
 |
 |
 |
 |
 |
Artigo
| HPLC-DAD based method for the quantification of flavonoids in the hydroethanolic extract of Tonina fluviatilis Aubl. (Eriocaulaceae) and their radical scavenging activity |
|
Marcelo R. de AmorimI; Daniel RinaldoII; Fabiano P. do AmaralIII; Wagner VilegasIII; Mara A. G. MagentaIV; Gerardo M. Vieira JrV; Lourdes C. dos SantosI,*
IDepartamento de Química Orgânica, Instituto de Química, Universidade Estadual Paulista, 14800-900 Araraquara - SP, Brasil Recebido em 03/10/2013 *e-mail: loursant@gmail.com This article describes the isolation and identification of flavonoids in the hydroethanolic extract of the aerial parts from Tonina fluviatilis and evaluation of their antiradical activity. A method based on HPLC-DAD was developed and validated for detecting and quantifying flavonoids in hydroethanolic extracts. The flavonoids identified and quantified in the extract were 6,7-dimethoxyquercetin-3-O-β-D-glucopyranoside (1), 6-hydroxy-7-methoxyquercetin-3-O-β-D-glucopyranoside (2), and 6-methoxyquercetin-3-O-β-D-glucopyranoside (3). The developed method presented good validation parameters, showing that the results obtained are consistent and can be used in ensuring the quantification of these constituents in the extracts. Compounds 2 and 3 showed strong antiradical activity when compared with the positive controls (quercetin and gallic acid). INTRODUCTION Secondary metabolites produced by plants constitute a source of bioactive substances. Scientific interest in these metabolites has increased recently because of the search for new drugs originating from plants. Eriocaulaceae is a pantropical, predominantly herbaceous monocotyledonous family, comprising around 1200 species in 10 genera. In Brazil, they are common and diagnostic components of the herbaceous rocky outcrop vegetation called "campos rupestres," which flourish at elevations exceeding 900 m above sea level.1 Taxonomic studies to delimit the genus, whose definition remains controversial, and the biological investigation of molecules isolated from Eriocaulaceae are of great importance, especially because several molecules possess antioxidant,2,3 cytotoxic, and mutagenic activities,4,5 and some extracts of the assayed plants show antiulcerogenic activity.6 Tonina, which belongs to family Eriocaulaceae, is a monospecific genus including only Tonina fluviatilis. T fluviatilis is an aquatic or amphibious species with broad geographic distribution, growing in South American, primarily in the Guianas (Guyana, Suriname, French Guiana) where it is widespread. In one medicinal use, the plant is ground up and mixed with a bark to make a liquid in which to bathe nursing infants. It is believed to give strength to the infant.1,7 The chemical constituents in T. fluviatilis is being investigated for the first time. A comparison of the HPLC-DAD profile of the hydroethanolic extract of the aerials parts of T. fluviatilis with that of other Eriocaulaceae species showed UV spectra typical of flavonols.8-11 In our laboratories, glycosylated acyl flavonoids and quercetin derivatives with one sugar unit have been isolated from Eriocaulaceae species.12 Flavonoids have frequently been cited in chemotaxonomic discussions of the Eriocaulaceae family, which possess a taxonomy complicated by the morphologic of its capitulae.13 The flavonoids are widespread, their patterns tend to be specific, they are relatively stable, and their biosynthesis/accumulation is largely independent of environmental influence.14 Therefore, this paper presents the isolation of flavonoids present in the aerial parts of T. fluviatilis, and the evaluation of the sensitivity, specificity, linearity, accuracy, and precision of the method based on HPLC-DAD for their detection along with their antiradical activity.
