
 |
 |
 |
 |
 |
Nova Técnica
| Stability-indicating RP-HPLC method for simultaneous determination of gatifloxacin and flurbiprofen in binary combination |
|
Islam Ullah KhanI; Syed Naeem Razzaq*,I; Irfana MariamIII; Muhammad AshfaqII; Syed Saleem RazzaqIV
IDepartment of Chemistry, Government College University, Lahore-54000, Pakistan Recebido em 09/05/2013 *e-mail: naeemraindrops@yahoo.com A stability-indicating RP-HPLC method is presented for determination of gatifloxacin and flurbiprofen in binary combination. Gatifloxacin, flurbiprofen and their degradation products were detected at 254 nm using a BDS Hypersil C8 (250 X 4.6 mm, 5 µm) column and mixture of 20 mM phosphate buffer (pH 3.0) and methanol 30:70 v/v as mobile phase. Response was linear over the range of 15-105 µg mL-1 for gatifloxacin (r2 > 0.998) and of 1.5-10.5 µg mL-1 for flurbiprofen (r2 > 0.999). The developed method efficiently separated the analytical peaks from degradation products (peak purity index > 0.9999). The method developed can be applied successfully for determination of gatifloxacin and flurbiprofen in human serum, urine, pharmaceutical formulations, and their stability studies. INTRODUCTION Flurbiprofen sodium is chemically designated as Sodium (±)-2-(2-fluorobiphenyl-4-yl) propionate. It is a non-steroidal anti-inflammatory drug used in musculoskeletal disorders, joint disorders such as ankylosing osteoarthritis, rheumatoid arthritis, post-operative pain and migraine, among other conditions. It is also used to control post-operative inflammation of the anterior segment of the eye. Gatifloxacin is chemically designated as 1-cyclopropyl-6-fluoro-8-methoxy-7-(3-methylpiperazin-1-yl)-4-oxo-quinoline-3-carboxylic acid (Figure 1). It is a broad spectrum antibiotic indicated for the treatment of bacterial conjunctivitis, keratitis, as well as for pre and post-operative infections of the eyes.1
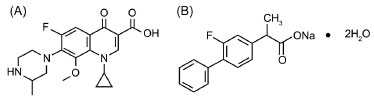 Figure 1. Chemical structure of Gatifloxacin (A) and Flurbiprofen Sodium (B)
Various techniques have been applied by different scientists to analyse flurbiprofen and gatifloxacin either alone or in combination with other drugs. Gatifloxacin has been determined by spectrophotometry,2,3 high performance liquid chromatography,4-6 atomic absorption spectroscopy, conductometry, colorimetry7 spectrofluorimetry,8 LC-MS,9 and bioassay,10 whereas flurbiprofen has been determined by capillary zone electrophoresis,11 spectrophotometry,12 and high performance liquid chromatography.13-15 The combination of flurbiprofen and gatifloxacin, although available in the market, has not yet been adopted by any official pharmacopoeia. Review of the literature found no stability-indicating HPLC method for simultaneous determination of these two drugs in pharmaceutical formulations, human serum and urine. Therefore, attempts were made to develop and validate a simple, fast, sensitive and isocratic stability-indicating HPLC method for simultaneous determination of both drugs. The developed method can be effectively applied for quality control analysis of both drugs in commercial preparations, stability studies, human serum and urine samples. We are currently engaged in binary combination analysis of different classes of drugs.16-32
EXPERIMENTAL Chemicals and reagents Reference standards of Gatifloxacin and Flurbiprofen with declared purity of 99.73 and 99.81%, respectively, were kindly gifted by Schazoo Zaka Laboratories (Lahore, Pakistan). Flubigat and Gatrex-F eye drops (composition 3 mg mL-1 of gatifloxacin and 0.3 mg mL-1 of flurbiprofen sodium) were used in this study. Acetonitrile (HPLC grade), HPLC methanol, triethylamine, sodium hydroxide, potassium dihydrogen phosphate, phosphoric acid, hydrochloric acid and hydrogen peroxide (analytical reagent grade) were from M.S Traders Lahore, Pakistan (Fluka origin). Double distilled water was used throughout method