
 |
 |
 |
 |
 |
Artigo
| Influence of chloride-mediated oxidation on the electrochemical degradation of the Direct Black 22 dye using boron-doped diamond and β-PbO2 anodes |
|
Douglas A. C. Coledam; José M. Aquino; Romeu C. Rocha-Filho*; Nerilso Bocchi; Sonia R. Biaggio
Departamento de Química, Universidade Federal de São Carlos, CP 676, 13560-970 São Carlos - SP, Brasil Recebido em 22/01/2014 *e-mail: romeu@ufscar.br The Direct Black 22 dye was electrooxidized at 30 mA cm−2 in a flow cell using a BDD or β-PbO2 anode, varying pH (3, 7, 11), temperature (10, 25, 45 ºC), and [NaCl] (0 or 1.5 g L−1). In the presence of NaCl, decolorization rates were similar for all conditions investigated, but much higher than predicted through a theoretical model assuming mass-transport control; similar behavior was observed for COD removal (at pH 7, 25 ºC), independently of the anode. With no NaCl, COD removals were also higher than predicted with a theoretical model, which suggests the existence of distinct dye degradation pathways. INTRODUCTION The contamination of surface waters by synthetic organic compounds is one of the main challenges to human society in the 21st century. Synthetic textile dyes are organic compounds that are of major concern because of their impact on aquatic and human lives.1 According to Hao et al.,2 it is estimated that the half-life of the hydrolyzed form of the Reactive Blue 19 dye is 46 years at neutral conditions and 25 ºC. Synthetic dyes may enter into the environment through their production, usage, and also when they are released from textile fibers to which they are bound. Large amounts of potable water are used during dyeing processes by textile industries, which are the main consumers of synthetic dyes.3 Consequently, significant volumes of textile dyeing effluents are generated; most of these effluents must be treated before their disposal according to regulations of governmental environmental agencies.3,4 Conventional (biological) treatment technologies are inefficient to treat effluents containing some synthetic textile dyes, since more toxic compounds can be generated during their biological treatment, such as aromatic amines.5 In this context, electrochemical methods might be an interesting treatment technology that is easily implemented and has potential for high (contaminant) removal rates.6 The electrooxidation of organic pollutants can be performed directly, when the contaminant exchanges electrons with the anode material itself, and indirectly, when the contaminant is oxidized through electrogenerated oxidants.7 Among these oxidizing species, the hydroxyl radical (•OH) is the most common; however, because of its high reactivity, high concentrations of the •OH species only occur very close to the anode surface.8 Consequently, when electrooxidation is mediated by the •OH species, a combination between high mass transfer rate and current density is required to attain high removal and energy efficiencies.9,10 Organic pollutants may also be oxidized by other electrogenerated oxidants, such as active chlorine (Cl2, HClO, and OCl−) and S2O82−, when chloride and sulfate containing solutions are electrolyzed, respectively. In the presence of chloride ions, the rate of organic removal under mass transport limitations during the electrooxidation of dyes11 or plasticizers10 was shown to be significantly improved by indirect processes mediated by active chlorine. One of the most important parameters in the electrooxidation of organics is the selection of adequate electrode materials.12 Boron-doped diamond (BDD) is one of the anode materials commonly used to degrade distinct classes of organic contaminants,10,11,13-15 because of its high oxidation power resulting from the significant production of quasi-free •OH radicals on its surface.16,17 The difficulty of finding adequate substrates for the BDD film deposition and the high prices of these electrodes are their main drawbacks.18 Alternatively, PbO2 films, especially in the beta form (β), are interesting electrode materials for organic pollutant removal,19,20 despite their somewhat lower oxidation power.16 The electrochemical performance of β-PbO2 anodes is mainly related to the substrate and electrodeposition bath conditions used in their preparation.21-24 Electrodes prepared with Ti-Pt substrates have proven to be highly stable for possible environmental applications, as the release of toxic Pb2+ ions is very low.25 Thus, considering the above and the fact that the electrooxidation of the Direct Black 22 (DB 22) dye has only been assessed directly with a BDD anode,11 the aim of the present study is to compare the influence of chloride-mediated oxidation on the performances of BDD and β-PbO2 anodes in the electrochemical degradation of a large model compound, such as the DB 22 dye molecule. These performances will be compared through the decay of the color (assessed through UV-vis spectrophotometry) and chemical oxygen demand (COD) of the dye solution. The electrooxidation process of a large model compound, the DB 22 dye molecule, will be compared to a theoretical model based on a purely mass-transport controlled process presented in the literature. Results will explicitly define the contribution of the electrochemical and chemical processes. Additionally, to the best of our knowledge, results on the electrooxidation of the DB 22 dye using a β-PbO2 anode and its superior efficiency relative to a BDD anode (in the absence of NaCl in solution) will be reported for the first time.