EXPERIMENTAL Chemicals and reagents HPLC grade methanol (Merck, Darmstadt, Germany) and trifluoroacetic acid (TFA; Tedia, OH, USA) were used for the HPLC analysis. Deionized water (18 MΩ cm) from a Direct Milli-Q purification system (Millipore Co., Bedford, MA, USA) was used. Sep-Pak RP18 cartridges (1000 mg/6 mL) for solid-phase extraction (SPE) were purchased from Phenomenex Co. (Torrance, CA, USA). Flavonoids standards (1-3) were isolated from the hydroethanolic extract of the aerial parts of T. fluviatilis according to the procedures described in "Extraction and isolation of the flavonoids" section and then used as external standards. HPLC-DAD analysis of these flavonoid standards revealed a purity of 98%. Quercetin and gallic acid were purchased from Sigma-Aldrich (St. Louis, MO, USA). All others chemicals used were analytical grade and from commercial sources. Preparation of T. fluviatilis crude extract The aerial parts of T. fluviatilis (capitulae, scapes, and leaves) were collected in Bertioga City, São Paulo, Brazil, 23º46'26.64"S, 45º58'24.29" W, and identified by Dr. Mara Angelina Galvão Magenta from University Santa Cecilia (Unisanta) in Santos City, São Paulo state, Brazil. A voucher specimen (M. Magenta 729) was deposited at the Herbarium of the Unisanta (HUSC). The dried aerial parts of T. fluviatilis (352 g) were powdered and extracted with ethanol:water, (7:3, v/v) by percolation, at room temperature. The solution was evaporated to dryness in vacuo to give to give the crude hydroethanolic extract (66.2 g, 18.8%), which was stored at 4 ºC. Isolation of the flavonoids The hydroethanolic extract (2.35 g) was solubilized in methanol:water (7:3, v/v, 20.0 mL) using ultrasound for 10 minutes and then centrifuged for 15 minutes. The precipitate (0.25 g) was separated from the supernatant (2.10 g). The supernatant was filtered and chromatographed on Sephadex LH-20 (85 × 2.5 cm, i.d.) and eluted with methanol:water (7:3, v/v) to give 160 fractions (10 mL each). Fractions were analyzed by silica gel TLC (chloroform:methanol:n-propanol:water, 5:6:1:4, v/v/v/v, organic phase). Fractions 75-78 (24 mg) were combined and purified by solid phase extraction (SPE) using Phenomenex Strata C18 cartridge (1000 mg of stationary phase) that was previously activated with MeOH (5 mL) and equilibrated with MeOH:H2O (7:3, v/v; 5 mL). The fractions eluted from cartridge using MeOH:H2O (7:3, v/v; 15 mL) afforded the compound identified as 6,7-dimethoxyquercetin-3-O-β-D-glucopyranoside (1, 19 mg). Fractions 97-160 (86 mg) were combined and further purified by semipreparative HPLC on a Varian (Walnut Creek, CA, USA) chromatograph, equipped with a photodiode array detector (330 model Varian) and injector with a loop (500 µL) A Phenomenex Luna (2) RP18 column (250 × 10 mm i.d., 10 µm) along with the protective guard column Phenomenex (4 cm × 3 mm) was used in the purification. The purification was performed using an isocratic mode, where the eluent 40% methanol (0.1% TFA) and 60% water (0.1% TFA), had a flow rate of 2 mL min-1. Monitoring the separation of compounds at 270 nm led to the isolation of 6-hydroxy-7-methoxyquercetin-3-O-β-D-glucopyranoside (2, 11.4 mg), 6-methoxyquercetin-3-O-β-D-glucopyranoside (3, 4.3 mg), and the compound 6,7-dimethoxyquercetin-3-O-β-D-glucopyranoside (1, 9.2 mg), which had been isolated earlier. The software ProStar was used to control the semipreparative system, data collection, and processing. Structural identification of the compounds Compounds were characterized by NMR (1H, 13C, and 2D) analysis on a Varian INOVA 500 operating at 500 MHz for 1H, and at 125 MHz for 13C (11.7 T) and 2D-NMR, with TMS as the internal standard and by comparison with the data reported previously for 6,7-dimethoxyquercetin-3-O-β-D-glucopyranoside (1),15,16 6-hydroxy-7-methoxyquercetin-3-O-β-D-glucopyranoside (2),12 and 6-methoxyquercetin-3-O-β-D-glucopyranoside (3).17 An LCQ Fleet ion trap mass spectrometer with Xcalibur (software version 2.1; Thermo Scientific) and equipped with an electrospray ionization (ESI) interface was used for MS and multi-stage (MSn) analysis. Each compound was dissolved in methanol and infused in the ESI source by a syringe pump (flow rate = 5 µL min-1). High purity nitrogen (N2) was used as the nebulizing gas and ultra-high purity helium (He) was used as the collision gas, at flow rate of 10 arbitrary units. Mass acquisition parameters in negative ion mode were as follows: spray voltage, 5 kV; capillary voltage, -25 V; tube lens offset voltage, -126 V; capillary temperature, 275 ºC. The first event, the full-scan MS data were recorded within range from m/z 50 to m/z 1000. The second event was the MS/MS experiment during which data-dependent scanning was initiated for identifying fragments