development and validation. Mobile phase was filtered using 0.45µm nylon filters by Millipore (USA). Equipment and chromatographic conditions The HPLC system consisted of a Shimadzu LC-20A (Kyoto, Japan) equipped with a model LC-20AT pump, SPD-M20A Diode array detector (set at 254 nm), DGU-20A5 online degasser and a Rheodyne injection valve with a 20 µL loop applied for method development. Peak areas were integrated using the Shimadzu LC solution (version 1.227) software program. The chromatographic experiments were optimized on a BDS Hypersil C8 column (250 X 4.6 mm, 5 µm) at room temperature. Mobile phase consisting of methanol and 20 mM phosphate buffer (pH 3.0) in the ratio of (70:30 v/v, respectively) was used. Phosphate buffer was prepared using 2.72 g of potassium dihydrogen phosphate in 1000 mL water. 1 mL triethylamine (TEA) was supplemented and pH adjusted to 3.0 with dilute phosphoric acid. Flow rate of the mobile phase was 1.5 mL min-1 and all chromatographic experiments were performed at room temperature (25 ºC ± 2 ºC). Preparation of stock solution (A) Stock solution (A) was prepared to decrease the number of recurring operations involved and hence, reduce the probability of human or experimental error. Furthermore, direct weighing of gatifloxacin (60 µg mL-1) and flurbiprofen (6 µg mL-1) to prepare the working standard solution cannot be carried out with sufficient accuracy. Standard stock solution of gatifloxacin (1500 µg mL-1) and flurbiprofen (150 µg mL-1) was prepared by accurately weighing 150 mg gatifloxacin and 15 mg flurbiprofen in a 100 mL volumetric flask and completing up to the mark with mobile phase. The contents were dissolved through sonication for 5 minutes. Working standard solutions of gatifloxacin and flurbiprofen were prepared from stock solution. Stock solution was also used to arrange working solutions for accuracy, precision, linearity, forced degradation studies and robustness. Preparation of working standard solution (B) 1 mL of stock solution (A) was diluted to 25 mL with mobile phase to prepare working standard solution (B) having a concentration equal to 60 µg mL-1 of gatifloxacin and 6 µg mL-1 of flurbiprofen. The solution was filtered through a 0.45 mm nylon filter before analysis. Preparation of sample solution 1 mL of commercial eye drops (composition 3 mg mL-1 gatifloxacin and 0.3 mg mL-1 flurbiprofen) was diluted to 50 mL with mobile phase to obtain a concentration equal to 60 µg mL-1 of gatifloxacin and 6 µg mL-1 of flurbiprofen. The solution was filtered through a 0.45 mm nylon filter before analysis. Preparation of human urine samples Human urine samples were injected directly after dilution in the mobile phase. 1250 µL of stock solution (A) was spiked with 1250 mL of human urine in a polypropylene tube. The solution was centrifuged at 4000 rpm for 10 minutes. A 200 µL volume of supernatant solution was then diluted with 2300 µL of mobile phase. This furnished a solution with a concentration equal to (60 µg mL-1 of gatifloxacin and 6.0 µg mL-1 of flurbiprofen). The resultant solution was filtered through a 0.45 µm nylon filter before analysis. Preparation of human serum samples Serum samples were injected after protein precipitation with mobile phase. 250 µL of human serum was spiked with 50 µL of stock solution (A) in a polypropylene tube. A 950 µL volume of mobile phase was added and the solution centrifuged at 4000 rpm for 10 minutes to precipitate plasma proteins. This furnished a solution concentration equal to (60 µg mL-1 of gatifloxacin and 6.0 µg mL-1 of flurbiprofen). The supernatant solution was separated in a polypropylene tube. The solution was filtered through a 0.45 µm nylon filter before analysis. Linearity Linear calibration plots of the developed method were obtained over concentration ranges of 15-105 µg mL-1 (15, 30, 45, 60, 75, 90 and 105 µg mL-1) for gatifloxacin and 