EXPERIMENTAL Chemicals All chemicals, including Pb(NO3)2 (a.r., Sigma-Aldrich), sodium lauryl sulfate, SLS (99%, Fisher Scientific), H2PtCl6 (99.9%, Aldrich), HCl (36,5%, Mallinckrodt), H2SO4 (98%, Mallinckrodt), NaCl (a.r., JT Baker), Na2SO4 (a.r., Qhemis), and DB 22 (Quimanil) were used without further purification. Doubly deionized water (Millipore Milli-Q system, ρ > 18.2 MΩ cm) was used for the preparation of all solutions. The chemical structure of the DB 22 dye can be seen in Figure 1S in the supplementary material file. Preparation of the electrodes The β-PbO2 films were galvanostatically electrodeposited on a platinized Ti substrate in a conventional cell, using two AISI-304 stainless steel plates as counter electrodes. The detailed procedures involving the Ti substrate platinization and the β-PbO2 film preparation are fully described in a previous work.26 The BDD electrode was purchased from Condias® (Germany). The BDD film (with a specified boron content range of 2000-4500 ppm) was grown on a niobium substrate by the hot filament chemical vapor deposition technique. Linear polarization measurements The linear polarization study of a naturally aerated 0.1 mol L−1 Na2SO4 solution in the presence or absence of 100 mg L−1 DB 22 dye was made in a conventional electrochemical cell at ambient temperature. The Nb/BDD anode was fixed on an acrylic holder through a Viton® O-ring (exposed area: 0.49 cm2), whereas the Ti-Pt/β-PbO2 anode (exposed area: 3.64 cm2) was directly immersed in the cell. An Ag/AgCl (3 mol L−1 KCl) electrode and a platinum foil were used as reference and counter electrode, respectively. Before the polarization assays, the anodes were pretreated for 15 min in a 0.1 mol L−1 Na2SO4 solution by applying 6 mA cm−2, which eliminated any adsorbed organic contaminants. All measurements were performed using a PGSTAT 20 Autolab potentiostat/galvanostat controlled by the GPES software. Electrochemical degradation of the DB 22 dye The electrochemical degradations of the DB 22 dye were performed galvanostatically in a one-compartment filter-press flow reactor composed of Nb/BDD (exposed area: 3.0 cm × 2.5 cm, each face) or Ti-Pt/β-PbO2 (exposed area: 3.1 cm × 2.7 cm, each face) as anode and two AISI 304 stainless steel plates as cathodes. More details about the experimental setup are described in a previous work.26 All experiments were performed using 0.4 L of a 100 mg L−1 aqueous solution of the DB 22 dye in 0.1 mol L−1 Na2SO4. The investigated variables for the degradation of the DB 22 dye were pH (3, 7, and 11), temperature (θ = 10, 25, and 45 ºC), and presence or absence of NaCl (1.50 g L−1) in the solution. Other variables were kept constant according to previous studies of our group:11,19,25-28 current density (j = 30 mA cm−2), and volumetric flow rate (qV = 7.0 L min−1). Before each electrolysis, adsorbed organic contaminants were eliminated by pretreating the respective anode for 15 min in a 0.1 mol L−1 Na2SO4 solution by applying 30 mA cm−2. Analyses Operational conditions were optimized for the electrochemical degradation of the DB 22 dye, following its decolorization. COD assays were performed at those optimized conditions to assess the attained degrees of oxidation. The color of the electrolyzed dye solution was monitored at specific electrolysis times by measuring the absorbance of a 3 mL sample of this solution from 200 nm to 800 nm with a Hewlett-Packard UV-vis spectrophotometer (model HP8452, with a diode array detector). COD assays used 2 mL of the electrolyzed dye solution, which was mixed in a glass tube with an oxidizing solution composed of an acidified dichromate solution from Hach®. The glass tubes were left at 150 ºC for 2 h to completely oxidize the organic matter, according to the closed reflux colorimetric method.29 Then, after ambient temperature was reached, the absorbance at 420 nm of each tube was measured using a Hach DR/2010 model spectrophotometer, and the corresponding COD value was determined.