of deprotonated molecules at normalized collision energy of 25 and activation time of 30 ms. These spectra are included in the Supplementary Material (Figures 1S-16S). Sample preparation Two samples of the hydroethanolic extract (50 mg) were separately dissolved in a solution of spectroscopy grade methanol:water (7:3, v/v; 5.0 mL) under the influence of ultrasound for 5 minutes. The samples were then filtered through a 0.22 µm polytetrafluoroethylene (PTFE) membrane filter and aliquots (10 µL) were directly injected into HPLC-DAD analytical system. This procedure was performed by two different analysts. All solutions were stored below 4 ºC. HPLC-DAD analysis The hydroethanolic extract obtained from aerial parts from T. fluviatilis and the isolated flavonoids (1-3) were analyzed on the HPLC equipment Jasco (Tokyo, Japan) equipped with a PU-2089 quaternary solvent pump with degasser, a MD-2018 DAD detector, and a Rheodyne AS-2055 sample injector with a 100 µL sample loop. The analytical column was a Phenomenex Synergi Hydro RP18 (Phenomenex, Torrance, CA, USA) (250 × 4.6 mm i.d.; 4 µm) equipped with a Phenomenex security guard column (4.0 × 2.0 mm i.d.). The composition of mobile phases was water (eluent A) and methanol (eluent B), both containing 0.1% TFA. The gradient program for analyzing the standards (1-3) was as follows: 36-60% B (0-30 min), 60-100% B (30-31 min), returning after 5 min at 100% B to the initial conditions. A flow rate of 1.0 mL min-1 was set and the volume of injection was 10 µL. All chromatographic assays were performed at 22 ºC and the separated compounds were monitored at 270 nm. The software ChromNav was used to control the analytical system, data collection, and processing. Validation of HPLC-DAD method To validate the method based on HPLC-DAD, specificity/selectivity, precision, limit of detection (LOD), limit of quantification (LOQ), linearity, and accuracy were determined following the recommendations of the International Conference on Harmonisation (ICH) for validation of analytical procedures.18-20 Linearity Compounds 6,7-dimethoxyquercetin-3-O-β-D-glucopyranoside (1), 6-hydroxy-7-methoxyquercetin-3-O-β-D-glucopyranoside (2), and 6-methoxyquercetin-3-O-β-D-glucopyranoside (3) were dissolved separately in spectroscopy grade methanol:water (7:3, v/v) in order to obtain stock solutions (1.0 mg mL-1 each). Each of the stock solution was diluted appropriately (10, 50, 75, 100, 250, 500, and 1000 µg mL-1), and each diluted solution was analyzed in triplicate. A linear least square regression of the peak areas as a function of the concentrations was performed to determine the correlation coefficients. The equation parameters (slope and intercept) of each standard curve were used to obtain the concentration values for the samples. Selectivity Selectivity was assessed by DAD detector comparing the peak spectrum obtained from the separation with a pattern and used it as an indication of the presence of pure compound.21 Limit of detection (LOD) and limit of quantitation (LOQ) The LOD and LOQ were calculated based on the standard deviation of the y-intercept (σ) and slope of the calibration curve (S) obtained from linear regression. LOD was calculated using the expression 3.3 σ/S and LOQ was calculated using 10 σ/S. Precision Six extract samples, prepared by different analysts on different days, were used to evaluate repeatability and intermediate precision. The precision was expressed as relative standard deviation (RSD). Accuracy Solutions containing three known concentrations of the three standards, compounds 1 (150, 400, and 650 µg mL-1), 2 (150, 400, and 650 µg mL-1), and 3 (40, 100, and 150 µg mL-1), were separately added to the hydroethanolic extract samples and then analyzed in triplicate (n = 9 for each compound). Antiradical activity determination DPPH (2,2-diphenyl-1-picrylhydrazyl) assays were used to measure the free-radical scavenging potential of the extracts.22 Solutions of the hydroethanolic extract and compounds 1-3 were separately prepared by dissolving 2.5 mg of each in H2O/MeOH (2:8, v/v; 10 mL). Each of these solutions were diluted to 6.25, 12.5, 25, 50, 100, and 200 µg mL-1 of sample, and each solution was analyzed for antiradical activity in triplicate. Solutions of DPPH (0.004%) in H2O/MeOH (2:8, v/v) were used to determine the antiradical activity. Aliquots (20 µL) of the sample solutions were added to the DPPH solutions (200 µL). After exactly 30 min of reaction, absorbance at 517 nm was recorded using a UV-vis spectrophotometer, Biotek, model Epoch. Standard solutions of quercetin and gallic acid were prepared and analyzed under identical conditions. Reported IC50 values denote the concentration of the sample required to decrease the free-radical activity of DPPH by 50%.