1.5-10.5 mg mL-1 for flurbiprofen (1.5, 3.0, 4.5, 6.0, 7.5, 9.0 and 10.5 µg mL-1). Each solution was prepared in triplicate. Accuracy For accuracy determination, known quantities (50,100 and 150 %) of gatifloxacin and flurbiprofen with recognized purity were spiked with placebo components (benzalkonium chloride and sodium chloride in aqueous base), human serum and urine. A synthetic mixture (100 % nominal analytical concentration) of gatifloxacin (60 µg mL-1) and flurbiprofen (6.0 µg mL-1) was prepared by mixing gatifloxacin (60 µg), flurbiprofen (6.0 µg), benzalkonium chloride (0.2 g) and sodium chloride (2.5g) in 1000 mL purified water for 30 minutes using a magnetic stirrer. Three levels of synthetic mixtures were prepared corresponding to 50,100 and 150 % of nominal analytical concentration (60 µg mL-1 of gatifloxacin and 6.0 µg mL-1 of flurbiprofen) and analysed by the developed method. Precision Repeatability was evaluated by determination of intra-day and inter-day precisions. Intra-day precision was determined by injecting five standard solutions of three different concentrations of gatifloxacin and flurbiprofen on the same day whereas inter-day precision was determined by injecting the same solutions for three consecutive days. Relative standard deviation of gatifloxacin and flurbiprofen peaks was then calculated to represent precision.
SPECIFICITY (STRESS TESTING) The specificity of the developed method was evaluated by using different ICH prescribed (acidic, basic, oxidative, thermal and photolytic) stress conditions. Acid degradation studies the acid degradation study was carried out in a versatile environmental test chamber (Sanyo, Japan) at 40 ºC/75% RH using 5 M HCl. For this purpose, 1 mL of stock solution (A) was taken and placed in a 25 mL volumetric flask. 1 mL of 5 M HCl was added to the flask and kept in the versatile environmental test chamber at 40 ºC/75% RH for 24 h. After completion of the stress testing, the solution was neutralized by using 5 M NaOH and completed up to the mark with mobile phase. Base degradation studies the base degradation study was performed at 40 ºC/75% RH using 5 M NaOH. For this purpose, 1 mL of stock solution (A) was taken and placed in three 25 mL volumetric flasks. 1 mL of 5 M NaOH was added to the flask and kept at 40 ºC/75% RH for 1, 3 and 7 days. After completion of the stress testing, solutions were neutralized by using 5 M HCl and completed up to the mark with mobile phase. Oxidative degradation studies The oxidative degradation study was performed at 40 ºC/75% RH using 6 % H2O2. For this purpose, 1 mL of stock solution (A) was taken and placed in a 25 mL volumetric flask. 1 mL of 6 % H2O2 was added to the flask and kept in the versatile environmental test chamber at 40 ºC/75% RH for 24 h. After completion of stress testing, the 25 mL flask was completed up to the mark with mobile phase. Thermal degradation studies The thermal degradation study was performed at 40 ºC/75% RH. For this purpose, 1 mL of stock solution (A) was taken and placed in three different 25 mL volumetric flasks and kept in the versatile environmental test chamber at 40 ºC/75% RH for 1, 3 and 7 days. After the specified times, the 25 mL flasks were completed up to the mark with mobile phase. Photolytic degradation studies For photolytic degradation, 1 mL of the stock solution (A) was taken and placed in three different 25 mL volumetric flasks and placed in direct sunlight for 1, 2 and 3 h. After completion of the stress testing, the 25 mL flasks were completed up to the mark with mobile phase. Robustness Premeditate variations were performed under the experimental conditions of the proposed method to assess method robustness. For this purpose, minor changes were made to mobile phase composition, flow rate and pH of buffer solution and the effect of these changes on chromatographic parameters such as retention time, tailing