RESULTS AND DISCUSSION Linear polarization Figure 1 shows linear polarization measurements with the BDD and β-PbO2 anodes in the absence and presence of the DB 22 dye in solution. The anodic electrochemical window of the BDD electrode is larger than that of the β-PbO2. However, distinct behaviors occur when the polarization occurs in the presence of the DB 22 dye: i) on the BDD anode, the current density for the oxygen evolution reaction (OER: E > 3.0 V) decreases with the number of scans (the same happens with the oxidation peak current of the DB 22 dye molecule at 1.1 V; see Fig. 2S in the supplementary material file), which is an indication that the active area of the electrode is being blocked by the DB 22 dye molecule or its oxidation products; ii) on the β-PbO2 anode, the OER current density gradually increases with the number of scans, which indicates that the oxidation of the DB 22 dye is leading to byproducts that are further oxidized, as two oxidation peaks appear at about 0.80 V and 1.30 V (for more details, see Fig. 2S in the supplementary material file). After the polarizations, the color of the surface of the β-PbO2 electrode became light brown, possibly due to a film that formed from oxidation byproducts. The oxidation current peaks observed for the BDD anode in the potential range of 1.70-2.50 V, whose intensities decrease with the number of linear scans, are related to the oxidation of sp2 carbon atoms (graphite).30
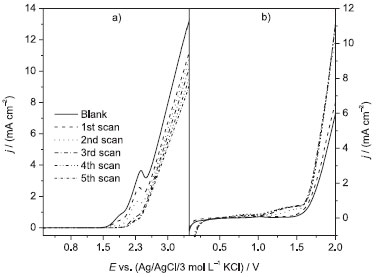 Figure 1. Linear polarization measurements (20 mV s−1) using a (a) Nb/BDD or (b) Ti-Pt/β-PbO2 anode in the absence (blank: 0.1 mol L−1 Na2SO4) and presence of 100 mg L−1 DB 22 dye in 0.1 mol L−1 Na2SO4, at 25 ºC. Previously, the anodes were pretreated for 15 min in a 0.1 mol L−1 Na2SO4 solution by applying 6 mA cm−2
Color removal Figure 2 shows the decay of the relative absorbance (Arel) as a function of the applied charge per unit volume of electrolyzed solution (Qapl) for the electrooxidation of the DB 22 dye with the BDD anode at 25 ºC and distinct pH values (similar data at 10 and 45 ºC can be seen in Figure 3S in the supplementary material file).
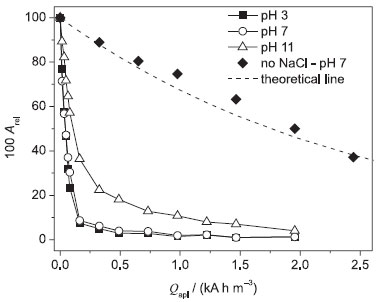 Figure 2. Relative absorbance (Arel) as a function of the applied charge per unit volume of electrolyzed solution (Qapl) for the electrooxidation of 100 mg L−1 DB 22 dye solutions (in 0.1 mol L−1 Na2SO4 + 1.50 g L−1 NaCl) of distinct pH values (indicated in the figure) using the Nb/BDD anode in a filter-press flow reactor at 25 ºC. Theoretical line (- - -) (see text). Electrolysis conditions: j = 30 mA cm−2; qV = 7.0 L min−1; V = 0.4 L
It is interesting to observe that the Qapl values needed for complete decolorization were about the same at pH 3 and 7, independently of the temperature: ca. 0.5 kA h m−3. Under alkaline conditions (pH 11), the Qapl value needed for complete decolorization increased for all investigated temperature values. This behavior is related to the distinct chloro oxidant species present in the bulk of the solution, which are mainly HOCl at pH 3 and 7, and OCl− at pH 11. According to Wang and Margerum,31 the predominance ratio of the Cl2/HOCl species is dependent on many parameters, such as the solution temperature, pH, ionic strength, and chloride ion concentration. The predominance ratio of the HOCl/OCl− pair is similarly dependent on those parameters (except chloride ion concentration), as reported by Morris32 in a study on the temperature dependence of the acidity constant of HClO. Thus, since the HOCl species has a higher oxidation potential than the OCl− species, a superior color removal is expected in neutral to acidic solutions. It is important to highlight that the gaseous nature of the HOCl species did not exert a significant influence on the attained decolorization, since its concentration might be low, and consequently could lead to decolorization levels comparable to the ones attained in the alkaline solution. When the electrolyses with the BDD anode were performed in alkaline medium, another important aspect is the possible electrogeneration of chloro species at higher oxidation states,33 such as ClO3− and ClO4−, which are inefficient oxidants for organics oxidation.34 These species are formed after successive oxidations of the OCl− species mediated by hydroxyl radicals (•OH) on the electrode surface, whereby available OCl− concentrations decrease. This might be the main reason for the lower decolorization level attained in the alkaline solution. Figure 3 shows the decay of Arel as a function of Qapl during the electrooxidation of the DB 22 dye solution with the β-PbO2 anode at 25 ºC and distinct pH values (similar data at 10 and 45 ºC can be seen in Figure 4S in the supplementary material file). Complete decolorization was attained at pH 3 and 7 in the three temperatures investigated after supplying a Qapl value of ca. 0.8 kA h m−3. Decolorization level under alkaline conditions was slightly superior to that attained using the BDD anode. As previously mentioned, this behavior might be due to the high oxidation power of the BDD anode that promotes the oxidation of the OCl− species to weaker oxidants. Moreover, at 45 ºC, it is possible that the β-PbO2 anode presents a similar behavior with respect to the loss of oxidants, leading to a lower decolorization level.
