
 |
 |
 |
 |
 |
Artigo
|
|
| Efeito do solvente na extração de ácidos clorogênicos, cafeína e trigonelina em Coffea arabica Solvent effects on extraction of chlorogenic acids, caffeine and trigonelline in Coffea arabica |
|
Ivanira Moreira; Guilherme Luiz Scheel; Pedro Henrique Hatumura; Ieda Spacino Scarminio*
Departamento de Química, Universidade Estadual de Londrina, CP 6001, 86051-990 Londrina - PR, Brasil Recebido em 07/03/2013 *e-mail: ieda@uel.br In this research work the effects of four solvents and their mixtures on the extraction of chlorogenic acids, caffeine and trigonelline in crude extracts of four coffee cultivars, traditional red bourbon, IAPAR59, IPR101 and IPR108 cultivars, were investigated by UV spectrophotometry and UV spectra obtained from RP-HPLC-DAD. The experimental results and the principal component analysis of UV spectra showed that the effect of solvent extraction of the metabolites does not depend on cultivars, because the spectral characteristics are similar, but the concentrations are different. The UV and UV-DAD spectra for four simplex centroid design mixtures were also similar but the concentrations of caffeine, trigonelline and the chlorogenic acids are different and depend on the solvent used in the extraction. INTRODUÇÃO Os grãos do café contêm proteinas, lipidios, ácidos nucleicos, carboidratos, clorofila, carotenóides, enzimas e hormônios. Quando torrados, estes constituintes são destruidos ou podem ser transformados em uma grande variedade de novos compostos.1 Os grãos do café verde também contém compostos biologicamente ativos como a cafeína, ácidos clorogenicos (ACG), trigonelina, entre outros. Estes metabólitos são alguns dos precursores do aroma do café.2 A trigonelina é rapidamente decomposta na torra do café em muitos componentes voláteis, mas mesmo assim é encontrada no café torrado em concentrações mais baixas.1 Os ACG são os principais compostos fenólicos encontrados no café, sendo ésteres de ácidos trans-cinâmico tais como, ácidos cafeico, ferúlico e p-cumárico com ácido quínico.3 São metabólitos secundários de grande interesse econômico porque também contribuem na formação de pigmentos e sabor dos grãos do café, determinando assim a qualidade e aceitação da bebida. A cafeína, por ser o principal metabólito fisiologicamente ativo e o alcalóide quantitativamente mais importante do café, foi e ainda é o mais estudado desde a sua descoberta e identificação.1 As duas variedades comerciais mais importantes de café são a Coffea arabica e a Coffea canephora syn. Coffea robusta, sendo C. arabica de qualidade superior. Por esta razão, vários trabalhos relatam métodos para discriminar as duas espécies.4-9 Embora existam milhares de artigos na literatura sobre cafés defeituosos e não-defeituosos,10,11 propriedades antioxidantes dos metabólitos,12 os efeitos do café na saúde,13-15 os efeitos da torrefação dos grãos de café,12 a qualidade da bebida,16-20 entre outros, poucos são os relatos na literatura sobre o efeito do solvente na extração de ácidos clorogênicos, cafeína e trigonelina, principalmente na variedade Coffea arabica. Nos programas de melhoramento genético do café o objetivo é desenvolver diferentes cultivares para promover ganhos na produtividade e também agregar características agronômicas de resistência às pragas e doenças. Por outro lado, a composição química e atributos sensoriais destes novos cultivares poderão ser afetados. Muitos métodos têm sido desenvolvidos para a determinação de cafeina em café e outras bebidas, sendo os mais frequentemente usados a cromatografia líquida de alta eficiência21 (CLAE) e as espectroscopias no infravermelho com transformada de Fourier22 e UV/Vis.23 Para os ACG os métodos mais usados são CLAE24 e eletroforese capilar.25 CLAE com espectroscopia UV/Vis é a técnica analítica hifenada mais popular baseada na cromatografia. O espectro UV-Vis de cada pico cromatográfico pode ser uma ferramenta útil para a identificação dos componentes da amostra. Atualmente, uma das abordagens mais poderosas para o controle