
 |
 |
 |
 |
 |
Artigo
|
|
| A influência da piperina na biodisponibilidade de fármacos: uma abordagem molecular The influence of piperine on the bioavailability of drugs: a molecular approach |
|
Ramon G. de OliveiraI; Edilson B. Alencar-FilhoII; Mário L. A. A. VasconcellosI, *
IDepartamento de Química, Universidade Federal da Paraíba, Campus I, 58059-900 João Pessoa - PB, Brasil Recebido em 15/05/2013 *e-mail: mlaav@quimica.ufpb.br Piperine is the major alkaloid of Piper nigrum Linn., used as a spice and in folk medicine. We present a molecular docking study supporting experimental data on the enhancement in bioavailability of propranolol, theophylline, phenytoin, nevirapine, nimesulide, pyrazinamide, carbamazepine, and spartein in the presence of piperine. The complex formed with piperine and CYP3A4 was shown to be the most stable of all, with a binding energy of -8.60 kcal/mol. This explains the related mechanism of drug-herb interaction, since the better anchoring of piperine in the active site of CYP3A4 can hinder the drug-enzyme interaction, thereby increasing the bioavailability of the drugs studied. INTRODUÇÃO Piperina (1-piperoil-piperidina, 1) é o alcalóide principal da pimenta-do-reino ou pimenta preta (Piper nigrum Linn.), a qual é comumente usada como condimento e também em vários preparos tradicionais da medicina popular.1 Frutos secos de Piper longum (pimenta longa), Piper nigrum e rizomas secos de Zingiber officinalis (gengibre) são usados na medicina popular indiana Ayurveda (conhecimento médico desenvolvido na Índia há cerca de 7 mil anos), pelo nome de "Trikatu".2 Entre as suas atividades biológicas, a piperina apresenta atividade antimicrobiana,3 anti-inflamatória,4 antiparasitária (doença de Chagas e leishmaniose),5,6 propriedades antioxidantes, entre outros.1 Considerando outros aspectos farmacológicos, a literatura descreve o aumento da biodisponibilidade de diversos fármacos, tais como o propranolol (2),7 fenitoína (3),8 nevirapina (4),9 esparteína (5),10 nimesulida (6),11 pirazinamida (7), carbamazepina (8)12 e teofilina (9),7 quando co-administrados com a piperina (1) (Figura 1). Em adição, alguns trabalhos relatam que a utilização de espécies de Piper spp. em geral, seja na dieta tradicional bem como em formulações à base de plantas, pode ser responsável pelo aumento da biodisponibilidade de fármacos, modulando seus metabolismos ao nível do complexo enzimático citocromo P450 (CYP450).13,14 Outros estudos têm demonstrado que a piperina (1) inibe a atividade constitutiva e induzida do CYP 450 in vitro e in vivo.15 O citocromo P450 (CYP450) corresponde a um conjunto de enzimas da superfamília do heme-tiolato, responsáveis pelo metabolismo oxidativo de xenobióticos.16 Em geral, os fármacos são metabolizados para evitar efeitos tóxicos de acumulação. As proteínas CYP1A2, CYP2C9, CYP2C19, CYP2D6 e CYP3A4 do CYP450 humano são as isoformas principais, as quais desempenham um papel importante no metabolismo oxidativo de 90% dos princípios ativos de medicamentos atualmente em uso clínico. Dentre essas isoformas, o CYP3A4 está envolvida no metabolismo de mais de 50% dos fármacos e outros xenobióticos.16
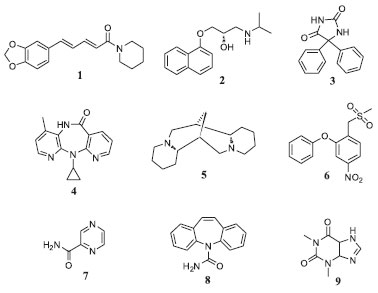 Figura 1. Moléculas estudadas neste trabalho: (1) piperina, (2) propranolol, (3) fenitoína, (4) nevirapina, (5) esparteína, (6) nimesulida, (7) pirazinamida, (8) carbamazepina e (9) teofilina
As interações do tipo erva-fármaco devem ser consideradas como fonte importante de alterações dos efeitos de medicamentos, visto que produtos vegetais são utilizados comumente como condimento na culinária ou na medicina popular. Um exemplo dessas interações é o caso com o suco de toranja (um híbrido, resultante do cruzamento do pomelo com a laranja), que acarreta em acentuadas interações erva-fármaco principalmente devido à inibição do CYP3A4 intestinal, resultando no aumento da concentração plasmática de felodipina, nitrendipina, saquinavir, ciclosporina, terfenadina e outros compostos.17,18 Mecanismos farmacocinéticos e/ou farmacodinâmicos têm sido considerados para explicar essas interações. As interações farmacodinâmicas são causadas ao nível da interação molécula-alvo de duas ou mais substâncias com efeitos similares ou opostos. Nesse caso, há uma alteração da resposta por parte do usuário sem afetar os processos farmacocinéticos de absorção, distribuição, metabolismo e excreção (ADME). As interações farmacocinéticas ocorrem