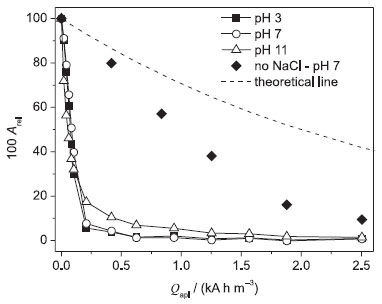 Figure 3. Relative absorbance (Arel) as a function of the applied charge per unit volume of electrolyzed solution (Qapl) for the electrooxidation of 100 mg L−1 DB 22 dye solutions (in 0.1 mol L−1 Na2SO4 + 1.50 g L−1 NaCl) of distinct pH values (indicated in the figure) using the Ti-Pt/β-PbO2 anode in a filter-press flow reactor at 25 ºC. Theoretical line (- - -) (see text). Electrolysis conditions: j = 30 mA cm−2; qV = 7.0 L min−1; V = 0.4 L
Another important characteristic that can be observed in Figures 2 and 3 is the comparison between an indirect decolorization process mediated by chloro oxidant species (dye oxidation that mainly occurs in the bulk of the solution) and a purely mass-transport (dye oxidation that occurs mainly on the anode surface) controlled process (see eq. 1), as the DB 22 dye molecules need to be transported from the bulk of the solution to the electrode surface to react with •OH radicals. In such a process, the main parameter is the mass-transfer coefficient, km, which was equal to 2.90 × 10−5 m s−1 at 25 ºC, when obtained by a simple electrochemical assay based on the Fe(CN)64−/Fe(CN)63− redox pair:35
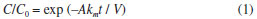
where C0 is the initial concentration of the DB 22 dye (based on absorbance) and C this concentration at the time of electrolysis t (s), A is the area (m2) of the working electrode, km the mass-transfer coefficient (m s−1), and V the electrolyzed solution volume (m3). The indirect (chloride-mediated) process led to greater decolorization independent of the electrode material; see below the values obtained for the respective apparent first-order kinetic constants (this comparison was done only at 25 ºC; the km value does depend on the solution temperature, but similar behavior is expected for the other two temperature values investigated). On the other hand, the Qapl value for decolorization at pH 7 and 25 ºC increased significantly in the absence of chloride ions (see Figures 2 and 3). These results suggest that •OH radicals attack the chromophore group of the dye when chloride ions are absent from the electrolyzed solution. This process, which is an initial and soft oxidation step before further conversions toward CO2, depends only on the hydrodynamic conditions used. It is interesting to note that the experimental data obtained using the BDD anode fit the theoretical line for a purely mass-transport controlled process (eq. 1). However, when the electrolysis was performed using the β-PbO2 anode, the decolorization levels were significantly greater than those attained using the BDD anode or theoretically predicted for a mass-transport controlled process. This behavior might be related to an intrinsic characteristic of β-PbO2 films, which lead to prompt oxidation of the DB 22 dye molecule, as discussed above for the linear polarization measurements, and also to the production of oxidants such as O3. Decay of the relative absorbance of the DB 22 dye solution, in the absence of chloride ions, using the BDD and β-PbO2 anodes can be seen in Figure 5S in the supplementary material file, which confirms the superior performance of the β-PbO2 anode in the decolorization of the DB 22 dye. Theoretical lines based on a purely mass-transport controlled process provide information on the rate of the electrochemical oxidation process. Any deviation from the theoretical model (increase of the kinetic constant) is very likely related to indirect processes, such as chemical reactions in the bulk of the solution promoted by electrogenerated oxidants. Figure 4 shows the values of the apparent first-order kinetic constants (kap) for the chloride-mediated electrooxidative decolorization of the DB 22 dye solution at distinct temperatures using the BDD and β-PbO2 anodes. In the temperature range investigated, the kap values are practically constant for the solutions of pH 3 and 7; however, the decolorization rate decreased when electrolysis occurred at 45 ºC. Considering that the mediated oxidation of the chromophore groups is the main process, this behavior might be because of the loss of oxidants, particularly OCl−, caused by the electrogeneration of chloro oxidants of higher oxidation states that are not efficient oxidizers of organics. Moreover, the obtained kap values for the indirect electrooxidation using BDD and β-PbO2 anodes were very similar, as expected for a process that occurs in the bulk of the solution and is responsible for a soft oxidation of the organic molecule (through attacks on the chromophore group by addition, substitution, or oxidation reactions).36 On the other hand, in the absence of chloride ions, the kap values for both anodes decreased significantly (around one order of magnitude), since, in this case, the decolorization process takes place mainly on the anode surface.