de qualidade de alimentos são as técnicas de impressão digital. Ela descreve uma variedade de métodos de análise que podem fornecer a identificação e quantificação aproximada de um grupo de metabólitos. As variações dos metabólitos são observadas principalmente por variações nos padrões espectroscópicos ou cromatográficos, sem o conhecimento prévio das identidades dos compostos investigados. Geralmente, amostras com impressões digitais espectroscopica ou cromatográfica semelhantes têm composições similares. Para a identificação de marcadores de qualidade para o café é necessário examinar os componentes que determinam as características aromáticas na matéria prima, juntamente com os componentes gerados durante o processo de torra. Esta não é uma tarefa fácil, porque na torrefação não ocorre somente a transformação dos compostos originais individuais, mas eles também se combinam de forma sinérgica e antagônica contribuindo para a determinação do aroma final da bebida. Mesmo assim, a análise química de cafeína e ACG nos grãos é usada como ferramenta adicional para avaliar a qualidade do café. Nos últimos anos, o nosso grupo tem mostrado que os planejamentos estatísticos de misturas permitem desenvolver procedimento de análise rigoroso mas econômico, para mostrar os efeitos das variações dos solventes na extração de metabólitos em material vegetal.26-31 Considerando a importância dos ácidos clorogênicos e cafeína como padrão de qualidade do café, nesta pesquisa foi utilizado um planejamento estatístico de mistura de quatro componentes etanol, acetato de etila, diclorometano e hexano para encontrar o solvente extrator mais adequado para a extração destes metabólitos. Foram comparados grãos de cafés verdes da cultivar Bourbon vermelho e outras três cultivares desenvolvidos no Instituto Agronômico do Paraná, são elas IPR 101, IPR 108 e IAPAR 59. Todas as cultivares foram produzidas sob as mesmas condições. O objetivo principal desta pesquisa foi usar planejamento estatístico de misturas e análise de componentes principais associados aos espectros UV para avaliar o efeito do solvente na extração de ACG, cafeína e trigonelina em quatro cutivares de café, Bourbon, IAPAR59, IPR101 e IPR108.
PARTE EXPERIENTAL Amostras de café Foram analisados os grãos verdes de quatro diferentes cultivares de Coffea arabica, a cultivar Bourbon vermelho, e três cultivares desenvolvidas pelo Instituto Agronômico do Paraná (IAPAR), IAPAR 59, IPR101, IPR108. Na cultivar Bourbon não há cruzamento com outras cultivares e foi usada como referência neste trabalho. A cultivar IAPAR 59 originou-se do cruzamento entre Coffea arabica, Villa Sarchi 971/10 e o Hibrido de Timor 832/2 (Sarchimor); a cultivar IPR101 originou-se do cruzamento cultivar Catuaí x Sarchimor (com genes SH2, SH3 resistentes a ferrugem); a cultivar IPR 108 originou-se do cruzamento cultivar IAPAR 59 x (Catuaí x Icatu). Todas as amostras foram cedidas pelo Instituto Agronômico do Paraná, IAPAR. Reagentes Todos os regentes utilizados na extração foram de grau analítico: álcool etílico comercial (dR), acetato de etila (Vetec), hexano (Vetec), diclorometano (F Maia), clorofórmio (Vetec), KBr (Synth), Para as análises cromatográficas foram usados acetonitrila e metanol grau cromatográfico (Vetec) e água ultrapura Milli-Q. Equipamentos Os equipamentos e aparelhos utilizados foram: balança Analítica Shimadzu AY220, evaporador rotativo Fisaton 801, aparelho de ultra-som (Unique, modelo Ultracleaner), moinho de disco Perten-3600, peneira Granutest ABNT 25 com abertura de 0,71 mm, purificador de água Millipore Plus, cromatógrafo a líquido de alta eficiência SPD-M10AV Finnigan Surveyor com detector de arranjo de fotodiodo (PDA) da Termo-Electro Corporation. As medidas de absorção foram realizadas em uma cubeta de quartzo de 1 cm, por meio de um espectrofotômetro UV-VIS Genesys2, com varredura no intervalo de 200-900 nm e resolução de 1 nm. Não houve necessidade de diluição. Os solventes puros, bem como suas misturas, apresentados na Tabela 1, foram usados como branco.