factor and number of theoretical plates measured was assessed. The following protocol was used to assess the method's robustness.33 pH of aqueous buffer in the mobile phase The pH of the aqueous phosphate buffer used in the mobile phase was adjusted to ±0.2 units. Ratio of components in the mobile phase The following adjustment limits were applied to the minor component (aqueous buffer) of the mobile phase. The amount of buffer was adjusted within ± 30 % relative not exceeding ± 10 % absolute. For example, for a specified ratio of 70:30 methanol: buffer the mobile phase ratio was adjusted within the range of 79:21 or 61:39, respectively. Flow rate The flow rate of the mobile phase was adjusted to ± 50% of 1.5 mL min-1 (1.0 mL min-1 to 2.0 mL min-1). Acceptance criteria The above-mentioned factors (pH of buffer, mobile phase ratio and flow rate) should have no significant effect on the potency (assay) results of gatifloxacin and flurbiprofen. Tailing factor (T) The tailing factor should be NMT 1.5. Resolution (R) The resolution between adjacent peaks should be NLT 1.5. Theoretical plates (N) The theoretical plates for analytical peaks should be NLT 2000. Limit of detection and limit of quantitation The signal-to-noise (S/N) ratio approach was used to determine the LOD and LOQ values. LOD and LOQ values were determined by using solutions of different concentrations prepared by spiking known quantities of gatifloxacin and flurbiprofen into excipients (benzalkonium chloride and sodium chloride), human serum and urine. Each solution was made according to defined protocol and analysed repeatedly to determine the S/N ratio. The average S/N ratio at each concentration level was used to calculate the final limit of quantitation and limit of detection. The concentration level that yields an S/N ratio of about 10:1, at which gatifloxacin and flurbiprofen can be readily quantified with accuracy and precision, was reported as the limit of quantitation. The concentration level that yields an S/N ratio of about 3:1, at which gatifloxacin and flurbiprofen can be readily detected, was reported as the limit of detection.
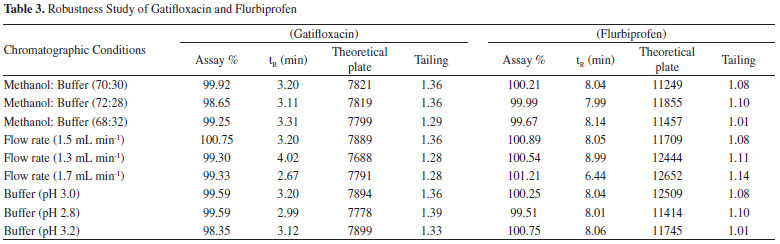
RESULTS AND DISCUSSION In this work we developed a simple, swift, and precise stability-indicating RP-HPLC method for simultaneous determination of gatifloxacin and flurbiprofen. Although many analytical methods with different techniques (spectrophotometery,2,3,12 HPLC,4-6,13-15 atomic absorption spectrometry, conductometry, colorimetry,7 spectrofluorimetry,8 LC-MS,9 bioassay,10 and capillary zone electrophoresis11) have been reported for the separate determination of gatifloxacin and flurbiprofen in pharmaceutical formulations or serum samples, no stability-indicating method is available for these combination drugs in pharmaceutical formulations, human serum or urine. The reported HPLC methods of gatifloxacin were developed and validated for routine quality control analysis of gatifloxacin in pharmaceutical formulations and were not applied for stability studies, degradation products, plasma or urine samples. Similarly, HPLC methods reported for analysis of flurbiprofen in pharmaceutical formulation are not stability-indicating and plasma methods involved a time consuming liquid-liquid extraction with ethyl acetate or addition of surfactant to extract the drug. For optimization of chromatographic conditions and to acquire symmetrical peaks with no peak contamination, different chromatographic conditions such as composition and pH of mobile phase, stationary phases with different packing materials (Hypersil BDS C8, Hypersil ODS C18, Hypersil BDS Phenyl-2, and Hypersil BDS Cyano) and configurations (10, 15, 25 cm columns) were applied to the gatifloxacin and flurbiprofen combination. Optimization of mobile phase and stationary phase A systematic approach was applied for optimization of the best possible