RESULTS AND DISCUSSION Analysis of the hydroethanolic extract of the aerial parts of T. fluviatilis by HPLC-DAD showed the presence of three flavonoids (Figure 1). The compounds were identified by comparison of the retention time and UV spectra of the peaks with those of the available reference standards and confirmed by spiking the hydroethanolic extract with isolated compounds (1-3). With DAD, it was possible to obtain the UV spectra (200-400 nm) of each peak, which permitted the selection of a suitable wavelength at which to maximize the detection of the constituents of hydroethanolic extract, thus minimizing any interference. According to the profile of the corresponding UV spectra, a wavelength of 270 nm was determined to be most appropriate to ensure maximum detection and simultaneous analyses of all compounds present in the hydroethanolic extract. The UV spectra of the peaks corroborated the presence of flavonoids with bands at 240-285 nm and 300-380 nm.23
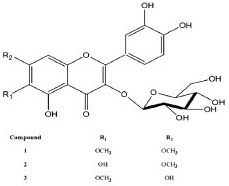 Figure 1. Structures of the flavonoids isolated
Compounds 1-3 were quantified at 10 mg mL-1. The samples were prepared and injected by two different analysts on different days. The samples were submitted to the same procedure as described in "Sample preparation" section. Optimization of HPLC condition for compounds (1-3) To ensure that the method which we established can be used widely, we chose DAD, which is sensitive and used widely, as the detector. We also decided to use a RP18 column based on its application in separation of wide types of compounds. Previous reports indicated that a mobile phase system composed of methanol:water containing 0.1% TFA can sharpen peaks in the chromatogram and, hence, was used as the mobile phase in this study. Optimal separation of the compounds 1, 2, and 3 was observed when the gradient of methanol (solvent B) in deionized water (both containing 0.1% TFA) was allowed to increase from 36% B to 60% B during the first 30 min and then from 60% B to 100% B in 1 min (Figure 2).
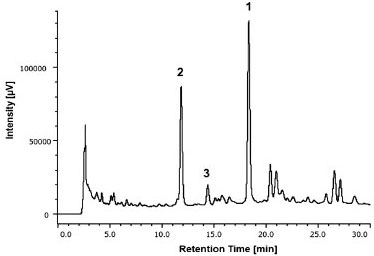 Figure 2. HPLC-DAD chromatogram for the quantification of the compounds 1-3 in the hydroethanolic extract of the aerial parts of T. fluviatilis monitored at 270 nm. A Phenomenex column (Synergi Hydro, Phenomenex, Torrance, CA, USA), RP18 (250 × 4.6 mm i.d.; 4 µm) equipped with a Phenomenex security guard column (4.0 × 2.0 mm i.d.) was used. The gradient program with methanol (B):water (A) (both with 0.1% TFA) flowing at a rate of 1.0 mL min-1 was: 36-60% B (0-30 min), 60-100% B (30-31 min). The volume of injection was 10 µL
Validation of HPLC-DAD method An important step in the development of any product that meets technical requirements for its commercialization is the validation of an analytical method. Selectivity The analysis of UV spectra (Figure 3) shows the peaks in the chromatograms (Figure 4) of isolated compounds in the HPLC-DAD at 254 nm, 270 nm, and 330 nm. In these spectra, it can be seen that there are no other (interfering) substances contributing significantly to the absorbance at the wavelengths monitored.