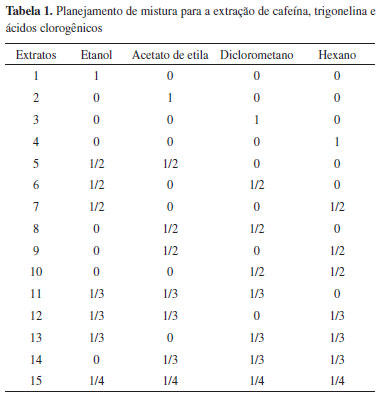
Preparo dos extratos Os grãos verdes dos cafés foram previamente imersos em nitrogênio líquido para facilitar a sua moagem e em seguida foram peneirados. As misturas de extração foram escolhidas de acordo com um planejamento centróide simplex para quatro componentes,32 totalizando 15 diferentes misturas, Tabela 1. Cada extrato foi preparado pesando-se 10 g com adição de 150 ml de uma das misturas listadas na Tabela 1. Estas misturas foram colocadas num banho de ultra-som durante 30 min. A água do banho foi trocada a cada 15 min para evitar o aquecimento. Os extratos foram filtrados em papel de filtro para separar a solução das amostras de café. Este procedimento foi repetido mais duas vezes. Após a extração foi retirada uma alíquota de 15 mL do extrato bruto que foram armazenadas sob refrigeração para as análises UV-VIS e CLAE. Os extratos foram concentrados em evaporador rotativo a uma temperartura de 60 ºC (±3 ºC) e mantidos sob ventilação forçada até atingir peso constante, para obtenção dos rendimentos em grama. Preparo das amostras para CLAE Pipetou-se uma alíquota de 20 µL de cada uma das soluções de extração de 15 mL e nesta adicionou-se 800 µL da fase móvel. As amostras foram filtradas em filtro Milipore Millex de 0,22 µm e analisadas em seguida. As condições cromatográficas foram: coluna cromatográfica Phenomenex C18, Kinetex 2,6 µm Hilic 100 Å, com dimensões de 150 mm x 4,6 mm, volume de injeção de 20 µL, vazão da fase móvel a 1,0 mL min-1 em modo isocrático. A eluição foi monitora nos comprimentos de onda de 210, 240 e 254 nm. Os dados foram processados usando o Software ChromQuest 4.2.
RESULTADOS E DISCUSSÃO Na primeira parte deste estudo foi avaliado o efeito das diferentes proporções dos solventes no rendimento do extrato bruto das quatro cultivares de café arábica, Figura 1. Ao se comparar os rendimentos do extrato bruto em g/10 g da amostra, nota-se que aqueles preparados em etanol puro apresentaram rendimentos maiores que aqueles com diclorometano, enquanto que em acetato de etila e hexano os rendimentos foram similares e menores que em etanol e diclorometano puros.