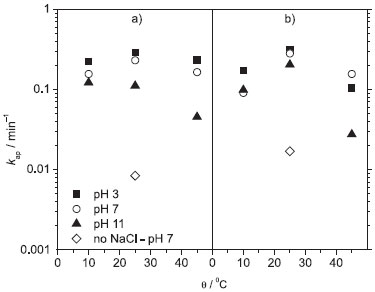 Figure 4. Apparent first-order kinetic constants (kap) as a function of solution temperature for the electrooxidative decolorization of 100 mg L−1 DB 22 dye solutions (in 0.1 mol L−1 Na2SO4 + 1.50 g L−1 NaCl) of distinct pH values (indicated in the figure) using the (a) Nb/BDD or (b) Ti-Pt/β-PbO2 anode in a filter-press flow reactor. Electrolysis conditions: j = 30 mA cm−2; qV = 7.0 L min−1; V = 0.4 L
Considering that there were no appreciable differences in the decolorization performances attained at pH 7 and 25 ºC, in the presence and absence of chloride ions, the investigations on COD removal were performed at these conditions. COD removal Figure 5 shows the relative COD removal as a function of Qapl for the electrooxidation of the DB 22 dye solution at pH 7 and 25 ºC, in the presence and absence of chloride ions, using the BDD and β-PbO2 anodes. As expected, the Qapl value needed for total COD removal, in the presence of chloride ions, was much higher than that for total decolorization because the oxidation of the DB 22 dye molecule is a more complex process. However, in the absence of chloride ions, the only difference was the initial lower COD removal rate as the Qapl needed for total color and COD removals were quite similar (compare the results shown in Figure 5 with those in Figure 5S in the supplementary material file). Moreover, the indirect oxidation process, in the presence of chloride ions, led to initial COD removal rates of near 100% efficiency (Figure 5). The Qapl values increased significantly when electrolysis was performed in the absence of chloride ions; then, complete COD removal only occurred after Qapl values of ca. 6.0 kA h m−3 and 4.0 kA h m−3 for the BDD and β-PbO2 anodes, respectively. These results could be ascribed to the absence of chloro oxidant species; however, it is very unlikely that these oxidants promoted complete conversion of the dye into CO2 because their oxidation powers are lower than that of the •OH radical. Thus, it is possible that the electrooxidation in the presence of chloro oxidant species was leading to recalcitrant intermediates that were not further oxidized by the dichromate ions.11 Baker et al.37 also reported on some organic compounds that were not oxidized by COD assays.
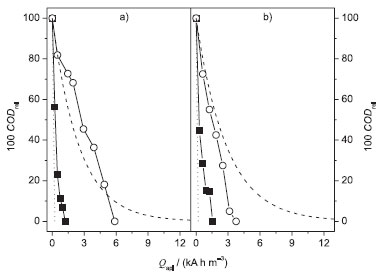 Figure 5. Relative COD removal (100 CODrel) as a function of the applied charge per unit volume of electrolyzed solution (Qapl) for the electrooxidation of 100 mg L−1 DB 22 dye solutions (in 0.1 mol L−1 Na2SO4) at pH 7 and 25 ºC, in the presence (■ - 1.5 g L−1) or absence of NaCl (○), using the (a)BDD or (b) Ti-Pt/β-PbO2 anode in a filter-press flow reactor. Theoretical line (- - -) (see text); 100 % COD efficiency line (•••). Electrolysis conditions: j = 30 mA cm−2; qV = 7.0 L min−1; V = 0.4 L
Another important aspect depicted in Figure 5 is that the Qapl value required for complete COD removal using the BDD anode in the absence of chloride ions is lower than that predicted by a theoretical model proposed by the group of Comninellis,38,39 which is based on a purely mass-transport controlled process. In this theoretical model, the main assumptions are that the electrochemical oxidation of organics is a rapid reaction mediated by the •OH radicals on the electrode surface and that no indirect oxidation mediated by electrogenerated oxidants occurs in the bulk of the solution. The mathematical expression for COD evolution under mass-transfer limitations is as follows:
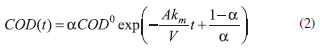
where COD0 and COD(t) are the COD (mol m−3) values at the times of electrolysis zero and t (s), α is the ratio between the applied current density (j) and the initial limiting current density ( j0lim):