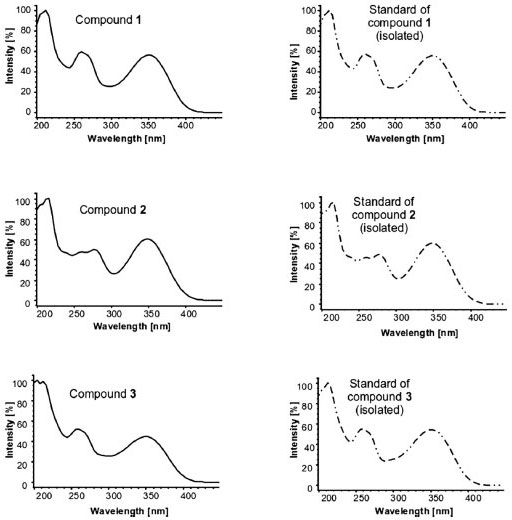 Figure 3. UV spectra of compounds 1-3 present in the hydroethanolic extract of the aerial parts of T. fluviatilis and the spectra of corresponding purified compound, used as standard
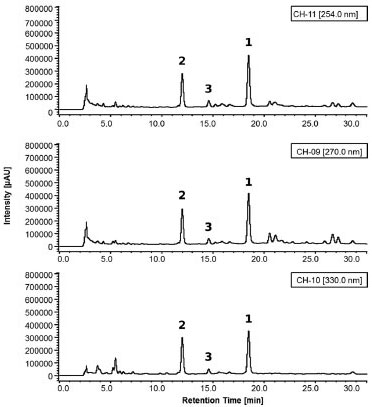 Figure 4. Representative HPLC-DAD chromatograms, monitored at 254 nm, 270 nm, and 330 nm, showing the separation of compounds 1-3 present in the hydroethanolic extract of T. fluviatilis. These chromatograms were used for quantification of compounds 1-3 (for chromatographic conditions, see Figure 2)
Linearity and sensitivity Analytical curves were constructed using the substances isolated (1, 2, and 3) from the hydroethanolic extracts of the aerial parts of T. fluviatilis. The analytical method was calibrated with a standard at seven different concentration levels (10, 50, 75, 100, 250, 500, and 1000 µg mL-1) in triplicate and the response absorption obtained at 270 nm. The areas under the chromatographic peaks were determined and cast as a function of concentration using linear regression. The linearity of the compounds (1-3) was obtained from the correlation coefficient of the linear regression within the range of concentrations studied. All analytes showed excellent linearity within the range studied, which could be verified by the values of the correlation coefficient (r) of standard curves (>0.999) and the sensitivity of the method was expressed by LOD and LOQ (Table 1).
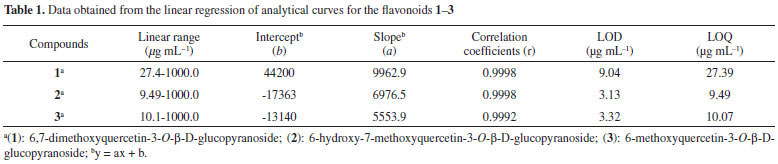
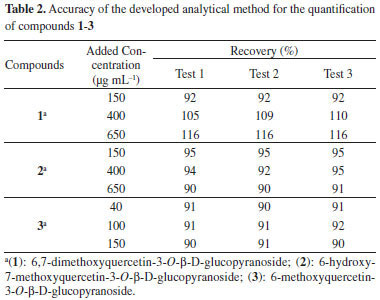
Accuracy The recovery of substances 1-3 were calculated to be >90% and <120%, indicating that the method was accurate and reliable (Table 2).
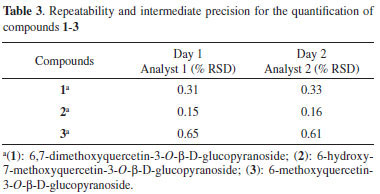
Precision The method showed excellent repeatability, with all relative standard deviation (RSD) less than 1%, and the difference between the mean values determined by the two analysts on different days was <1.1% (Table 3).
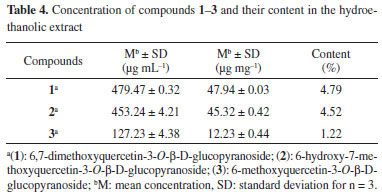
Analysis of the hydroethanolic extract Concentration of the three compounds of interest and content in the hydroethanolic extract are shown in Table 4. Through these values, concentrations were obtained (µg mL-1) from the calibration curves.
Compounds identified in T. fluviatilis revealed the presence of flavonol 6-oxygen. When these data are compared with those in the literature, it is found that naphthopyranones and flavonols are the major compounds of many species from the genus Paepalanthus and Eriocaulon.8,10,11,24 These classes of secondary metabolites are absent in species belonging to both Syngonanthus and Leiothrix genera.9,25 From the evolutionary point of view, it is considered that Leiothrix and Syngonanthus have originated from Paepalanthus, a more advanced position than Eriocaulon,12 since the substitution of flavonols by flavones.26,27 Therefore, this data can contribute towards rationalizing the taxonomy of Tonina and other Eriocaulaceae genus. Antiradical activity The radical scavenging capacity of the hydroethanolic extract, compounds (1-3) from T. fluviatilis, and positive controls (quercetin and gallic acid) are shown in Figure 5.