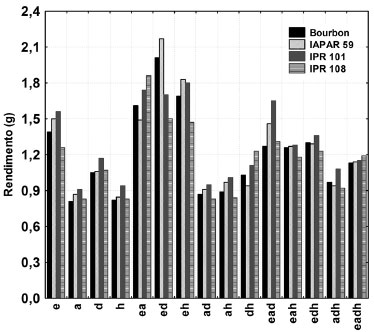 Figura 1. Rendimento dos extratos brutos em g/10g da amostra nas diferentes proporçoes dos solventes etanol, acetato de etila, diclorometano e hexano, para o café Bourbon, IAPAR 59, IPR 101e IPR 108
Observou-se que os maiores rendimentos foram obtidos em extratos preparados nas misturas binárias etanol:acetato de etila, etanol:diclorometano e etanol:hexano do que nos solventes puros. Para a mistura etanol:acetato de etila o maior rendimento foi obtido para o IPR 108, enquanto que para a mistura etanol:diclorometano o maior rendimento foi para o IAPAR 59 e o menor para o IPR 108. Na mistura binária etanol:hexano os rendimentos foram similares para o IAPAR 59 e IPR 101. Os efeitos dos solventes foram calculados no nosso trabalho anterior e mostraram-se significativos.33 Embora a espectroscopia de absorção UV não seja uma ferramenta para elucidação estrutural, o espectro traz informação relacionada à estrutura dos produtos naturais. Estas estruturas contém muitas ligações duplas e absorvem na luz ultravioleta (200-400 nm) ou na luz visível (400-800 nm). Os espectros de absorção nas diferentes proporções dos solventes para as amostras do café Bourbon foram submetidas à análise de componentes principais (ACP).34 Para ACP os dados espectrais foram colocados na forma de uma matriz 15 linhas e 201 colunas (240 a 400 nm), onde cada linha corresponde a um espectro de um extrato do planejamento e as colunas são as absorbâncias nos diferentes comprimentos de onda. Esta faixa foi escolhida devido ao ruído em alguns espectros abaixo de 240 nm e não conter informação acima de 400 nm. Os dados foram autoescalonados antes da análise das componentes principais. As três primeiras componentes principais explicam 93,5% da variância total dos dados. A Figura 2 mostra o gráfico dos escores da CP1×CP3 (~83% de variância). Observam-se dois grandes grupos ao longo da CP1. O grupo 1 é formado com os espectros dos extratos que contem etanol puro ou misturas com etanol, enquanto que o grupo 2 contem os espectros dos extratos preparados em acetato de etila e diclorometano puros, suas misturas bem como misturas com hexano. O espectro preparado em hexano é discriminado na CP1 e CP3. Como os pesos característicos na região de absorção de ACG foram positivos na CP1, então quanto mais para a direita (mais positivo) na CP1 maior a concentração de ACG. Este resultado fica mais fácil de interpretar quando se associa estas informações com os espectros originais, Figura 3.
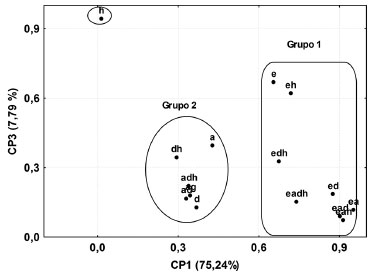 Figura 2. Gráfico dos escores da CP1×CP3 dos espectros nas diferentes proporçoes dos solventes para o café Bourbon
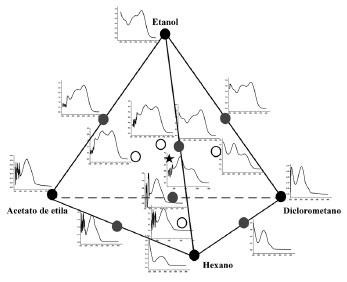 Figura 3. Espectros UV nas diferentes proporçoes dos solventes etanol, acetato de etila, diclorometano e hexano, para o café Bourbon