with α > 1 and j> j0lim. When the BDD anode was used, theoretical COD removal values were higher than the experimental ones during most of the electrolysis duration (Qapl < 5 kA h m−3). Therefore, it is possible that in this case the DB 22 dye oxidation did not exhibit a rapid electrochemical reaction on the electrode surface (i.e., the dye molecule oxidation occurs through successive steps to gradually have its structure broken down) since its complexity and number of carbon atoms were high in comparison to the model compounds used in the theoretical model. On the other hand, the COD removal values attained using the β-PbO2 anode were higher than those predicted by the theoretical model or obtained using the BDD anode. This might be due to a different oxidation pathway on the β-PbO2 anode, as previously discussed in the analysis of the linear polarization curves (Figure 1). Finally, the COD removal levels attained using BDD and β-PbO2 anodes in the presence of chloride ions were quite similar, which indicates that the DB 22 dye oxidation, mainly promoted in the bulk of the solution by chloro oxidant species, occurs through similar pathways. It is important to highlight that no film detachment was observed after the electrolysis experiments using the Nb/BDD or the Ti-Pt/β-PbO2 anode. Instantaneous current efficiency and energy consumption The instantaneous current efficiency (εinst) for the electrooxidation of the DB 22 dye solution was calculated according to
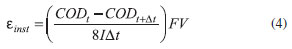
where CODt and CODt+Δt are the COD values (mol m−3) at times t and t + Δt (s), respectively, F is the Faraday constant (96485 C mol−1), V the solution volume (m3), I the applied electric current (A), and 8 is a conversion factor. Figure 6 shows the εinst values as a function of the relative COD decay using BDD and β-PbO2 anodes, in the absence and presence of chloride ions, at pH 7 and 25 ºC. The εinst values were higher, for both anodes, when the electrooxidation was performed in the presence of chloride ions. Indirect oxidation mediated by chloro oxidant species contributed to the reaction. On the other hand, in the absence of chloride ions, the εinst values remained low and very close to theoretically predicted values (εtinst) according to the following relation:
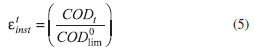
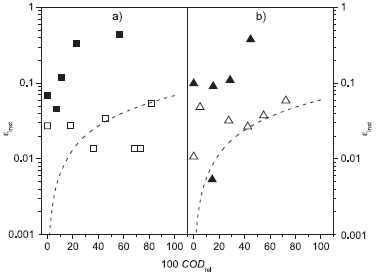 Figure 6. Instantaneous current efficiency (εinst) as a function of the relative COD for the electrooxidation of 100 mg L−1 DB 22 dye solutions (in 0.1 mol L−1 Na2SO4) at pH 7 and 25 ºC, in the absence (empty symbols) and presence (full symbols) of 1.5 g L−1 NaCl, using the (a) BDD or (b) Ti-Pt/β-PbO2 anode in a filter-press flow reactor. Theoretical line (- - -) (see text). Electrolysis conditions: j = 30 mA cm−2; qV = 7.0 L min−1; V = 0.4 L
where COD0lim is the initial COD limiting value calculated by eq. 3. The low εinst values obtained in the absence of chloride ions are due to the higher applied current density with respect to the limiting one. Thus, a large amount of the supplied charge was lost to the parasitic O2 evolution reaction. These results demonstrate the significant contribution of indirect oxidation to improve the efficiency of the electrooxidation process under mass-transfer limitation. Moreover, it is important to highlight that the εinst values were quite similar for both anodes, a characteristic also previously described for the COD removal. The decrease of the εinst values in the final stages of the electrooxidation may be because of increased O2 evolution reaction that occurred because the organic load was being completely oxidized. The electric energy consumption per unit volume of electrolyzed dye solution (w) was obtained according to

where U is the cell potential (V). Figure 7 shows the relative COD decay as a function of the obtained w values at pH 7 and 25 ºC, in the presence and absence of chloride ions, using the BDD and β-PbO2 anodes. The w values in the presence of chloride ions were much lower than those in their absence, due to the mediated oxidation promoted by chloro oxidant species; however, it is important to emphasize that when electrolysis occurred in the presence of chloride ions, the possible chlorination of the organic compounds might lead to erroneous COD values, as previously discussed. Thus, it is likely that the true w values required for the complete oxidation of such a large model compound are better represented by those attained in the absence of chloride ions. In a recent work, Aquino et al.11 showed that the COD removal values attained during the electrooxidation of synthetic dyes in the presence of chloride ions using a Si/BDD anode did not lead to complete conversion into CO2, as the total organic carbon content was not completely removed. On the other hand, in the absence of chloride ions, the COD assays could be used as an indicator of the dye conversion into CO2.