chromatographic conditions. The method development process was started with different ratios (20:80, 30:70, 40:60 and 50:50) of methanol and water using four different stationary phases (Hypersil BDS C8, Hypersil ODS C18, Hypersil BDS Phenyl-2, and Hypersil BDS Cyano). With the methanol and water mobile phase, tailed (asymmetrical) peaks of gatifloxacin and flurbiprofen were obtained on Hypersil ODS C18, Hypersil BDS Phenyl-2, and Hypersil BDS Cyano columns. The Hypersil BDS C8 column provided a symmetrical (tailing factor 1.36 with retention time 5.395 min) peak of flurbiprofen and asymmetrical peak of gatifloxacin (tailing factor 1.89) with a long retention time 19.122 min. Increase in temperature of the column oven to 50 ºC did not improve peak tailing, broadening, and long retention of gatifloxacin. The asymmetrical peak and long retention of gatifloxacin was due to the chelation of the carboxyl group of C-3 and oxygen atom of C-4 of gatifloxacin with the metal ion impurities present in the column or caused by superfluous interactions between nitrogen atoms of gatifloxacin and silanol residues of stationary phases. In order to overcome these unnecessary interactions, the ionic strength of the mobile phase was increased by using potassium phosphate buffer in the mobile phase composition and further chromatographic experiments were performed using methanol: 0.02 M phosphate buffer (70:30) at pH 3.0 as the mobile phase. The problems of peak broadening and long retention of gatifloxacin were resolved but the peak tailing problem (1.58) persisted and was not resolved even after changing the organic modifier from methanol to acetonitrile. In order to overcome the peak tailing problem of gatifloxacin it was decided to add silanol blocker (TEA) to the mobile phase and further chromatographic experiments were performed using methanol: 0.02 M phosphate buffer (70:30) at pH 3.0 containing 0.1 % triethylamine (TEA) as the silanol blocker. Highly symmetrical and sharp peaks of gatifloxacin and flurbiprofen were obtained with methanol: 0.02 M phosphate buffer containing 0.1% v/v triethylamine (70:30, v/v) on the Hypersil BDS C8 column (with better resolution, capacity factor, peak shapes and theoretical plates) as compared to the other stationary phases (Hypersil ODS C18, Hypersil BDS Phenyl-2, and Hypersil BDS Cyano). The variations in composition of mobile phase and dissimilar stationary phases had substantial influences on peak shape, tailing factor, retention factor, theoretical plates and resolution. Optimization of pH of phosphate buffer In order to optimize the appropriate pH of phosphate buffer, chromatographic experiments were performed at four different pH (3.0, 4.0, 5.0 and 6.5) of buffer solution. Mobile phase was prepared at the ratio of methanol: phosphate buffer (70: 30, v/v) containing 0.1 % TEA and the Hypersil BDS C8 column was used as the stationary phase. During this study, retention time of gatifloxacin was unaffected by buffer pH. This is due to the overall neutral charge on gatifloxacin (containing both acidic - COOH and basic - NH group) when pH increased from 3.0 to 6.5. However, the retention time of flurbiprofen showed a considerable decrease with increasing pH. Due to the acidic nature of flurbiprofen it tends to become less ionized (protonated) as pH decreases, therefore it becomes less polar (more hydrophobic), thus exhibiting an increased preference for bonding to the stationary phase. In addition, as the pH of the buffer increased from pH 3 to pH 6.5, the structure of flurbiprofen changed from a less ionized to an ionized form (polar or more hydrophilic) and the resolution between flurbiprofen and gatifloxacin was compromised with considerable overlapping of peaks observed at pH 6.5, rendering quantitation difficult. In contrast, when a buffer of pH 3 was used, peaks were well resolved, sharp and symmetrical (Figure 2).