Os espectros preparados em etanol puro ou misturas contendo mais que 50% de etanol (parte superior do tetraedro) mostram forte absorção na região de 225-375 nm. Os espectros com forte absorção entre 220-250 nm indicam a presença de duas ligações insaturadas conjugadas, enquanto que a absorção entre 250-290 nm indica a existência de benzeno ou heterociclico aromático. Absorção acima de 300 nm indica a existência de um sistema conjugado longo.35 Para amostras de café, estas regiões estão associadas às transições eletrônicas das moléculas de trigonelina, cafeína e ácidos clorogênicos. Os espectros das soluções de diclorometano ou misturas contendo mais do que 50% de diclorometano (lado direito do tetraedro) apresentam altos valores de absorção entre 225-325 nm, mas com máximo em torno de 272 nm indicando maior contribuição de cafeína e/ou trigonelina. Os espectros das soluções de acetato de etila puro ou misturas binárias com mais de 50% de acetato de etila (lado esquerdo do tetraedro) apresentam máximos em torno de 280 nm sugerindo presença de ácidos benzóicos de baixo peso molecular. Os comprimentos de onda para análise dos principais metabólitos do café são 326 (max) e 300 nm atribuídos aos ácidos clorogênicos,36 272 nm à cafeína,37 265 (max) e 272 nm à trigonelina.38 Comparando-se a razão da absorbância em λ = 326 nm para a mistura binária etanol:acetato de etila em relação ao etanol puro, ela é 2,5 vezes maior para a mistura binária, enquanto que nas misturas etanol:diclorometano e etanol:acetato de etila:hexano em relação ao etanol puro esta razão é 2,08 vezes maior. Considerando que maior o sinal analítico, maior a concentração, este resultado segue a mesma proporção. É interessante observar que misturas contendo diclorometano puro ou misturas com mais de 50% de diclorometano apresentam maiores valores de absorção em 272 nm associado à cafeína e/ou à trigonelina. A razão entre a absorbância em λ = 272 nm da mistura ternária etanol:diclorometano:hexano é 2,26 vezes maior que para o diclorometano puro. Os espectros das misturas contendo acetato de etila apresentam um máximo em λ = 280 nm, associados aos ácidos de baixo peso molecular. A Figura 4 mostra o gráfico das componentes principais CP1×CP2 incluindo todos os espectros dos extratos das quatro cultivares. Observa-se o mesmo padrão nos grupos da Figura 2, dado que o grupo 1 contém os extratos preparados em etanol puro ou misturas contendo etanol, no grupo 3 estão os extratos preparados em hexano e no grupo 2 os demais extratos. O gráfico dos escores das componentes 2 e 3, embora com menor variância explicada, tem uma distribuição de pontos exatamente igual àquele da Figura 2, mais positivo na CP2, maior a concentração do ACG.
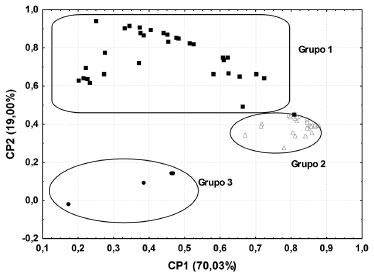 Figura 4. Gráfico dos escores da CP1×CP2 dos espectros nas diferentes proporçoes dos solventes etanol, acetato de etila, diclorometano e hexano, para o café Bourbon, IAPAR 59, IPR 101e IPR 108
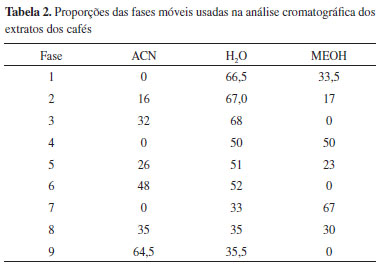
Para obter mais informação sobre os metabólitos predominantes nos diferentes extratos, foi usada a cromatografia líquida em fase reversa. Nove diferentes composições de fase móvel foram testadas na separação cromatográfica, Tabela 2. Estas proporções foram escolhidas usando um planejamento de misturas de três componentes para se obter um aumento gradativo na força do solvente e variação na polaridade. Três comprimentos de onda foram usados para investigar o número de picos em cada extrato, 210, 240 e 254 nm. Em geral a fase móvel preparada com 51:23:26 (v/v/v) em água:metanol:acetonitrila apresentou maior número de picos em 210 nm, sendo assim, esta fase móvel foi selecionada neste estudo.