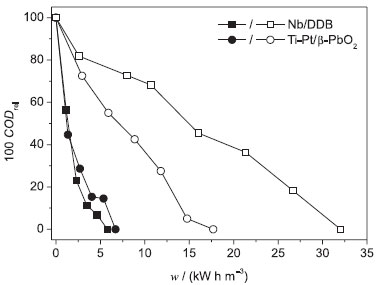 Figure 7. Relative COD removal (100 CODrel) as a function of the electrical energy per unit volume of electrolyzed solution (w) for the electrooxidation of 100 mg L−1 DB 22 dye solutions (in 0.1 mol L−1 Na2SO4) at pH 7 and 25 ºC, in the absence (empty symbols) and presence (full symbols) of 1.5 g L−1 NaCl, using the (a) BDD or (b) Ti-Pt/β-PbO2 anode in a filter-press flow reactor. Electrolysis conditions: j = 30 mA cm−2; qV = 7.0 L min−1; V = 0.4 L
Figure 7 also shows that the w values required for the complete oxidation of the DB 22 dye, in the presence of chloride ions, were independent of the anode, as observed for the values of COD abatement and calculated εinst, because the oxidation pathway of DB 22 dye was the same for both anodes. Previously, we reported similar values of w for the electrooxidation of other synthetic dyes.23,27 On the other hand, the degradation pathway of the DB 22 dye, in the absence of chloride ions, was different for the two anodes as reflected by the respective values of w. When the β-PbO2 anode was used, the dye molecule was more promptly attacked, which resulted in lower w values.
CONCLUSIONS When in the presence of chloride ions, the electrooxidative color and COD removals of solutions containing the DB 22 dye were successfully attained using BDD and β-PbO2 anodes because of the electrogeneration of chloro oxidant species. These species considerably improved the attack on the dye chromophore group in the bulk of the solution relative to the electrode surface associated with a purely mass-transport controlled process. The solution pH and temperature did not significantly affect the attained decolorization rates, except under alkaline conditions at 45 ºC, because of the parasitic reactions of loss of oxidants. These reactions are more likely to occur during electrooxidation using anodes of high oxidation power, such as the BDD electrode. The values of COD removal attained in the presence of chloride ions were very similar for both anodes, when a similar oxidation pathway was mediated by chloro oxidant species; moreover, these values were much higher than those predicted by a theoretical model based on a purely mass-transport controlled process. In the absence of chloride ions (at 25 ºC and pH 7), the Qapl values required for complete COD removal were higher than the Qapl values when chloride ions were present. Under these conditions, the β-PbO2 anode was more efficient for COD removal than the BDD anode, which suggests that distinct oxidation pathways take place on the surface of those anodes. Finally, indirect (chloride-mediated) oxidation clearly contributed to increase the efficiency values with respect to both εinst and w, independent of the anode material.
SUPPLEMENTARY MATERIAL The online version of this article contains supplementary material, including the chemical structure of the DB 22 dye molecule and relative absorbance decay curves as a function of Qap for distinct pH and temperature values. This material is available at http://quimicanova.sbq.org.br.
ACKNOWLEDGEMENTS The authors gratefully acknowledge financial support and scholarships from the Brazilian funding agencies Sao Paulo Research Foundation-FAPESP (grant no. 2012/13587-7), CNPq, and CAPES. Quimanil is acknowledged for supplying the DB 22 dye samples. Professors Antonio A. Mozeto, Pedro S. Fadini, and Alzir A. Batista (DQ-UFSCar) are gratefully acknowledged for granting access to different apparatuses. Gabriel F. Pereira is also gratefully acknowledged for lending a hand with some experiments.