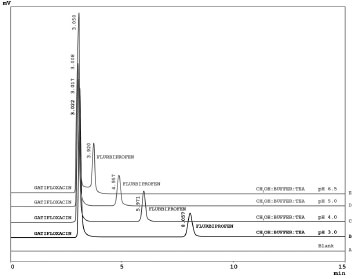 Figure 2. Chromatograms of Gatifloxacin and Flurbiprofen at Different pH
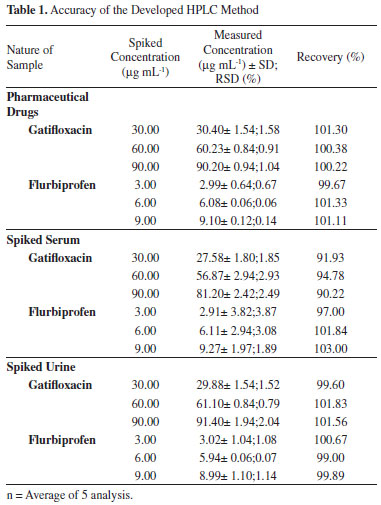
Finally, methanol: phosphate buffer 0.02M, pH 3.0 (70:30, v/v) containing 0.1 % TEA was selected, providing symmetrical peaks with acceptable peak purity index of gatifloxacin and flurbiprofen using the Hypersil BDS C8 column. Under the chromatographic conditions cited, highly symmetrical and sharp peaks of gatifloxacin and flurbiprofen were obtained at retention times of 3.202 and 8.057 min, respectively. Analytical method validation The developed method was validated using ICH guidelines.34 Validation parameters included linearity, accuracy, precision, robustness, specificity, and limits of detection and quantitation. Linear calibration plots of the developed method were obtained in concentration ranges of 15 to 105 µg mL-1 for gatifloxacin (15, 30, 45, 60, 75, 90 and 105 µg mL-1) and 1.5 to 10.5 µg mL-1 for flurbiprofen (1.5, 3.0, 4.5, 6.0, 7.5, 9.0 and 10.5 µg mL-1). The linear regression equation for gatifloxacin was found to be Y= 68441X + 47999, whereas for flurbiprofen this was Y= 20685X + 15285, with values of correlation coefficients equal to 0.998 and 0.999, respectively. The limits of detection (LOD) and of quantification (LOQ) were determined by making serial dilutions. LOD was found to be 0.09 µg mL-1 and 0.03 µg mL-1 for gatifloxacin and flurbiprofen, respectively (signal to noise ratio of 3:1). LOQ was found to be 0.31 µg mL-1 and 0.09 µg mL-1 for gatifloxacin and flurbiprofen, respectively (signal to noise ratio of 10:1). Accuracy of the developed method was determined by the synthetic mixture technique. Three levels of solutions (50, 100 and 150%) of nominal analytical concentration were prepared and analysed by the developed method. Percentage recoveries along with standard deviation and relative standard deviation for each analyte are given in (Table 1). Recovery studies showed the method to be highly accurate and suitable for the intended use.
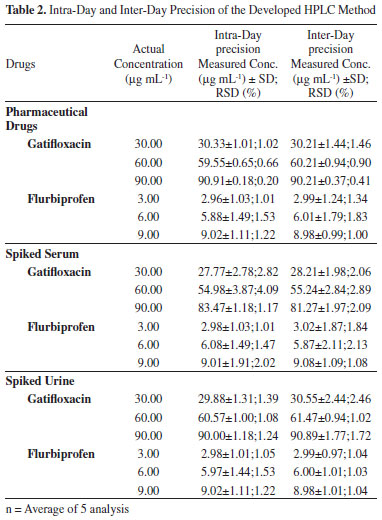
Intra-day precision was determined by injecting five standard solutions of three different concentrations on the same day whereas inter-day precision was determined by injecting the same solutions for three consecutive days. Relative standard deviation of gatifloxacin and flurbiprofen peaks was calculated to represent precision. The results of intra-day and inter-day precision are given in (Table 2).
Robustness of the developed method was performed by slightly varying the chromatographic conditions. The results showed that slight variations in chromatographic conditions had a negligible effect on the chromatographic parameters (Table 3).
Specificity of the developed method was evaluated by applying different stress conditions (acid, base, oxidation, thermal and photolytic) to gatifloxacin and flurbiprofen in combination form. The representative chromatograms under acidic and oxidative stress conditions are showed in (Figure 3). The results of stress studies are given in (Table 4).