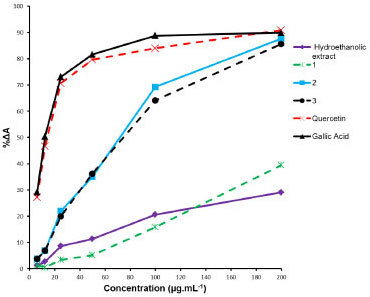 Figure 5. Antiradical activity of the hydroethanolic extract, flavonoids 1-3 isolated from T. fluviatilis, and the standards (gallic acid and quercetin) determined by DPPH assay
Quantities of analyte needed to decrease the DPPH free-radical activity by 50%, IC50, in case of compounds 2 (IC50 = 71.93 ± 1.31 µg mL-1) and 3 (IC50 = 74.47 ± 1.19 µg mL-1) were similar. With compound 1 and the hydroethanolic extract, IC50 values were not attained even at the highest concentration assayed (200 µg mL-1). The radical scavenging activity of the positive controls quercetin (IC50 = 14.11 ± 0.40 µg mL-1) and gallic acid (IC50 = 11.92 ± 0.87 µg mL-1) could be readily determined. Observed high antiradical activity of flavonoids, such as quercetin, is due to the presence of a free hydroxyl at position 3, a double bond between positions 2 and 3, and a keto group at position 4. Such an arrangement of functional groups allows for tautomerization, leading to the existence of a 3-keto form of the molecule.28 The flavonoids (1-3) identified in this study are all substituted at the position 3 of the aglycone; furthermore, the flavonoid 1 has both the hydroxyl groups (at positions 6 and 7) methoxylated, making it difficult for a free-radical to stabilize. Therefore, it is justified that the flavonoids present in the extracts of T. fluviatilis show a lower activity than the reference standards, quercetin and gallic acid.
CONCLUSIONS The chemical study performed with the hydroethanolic extract of the aerial parts of T. fluviatilis, resulted in the isolation of three flavonols (1-3). These substances were identified for the first time in the genus Tonina and the results will allow for the comparison of Tonina with others genus of Eriocaulaceae. This justifies the development of a method for quantification of these compounds in the extract. The method presented good validation parameters, showing that the results obtained are consistent and can be used in ensuring the quantification of these constituents in the extracts. The antiradical activity of the hydroethanolic extract and flavonoids 1-3 were evaluated using the DPPH assay, with gallic acid and quercetin as antiradical reference compounds. The study suggests that the antiradical activity of extracts and flavonoids from T. fluviatilis can be attributed to the flavonoids present in this plant. Therefore, the flavonoids 2 and 3 showed high activity when isolated and compared with the antiradical activity of the crude extract.
SUPPLEMENTARY MATERIAL The figures 1S-16S are available at http://quimicanova.sbq.org.br in PDF format with free access.
ACKNOWLEDGEMENTS The authors wish to thank the São Paulo Research Foundation (FAPESP) for fellowships to M. R. Amorim (2010/10676-3) and for providing financial aid to L. C. Santos (2012/03882-1). We also thank Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for grants to L. C. Santos and W. Vilegas.