REFERENCES 1. Carneiro, P. A.; Umbuzeiro, G. A.; Oliveira, D. P.; Zanoni, M. V. B.; J. Hazard. Mater. 2010, 174, 694. 2. Hao, O. J.; Kim, H.; Chang, P. C.; Crit. Rev. Environ. Sci. Technol. 2000, 30, 449. 3. Singh, K.; Arora, S.; Crit. Rev. Environ. Sci. Technol. 2011, 41, 807. 4. Hessel, C.; Allegre, C.; Maisseu, M.; Charbit, F.; Moulin, P.; J. Environ. Manage. 2007, 83, 171. 5. Pandey, A.; Singh, P.; Iyengar, L.; Int. Biodeterior. Biodegrad. 2007, 59, 73. 6. Cañizares, P.; Paz, R.; Sáez, C.; Rodrigo, M. A.; J. Environ. Manage. 2009, 90, 410. 7. Panizza, M.; Cerisola, G.; Chem. Rev. 2009, 109, 6541. 8. Kapałka, A.; Fóti, G.; Comninellis, C.; Electrochim. Acta 2009, 54, 2018. 9. Aquino, J. M.; Pereira, G. F.; Rocha-Filho, R. C.; Bocchi, N.; Biaggio, S. R.; J. Hazard. Mater. 2011, 192, 1275. 10. Pereira, G. F.; Rocha-Filho, R. C.; Bocchi, N.; Biaggio, S. R.; Chem. Eng. J. 2012, 198-199, 282. 11. Aquino, J. M.; Rodrigo, M. A.; Rocha-Filho, R. C.; Sáez, C.; Cañizares, P.; Chem. Eng. J. 2012, 184, 221. 12. Panizza, M.; Cerisola, G.; Appl. Catal., B 2007, 75, 95. 13. Polcaro, A. M.; Vacca, A.; Mascia, M.; Palmas, S.; Electrochim. Acta 2005, 50, 1841. 14. Rocha, J. H. B.; Gomes, M. M. S.; Fernandes, N. S.; da-Silva, D. R.; Martínez-Huitle, C. A.; Fuel Process. Technol. 2012, 96, 80. 15. Tissot, G. B.; Anglada, A.; Dimitriou-Christidis, P.; Rossi, L.; Arey, J. S.; Comninellis, C.; Electrochem. Commun. 2012, 23, 48. 16. Kapałka, A.; Fóti, G.; Comninellis, C.; J. Appl. Electrochem. 2008, 38, 7. 17. Martínez-Huitle, C. A.; Andrade, L. S.; Quim. Nova 2011, 34, 850. 18. Comninellis, C.; Chen, G.; Electrochemistry for the Environment, Springer: New York, 2010. 19. Andrade, L. S.; Tasso, T. T.; Silva, D. L.; Rocha-Filho, R. C.; Bocchi, N.; Biaggio, S. R.; Electrochim. Acta 2009, 54, 2024. 20. Ciríaco, L.; Anjo, C.; Correia, J.; Pacheco, M. J.; Lopes, A.; Electrochim. Acta 2009, 54, 1464. 21. Andrade, L. S.; Rocha-Filho, R. C.; Bocchi, N.; Biaggio, S. R.; Iniesta, J.; García-Garcia, V.; Montiel, V.; J. Hazard. Mater. 2008, 153, 252. 22. Andrade, L. S.; Ruotolo, L. A. M.; Rocha-Filho, R. C.; Bocchi, N.; Biaggio, S. R.; Iniesta, J.; García-Garcia, V.; Montiel, V.; Chemosphere 2007, 66, 2035. 23. Aquino, J. M.; Irikura, K.; Rocha-Filho, R. C.; Bocchi, N.; Biaggio, S. R.; Quim. Nova 2010, 33, 2124. 24. Li, X.; Pletcher, D.; Walsh, F. C.; Chem. Soc. Rev. 2011, 40, 3879. 25. Aquino, J. M.; Rocha-Filho, R. C.; Bocchi, N.; Biaggio, S. R.; Journal of Environmental Chemical Engineering 2013, 1, 954. 26. Aquino, J. M.; Rocha-Filho, R. C.; Bocchi, N.; Biaggio, S. R.; J. Braz. Chem. Soc. 2010, 21, 324. 27. Aquino, J. M.; Rocha-Filho, R. C.; Bocchi, N.; Biaggio, S. R.; J. Appl. Electrochem. 2010, 40, 1751. 28. Aquino, J. M.; Rocha-Filho, R. C.; Rodrigo, M. A.; Sáez, C.; Cañizares, P.; Water Air Soil Pollut. 2013, 224, 1. 29. Eaton, A. D.; Clesceri, L. S.; Greenberg, A. E.; Standard Methods for the Examination of Water and Wastewater, 19th ed., United Book Press: Baltimore, 1995. 30. Medel, A.; Bustos, E.; Apátiga, L. M.; Meas, Y.; Electrocatalysis 2013, 4, 189. 31. Wang, T. X.; Margerum, D. W.; Inorg. Chem. 1994, 33, 1050. 32. Morris, C.; J. Phys. Chem. 1966, 70, 3798. 33. Sánchez-Carretero, A.; Sáez, C.; Cañizares, P.; Rodrigo, M. A.; Chem. Eng. J. 2011, 166, 710. 34. Cañizares, P.; Martínez, L.; Paz, R.; Sáez, C.; Lobato, J.; Rodrigo, M. A.; J. Chem. Technol. Biotechnol. 2006, 81, 1331. 35. Cañizares, P.; García-Gómez, J.; de-Marcos, I. F.; Rodrigo, M. A.; Lobato, J.; J. Chem. Educ. 2006, 83, 1204. 36. Deborde, M.; von-Gunten, U.; Water Res. 2008, 42, 13. 37. Baker, J. R.; Milke, M. W.; Mihelcic, J. R.; Water Res. 1999, 33, 327. 38. Panizza, M.; Michaud, P. A.; Cerisola, G.; Comninellis, C.; J. Electroanal. Chem. 2001, 507, 206. 39. Rodrigo, M. A.; Michaud, P. A.; Duo, I.; Panizza, M.; Cerisola, G.; Comninellis, C.; J. Electrochem. Soc. 2001, 148, D60. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access