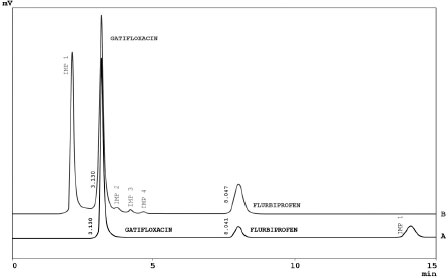 Figure 3. Chromatograms of Gatifloxacin and Flurbiprofen under Acidic (A) and Oxidative stress (B)
Comparison of the two drugs showed that under acidic stress gatifloxacin is more stable as compared to flurbiprofen while under oxidative stress flurbiprofen is more stable than gatifloxacin. Under acidic conditions flurbiprofen degraded by up to 60 % and gatifloxacin was found to be stable. Under oxidative stress gatifloxacin degraded by up to 8.45 % whereas flurbiprofen was found to be stable. Under basic, thermal and photolytic stresses both drugs were found to be highly stable, not degrading even after 7 days' treatment at 40 ºC/75 % RH. From these stress studies it is thus concluded that flurbiprofen and gatifloxacin drugs are not stable under acidic and oxidative stress conditions, whereas both drugs are highly stable under basic, thermal and photolytic stress conditions. Application of the developed method was checked by analyzing the gatifloxacin and flurbiprofen in commercially available pharmaceutical products, human plasma and urine samples (Figure 4).
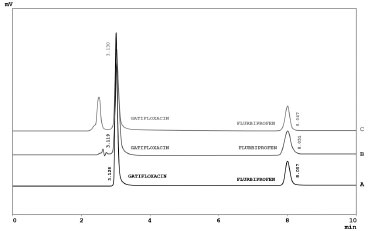 Figure 4. Chromatograms of Gatifloxacin and Flurbiprofen in Pharmaceutical Formulations (A), Human Serum (B) and Urine (C)
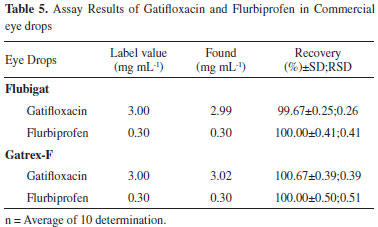
The results showed high percentage recoveries and low relative standard deviation values for both analytes (Table 5).
CONCLUSION A simple, fast and reliable stability-indicating RP-HPLC method is described for simultaneous determination of gatifloxacin and flurbiprofen in pharmaceutical formulations, human serum and urine. Method validation was performed by testing its linearity, accuracy, precision, limits of detection and of quantitation as well as specificity. The method is simple and fast, without ion pair or derivatization reagent. Sample preparation is very simple without the use of laborious liquid-liquid extraction or expensive solid phase extraction techniques. The developed method separated the peaks of active pharmaceutical ingredients (APIs) from degradation products. The method can be successfully used for any kind of stability and validation studies.