A Figura 5 mostra os espectros DAD correspondentes aos picos cromatográficos predominantes nos cromatogramas para o café Bourbon. Comparando-se os espectros de absorção UV apresentados em cada ponto do planejamento da Figura 2 com os espectros DAD dos picos predominantes dos cromatogramas apresentados na Figura 5, observamos um efeito da fase móvel nos extratos que contem acetato de etila, que não acontece com os outros solventes. A adição da fase móvel provocou uma diminuição da absovância e mudança no comprimento de onda de absorção, inicialmente na região de 250-325 nm para 200-240 nm. Este efeito geralmente é produzido por mudanças estruturais,39 efeito do solvente e efeito do pH.40 É bem conhecido que a intensidade e a posição do comprimento de onda λ é afetada pela diferença entre os níveis de energias eletrônicas. Pelo princípio de Franck-Condon as transições eletrônicas envolvem o movimento dos elétrons, incluindo aqueles do solvente, mas não o movimento dos átomos. O aumento da polaridade do solvente provoca um desvio do espectro de absorção para o vermelho (deslocamento batocrômico) ou para o azul (deslocamento hipsocrômico). Em geral, mas nem sempre solventes polares produzem transições π→π* com deslocamentos para o vermelho, em comprimentos de onda maiores ou seja um deslocamento batocrômico.39 Por outro lado, no caso da transição n→π* de uma cetona, a interação do par isolado de elétrons da carbonila no estado fundamental com um solvente polar diminui a energia do orbital n do par isolado, e assim aumenta a energia requerida para promover um elétron para o nível de energia π*.40 Consequentemente ocorre um deslocamento para comprimento de onda menor, ou seja, um deslocamento hipsocrômico.
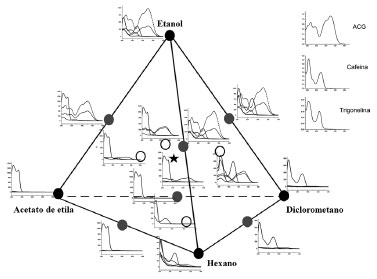 Figura 5. Espectros UV-DAD nas diferentes proporçoes dos solventes etanol, acetato de etila, diclorometano e hexano, para o café Bourbon
A Figura 6 mostra os espectros DAD dos picos predominantes dos cromatogramas dos extratos do café IAPAR 59. Observa-se que a Figura 6 apresenta um perfil semelhante ao da Figura 5, inclusive o mesmo deslocamento hipsocrômico para os espectros DAD dos extratos que contem acetato de etila. Este deslocamento foi observado também para as cultivares IPR 101 e IPR 108. Os espectros DAD para os quatro planejamentos são similares ocorrendo apenas variações nas concentrações de cafeína, ACG e trigonelina. Esta diferença depende do solvente usado na extração e era esperada, porque no melhoramento genético pode mudar a composição química e atributos sensoriais do café. Com exceção do IPR 108 o melhor solvente extrator para os ácidos clorogênicos foi o etanol, por outro lado também extrai uma quantidade razoável de cafeína e trigonelina. Para o IPR 108 o melhor solvente extrator para ACG é a mistura binária etanol:diclorometano (1:1 v/v). Mistura de etanol:acetato de etila:diclorometano (1:1:1 v/v/v) também é uma opção para extração de ácidos clorogênicos. Para a cafeína os melhores extratores são o diclorometano puro ou a mistura diclorometano:hexano (1:1 v/v) e para a trigonelina o etanol puro e as misturas etanol:diclorometano (1:1 v/v) e etanol:hexano (1:1 v/v).