REFERENCES 1. Giulietti, A. M.; Hensold, N.; Parra, L. R.; Andrade, M. J. G,; Van Den Berg, C.; Harley, R. M.; Phytotaxa 2012, 60, 50. 2. Devienne, K. F.; Calgaro-Helena, A. F.; Dorta, D. J.; Prado, I. M. R.; Raddi, M. S. G.; Vilegas, W.; Uyemura, S.A.; Santos, A. C.; Curti, C.; Phytochemistry 2007, 68, 1075. 3. Santos, L. C.; Piacente, S.; Montoro, P.; Pizza, C.; Vilegas, W.; Rev. Bras. Farmacogn. 2003, 13, 67. 4. Silva, M. A.; Oliveira, A. P. S.; Sannomiya, M.; Sano P. T.; Varanda, E. A.; Vilegas, W.; Santos, L. C.; Chem. Pharm. Bull. 2007, 55, 1635. 5. Varanda, E. A.; Raddi, M. S. G.; Dias, F. L. P.; Araujo, M. C. S.; Gibran, S. C. A.; Takahashi, C. S.; Vilegas, W.; Teratog., Carcinog., Mutagen. 1997, 17, 85. 6. Coelho, R. G.; Batista, L. M.; Santos, L. C.; Brito, A. R. M. S.; Vilegas, W.; Rev. Bras. Cienc. Farm. 2006, 42, 413. 7. Giulietti, A. M.; Hensold, N.; Acta Bot. Bras. 1990, 4, 133. 8. Amaral, F. P.; Napolitano, A.; Masullo, M.; Santos, L. C.; Festa, M.; Vilegas, W.; Pizza, C.; Piacente, S.; J. Nat. Prod. 2012, 75, 547. 9. Pacifico, M.; Napolitano, A.; Masullo, M.; Hilario, F.; Vilegas W.; Piacente S.; Santos L. C.; Ind. Crops Prod. 2011, 33, 488. 10. Santos, L. C.; Silva, M. A.; Rodrigues, C. M.; Carbone, V.; Napolitano, A.; Bassarello, C.; Mari, A.; Piacente, S.; Pizza, C.; Vilegas, W.; Nat. Prod. Commun. 2009, 4, 1651. 11. Zanutto, F. V.; Boldrin, P. K.; Varanda, E. A.; Souza, S. F.; Sano, P. T.; Vilegas, W.; Santos, L. C.; Molecules 2013, 18, 244. 12. Vilegas, W.; Nehme, C. J.; Dokkedal, A. L.; Piacente, S.; Rastrelli, L.; Pizza, C.; Phytochemistry 1999, 51, 403. 13. Dokkedal, A. L.; Santos, L. C.; Sano, P. T.; Vilegas, W.; Z. Naturforsch. 2008, 63c, 169. 14. Markham, K. R.; Techniques of Flavonoid Identification, Academic Press: London, 1982. 15. Ulubelen, A.; Timmermann, B. N.; Mabry, T. J. Phytochemistry 1980, 19, 905. 16. Schalangen, K.; Miosic, S.; Castro, A.; Freudmann, K.; Luckziewicz, M.; Vitzthum, F.; Schwab, W.; Gamsjaeger, S.; Musso, M.; Halbwirth, H. Phytochemistry 2009, 70, 889. 17. Thomas, M. B.; Mabry, P. J. Phytochemistry 1968, 7, 787. 18. ICH - International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use; Validation of Analytical Procedures: Text and Methodology Q2(R1), 2005. 19. Peters, F. T.; Drummer, O. H.; Mosshoff, F.; Forensic. Sci. Int. 2007, 165, 216. 20. Snyder, L. R.; Kirkland, J. J.; Dolan, J. W.; Introduction to Modern Liquid Chromatography, 3rd ed., John Wiley & Sons: New Jersey, 2010. 21. Ribani, M.; Botolli, C. B. G.; Collins, C. H.; Jardim, I. C. S. F.; Melo, L. F. C.; Quim. Nova 2004, 27, 771. 22. Brand-Williams, M.; Cuvelier, M. E.; Berset, C.; Lebensm.-Wiss. Technol. 1995, 28, 25. 23. Merken, H. M.; Beecher, G. R.; J. Agric. Food Chem. 2000, 48, 577. 24. Dokkedal, A. L.; Salatino, A. Biochem. Syst. Ecol. 1992, 20, 31. 25. Ricci, C. V.; Patricio, M. C.; Salatino, M. L.; Salatino, A.; Giulietti, A. M. Biochem. Syst. Ecol. 1996, 24, 577. 26. Santos, L. C.; Rodrigues, C. M.; Silva, M. A.; Coelho, R. G.; Sannomiya, M.; Vilegas W. Biochem. Syst. Ecol. 2005, 33, 1159. 27. Harborne, J. B. The Flavonoids: Advances in Research since 1986. Chapman and Hall: London, 1996. 28. Sousa, C. M. M.; Silva, H. R.; Vieira-Júnior, G. M.; Ayres, M. C. C.; Costa, C. L. S.; Araújo, D. S.; Cavalcante, L. C. D.; Barros, E. D. S.; Araújo, P. B. M.; Brandão, M. S.; Chaves, M. H.; Quim. Nova 2007, 30, 351. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access