REFERENCES 1. Reynolds, J. E. F.; Martindale, The Extra Pharmacopoeia, 36th ed., Pharmaceutical Press: London, 2009, p.74, 281. 2. Venugopal, K.; Saha, R.N.; Il Farmaco 2005, 60, 906. 3. Paramane, S.; Kothapalli, L.; Thomas, A.; Deshpande, A.D.; Indian J. Pharm. Sci. 2006, 68, 819. 4. Santoro, M.I.R.M.; Kassab, N.M.; Singh, A.K.; Kedor-Hackmam, E.R.M.; J. Pharm. Biomed. Anal. 2006, 40, 179. 5. Lopes, C.C.G.O.; Salgado, H.R.N.; Quim Nova 2008, 31, 1831. 6. Salgado, H.R.N.; Lopes, C.C.G.O.; J. AOAC Int. 2006, 89, 642. 7. Al-Ghannam, S.M.; Spectrochim. Acta, Part A 2008, 69, 1188. 8. Ocana, J.A.; Barragan, F.J.; Callejon, M.; J. Pharm. Biomed. Anal. 2005, 37, 327. 9. Bao, Y.X.X.; Acta Pharm. Sinica 2002, 37, 462. 10. Salgado, H.R.N.; Lopes, C.C.G.O.; Lucchesi, M.B.B.; J. Pharm. Biomed. Anal. 2006, 40, 443. 11. Hamoudova, R.; Pospisilova, M.; J. Pharm. Biomed. Anal.. 2006, 41, 1463. 12. Sajeev, C.; Jadhav, P.R.; RaviShankar, D.; Saha, R.N.; Anal. Chim. Acta. 2002, 463, 207. 13. Sun, Y.; Takaba, K.; Kido, H.; Nakashima, M.N.; Nakashima, K.; J. Pharm. Biomed. Anal. 2003, 30, 1611. 14. Han, F.; Yin, R.; Shi, X.; Jia, Q.; Liu, H.; Yao, H.; Xu, L.; Li, S.; J. Chromatogr. B 2008, 64, 868. 15. Nawaz, M.; Quim Nova 2012, 35, 939. 16. Razzaq, S.N.; Mariam, I.; Khan, I.U.; Ashfaq, M.; J. Liq. Chromatogr. Related Technol. 2012, 35, 651. 17. Ashfaq, M.; Khan, I.U.; Qutab, S.S.; Razzaq, S.N.; J. Chil. Chem. Soc. 2007, 52, 1220. 18. Qutab, S.S.; Razzaq, S.N.; Khan, I.U.; Ashfaq, M.; Shuja, Z.A.; J. Food Drug Anal. 2007, 15, 139. 19. Ashfaq, M.; Khan, I.U.; Asghar, M.N.; J. Chil. Chem. Soc. 2008, 53, 1617. 20. Khan, I.U.; Sharif, S.; Ashfaq, M.; Asghar, M.N.; J. AOAC Int. 2008, 91, 744. 21. Qutab, S.S.; Razzaq, S.N.; Ashfaq, M.; Khan, I.U.; Mumtaz, A.M.; J. Chil. Chem. Soc. 2008, 54, 234. 22. Khan, I.U.; Jillani, S.M.; Ashfaq, M.; Lat. Am. J. Pharm. 2010, 29, 1383. 23. Khan, I.U.; Kausar, T.; Ashfaq, M.; Sharif, S.; J. Chil. Chem. Soc. 2010, 55, 461. 24. Sharif, S.; Khan, I.U.; Ashfaq, M.; Iqbal, M.S.; Ahmad, S.; J. Anal Chem. 2010, 65, 1029. 25. Qutab, S.S.; Razzaq, S.N.; Ashfaq, M.; Shuja, Z.A.; Khan, I.U.; Acta Chromatogr. 2007, 19, 119. 26. Razzaq, S.N.; Ashfaq, M.; Khan, I.U.; Mariam, I.; Quim. Nova 2012, 35, 1216. 27. Razzaq, S.N.; Ashfaq, M.; Khan, I.U.; Mariam, I.; Anal. Methods 2012, 4, 2121. 28. Khan, I.U.; Ashfaq, M.; Razzaq, S.N.; Mariam, I.; J. Liq. Chromatogr. Related Technol. 2013, 36, 1437. 29. Razzaq, S.N.; Ashfaq, M.; Khan, I.U.; Mariam, I.; J. Chil. Chem. Soc. 2012, 4, 57. 30. Razzaq, S.N.; Khan, I.U.; Mariam, I.; Razzaq, S.S.; Chem. Cent. J. 2012, 6, 94. 31. Razzaq, S.N.; Ashfaq, M.; Mariam, I.; Khan, I.U.; Razzaq, S.S.; Braz. J. Pharm. Sci. 2013, 49, 301. 32. Razzaq, S.N.; Mariam, I.; Khan, I.U.; Ashfaq, M.; J. Liq. Chromatogr. Related Technol. 2012, 35, 651. 33. United States Pharmacopoeia, 32th ed., 2009, p. 236. 34. ICH (Q2B); Note for guidance on validation of analytical procedures: methodology, International conference on Harmonization, IFPMA: Geneva 1996. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access