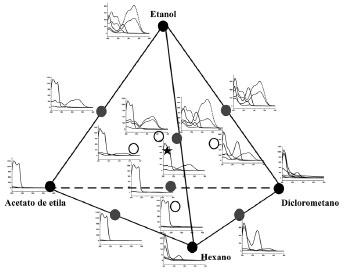 Figura 6. Espectros UV-DAD nas diferentes proporçoes dos solventes etanol, acetato de etila, diclorometano e hexano, para o café IAPAR 59
CONCLUSÃO As diferenças nas concentrações dos ácidos clorogênicos, cafeína e trigonelina nos extratos das quatro cultivares de café são fortemente dependentes dos solventes usados na extração. Em geral o melhor solvente extrator para os ácidos clorogênicos foi o etanol ou a mistura ternária etanol:acetato de etila:diclorometano (1:1:1v/v/v). Para a cafeína os melhores extratores são o diclorometano puro ou a mistura diclorometano:hexano (1:1 v/v) e para a trigonelina o etanol puro e as misturas etanol:diclorometano (1:1 v/v) e etanol:hexano (1:1 v/v). O acetato de etila mostrou-se eficiente na extração de ácidos fenólicos de baixo peso molecular mas não na extração dos metabólitos de interesse. Além de extrair baixissima quantidade de ácidos clorogênicos, a adição de água:metanol:acetonitrila (51:23:26 v/v/v) provoca modificação estrutural em alguns metabólitos, gerando um deslocamento hipsocrômico no espectro.
AGRADECIMENTOS Os autores agradecem os apoios financeiros do CNPq e da Fundação Araucária bem como as bolsas concedidas. Ao Instituto Agronômico do Paraná e à Dra. M. B. dos S. Scholz pelo fornecimento das amostras de grãos de cafés usados neste trabalho.
REFERÊNCIAS 1. Arnaud, M. J. Em Coffee: Physiology; Clarke, R. J.; Macrae, R., eds.; Elsevier: New York, 1988, caps. 2 e 9. 2. Ky, C-L.; Louarn, J.; Dussert, S.; Guyot, B.; Hamon, S.; Noirot, M.; Food Chem. 2001, 75, 223. 3. Cliffo M. N.; J. Sci. Food Agric. 2000, 80, 1033. 4. Casal, S.; Oliveira, M. B.; Alves, M. R.; Ferreira, M. A.; J. Agric. Food Chem. 2000, 48, 3420. 5. González, A. G.; Pablos, F.; Martin, M. J.; León-Camacho, M.; Valdenebro, M. S.; Food Chem. 2001, 73, 93. 6. Bicchi, C. P.; Binello, A. E.; Pellegrino, G. M.; Vanni, A.C.; J. Agric. Food Chem. 1995, 45, 1549. 7. Alves, R. C.; Casal, S.; Alves, M. R.; Oliveira, M. B.; Food Chem. 2009, 114, 295. 8. Casal, S.; Alves, M. R.; Mendes, E.; Beatriz, M.; Oliveira, P. P.; Ferreira, M. A.; J. Agric. Food Chem. 2003, 51, 6495. 9. Martin, M. J.; Pablos, F.; Gonzalez, A. G.; Food Chem. 1999, 66, 365. 10. Craig, A. P.; Franca, A. S.; Oliveira, L. S.; Food Chem. 2012, 132, 1368. 11. Craig, A. P.; Franca, A. S.; Oliveira, L. S.; LWT Food Sci. Technol. 2012, 47, 505. 12. Nicole, M. C.; Anese, M.; Manzocco, L.; Lerici, C. R.; LWT Food Sci. Technol. 1997, 30, 292. 13. Mattioli, A.V.; Farinetti, A.; Mirolo, C.; Pedrazzi, P.; Mattioli, G.; Nutr. Metab. Cardiovasc. Dis. 2011, 21, 412. 14. Gallus, S.; Tavani, A.; Negri, E.; La Vecchia, C.; Ann. Epidemiol. 2002, 12, 202. 