InTech: Rijeka, 2012. 43. Yifeng, D.; Hongliang, S.; Lixia, Q.; J. Phys.: Condens. Matter 2008, 20, 175210. 44. Han, F.; A Modern Course the Quantum Theory of Solids, 1st ed., World Scientific Publisher: Hackensack, 2013. 45. Leite, E. R.; Santos, L. P. S.; Carreño, N. L. V.; Longo, E.; Paskocimas, C. A.; Varela, J. A.; Lanciotti, F.; Campos, C. E. M.; Pizani, P. S.; Appl. Phys. Lett. 2001, 78, 2148. 46. Galasso, F. G.; Perovskites and High Tc Superconductors. 1nd ed., Science Publishers: New Hampshire, 1990. 47. Kroger, F. A.; Vink, H. J.; Solid State Physics, 1st ed., Academic Press: New York, 1956. 48. Moreira, M. L.; Longo, V. M.; Avansi, W.; Ferrer, M. M.; Andrés, J.; Mastelaro, V. R.; Varela, J. A.; Longo, É.; J. Phys. Chem. C 2012, 116, 24792. 49. Oliveira, L. H.; Paris, E. C.; Avansi, W.; Ramirez, M. A.; Mastelaro, V. R.; Longo, E.; Varela, J. A.; J. Am. Ceram. Soc. 2013, 96, 209. 50. Longo, V. M.; Gracia, L.; Stroppa, D. G.; Cavalcante, L. S.; Orlandi, M.; Ramirez, A. J.; Leite, E. R.; Andrés, J.; Beltrán, A.; Varela, J. A.; Longo, E.; J. Phys. Chem. C 2011, 115, 20113. 51. Moreira, M. L.; Andres, J.; Mastelaro, V. R.; Varela, J. A.; Longo, E.; CrystEngComm 2011, 13, 5818. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access