15. Frost-Meyer, N. J.; Logomarsino, J. V.; J. Funct. Foods 2012, 4, 819. 16. Araújo, J. M. A.; Sandi D.; Food Chem. 2007, 101, 1087. 17. Tello, J.; Viguera, M.; Calvo, L.; J. Supercrit. Fluids 2011, 59, 53. 18. Mussatto, S. I.; Ballesteros, L. F.; Martins, S.; Teixeira; Sep. Purif. Technol. 2011, 83, 173. 19. Rodrigues, C. I.; Marta, L.; Maia, R.; Miranda, M.; Ribeirinho, M.; Máguas, C.; J. Food Compos. Anal. 2007, 20, 440. 20. Zhang, C.; Linforth, R.; Fisk Ian I. D.; Food Res. Int. 2012, 49, 27 21. Casal, S.; Oliveira, M. B.; Ferreira, M. A.; Food Chem. 2000, 68, 481. 22. Boushain, Z.; Garrigues, J. M.; Garriques; Garrigues, S.; Guardia, M.; Vib. Spectrosc. 1999, 21, 143. 23. Belay, A.; Ture, K.; Redi, M.; Asfaw, A.; Food Chem. 2008, 108, 310. 24. Pedrosa, M. M.; Muzquiz, M.; Garcia Vallejo, C.; Burbano, C.; Cuadrado, C.; Ayet, G.; Robredo, L. M.; J. Sci. Food Agric. 2000, 80, 459 25. Polnsek, M.; Petriska, I.; Pospisilova, M.; Jahodar, L.; Talanta 2006, 69, 192. 26. Souza, E. B. R. de; Silva, R. R. da; Afonso, S.; Scarminio, I. S.; J. Sep. Sci. 2009, 32, 4176. 27. Garcia, L. M. Z.; Daiane, E. P.; Câmara, C. A. P.; Scarminio, I. S; Nixdorf, S. L.; J. Chromatogr. Sci. 2009, 47, 825. 28. Soares P. K.; Bruns, R. E.; Scarminio I.S.; J. Sep. Sci. 2009, 32, 644. 29. Soares P. K.; Scarminio I. S.; Phytochem. Anal. 2008, 19, 78. 30. Delaroza, F.; Scarminio, I. S.; J. Sep. Sci. 2008, 31, 1034. 31. Garcia, L. M. Z.; Bruns, R. E.; Scarminio, I. S.; Chemom. and Intell. Lab. Sys. 2010, 103, 1. 32. Neto, B. B; Scarminio, I. S.; Bruns, R. E.; Como Fazer Experimentos: pesquisa e desenvolvimento na ciência e na indústria, 4ª. Ed., Bookman: Porto Alegre, 2010. 33. Moreira, I.; Scarminio, I.S.; Talanta 2013, 107, 416. 34. Rencher, A. C.; Methods of Multivariate Analysis, 2nd Ed., John Wiley &Sons: New York, 2002. 35. Liu, W. J. H.; Em Traditional Herbal Medicine Research Methods: Identification, Analysis, Bioassay and Pharmaceutical and Clinical Study; Liu, W. J. H., ed.; Wiley: Hoboken, 2011, cap. 5 36. Waksmundzka-Hajnos, M.; Sherma, J.; High Performance Liquid Chromatography in Phytochemical Analysis, CRC Press, 2011. 37. Macrae, R.; Beaumont, J. Em Coffee: Related Beverages; Clarke, R. J.; Macrae, R., eds.; Elsevier: London, 1987, cap. 4. 38. Amuta, O. P.; Nnamani, P. O.; Musa, A. D.; Nwodo, O. F. C.; Der Chemica Sinica 2011, 2, 42. 39. Fergunson, L. N. In Advances in Analytical Chemistry and instrumentation; Reilley C. N., ed.; Interscience: New York, 1965. 40. Anderson, R. J.; Bendell, D. J.; Groundwater, P. W.; Organic spectroscopy analysis, The Royal Society of Chemistry: Cambridge, 2004. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access