quando um fármaco consegue alterar a velocidade ou extensão de processos de ADME de outro fármaco. No que concerne às alterações farmacocinéticas relacionadas ao metabolismo, o sistema microssomal hepático é uma via de inibição constante por xenobióticos, os quais podem alterar a biodisponibilidade dos fármacos no organismo.19 O docking molecular, também chamado de ancoramento molecular, é um procedimento computacional utilizado no desenvolvimento de novos fármacos, o qual visa estabelecer as características de interação entre um ligante (micromolécula) e um alvo biológico (biomacromolécula).20,21 O processo de ancoramento envolve basicamente duas etapas: a predição de conformações e orientações de ligantes no sítios de ligação dos biorreceptores, bem como a atribuição de valores de energia de interação dos complexos, baseados no cálculo de potenciais clássicos. 22 Deste modo, a técnica pode ser usada no entendimento do metabolismo de fármacos, o qual envolve interações do tipo ligante-proteína. No melhor dos nossos conhecimentos, não existe até o momento um estudo teórico via ancoramento molecular que coadune para uma proposta mecanística sobre a interação seletiva entre piperina e outros fármacos, ao nível da CYP3A4. Assim, no presente trabalho, descrevemos um estudo de ancoramento molecular entre vários fármacos e a piperina nesta enzima, objetivando corroborar os dados experimentais amplamente descritos na literatura sobre o aumento na biodisponibilidade destes compostos (2-9, Figura 1) em presença da piperina (1).
METODOLOGIA COMPUTACIONAL A estrutura cristalográfica do alvo enzimático citocromo P450-3A4 (CYP3A4) foi obtida da base de dados Protein Data Bank [PDB ID: 1TQN].23 Esta estrutura foi utilizada previamente em um outro estudo de ancoragem, portanto, foi escolhida para a abordagem aqui apresentada.16 A enzima foi elucidada por cristalografia de raios-X, com resolução de 2,05 Å.24 Um conjunto de nove moléculas (1-9, Figura 1) foram escolhidas para o estudo, de acordo com relatos na literatura sobre o aumento de suas biodisponibilidades quando co-administradas com a piperina. As moléculas foram: propranolol [DrugBank: DB00571], fenitoína [DrugBank: DB00252], teofilina [DrugBank: DB00277], nimesulida [DrugBank: DB04743], nevirapina [DrugBank: DB00238], esparteína [DrugBank: DB06727], pirazinamida [DrugBank: DB00339], carbamazepina [DrugBank: DB00564] e a própria piperina. Os ligantes foram desenhados e passaram por uma otimização de geometria inicial usando o software Avogadro25 com método MMFF94 (gradiente de RMS 0.100). O software AutoDock 4.0 26 foi utilizado como escolha para conduzir os estudos no alvo CYP3A4. O módulo AutoDock Tools foi utilizado para preparar e analisar as simulações computacionais. Cargas Gasteiger e hidrogênios polares necessários aos cálculos de potencias foram adicionados considerando a estrutura do alvo, sendo as moléculas de água retiradas. As cargas Gasteiger foram atribuídas também aos ligantes, sendo os hidrogênios não polares suprimidos. As ligações rotacionáveis de cada ligante foram definidas automaticamente e as ligações amídicas foram mantidas rígidas (devido a sua barreira conformacional relativamente alta). O AutoDock necessita de mapas de tridimensionais pré-calculados, dispostos em uma caixa composta por uma grade tridimensional de pontos (grid maps), em uma região definida na macromolécula (sítio alvo). O programa AutoGrid 4.0 foi utilizado para gerar os mapas para os ligantes. A caixa foi posicionada na região catalítica da enzima entre o grupamento o grupo prostético heme e a serina-119 do CYP3A4.16 As dimensões da caixa no eixo X-, Y- e Z- foram, respectivamente, 58 Å 72 Å 70 Å com espaçamento de 0,375 Å. Essas definições foram feitas com base em artigo prévio usando mesma enzima.16 O algoritmo Genético Lamarckiano (GA-LS) foi escolhido para busca das melhores conformações com 100 corridas para cada ligante (algoritmo genético com busca local). Durante o processo de busca, a enzima foi mantida rígida, ao passo que os ligantes foram mantidos flexíveis. A população inicial foi definida como 150 e o processo de busca se deu através de conformações iniciais aleatórias. O valor máximo de avaliações de energia escolhido foi 25.000.000. O número máximo de gerações foi 27.000. O número de elitismo escolhido foi 1. As taxas de mutação de gene e crossover foram definidas respectivamente como 0,02 e 0,80. Ao fim dos cálculos, 100 diferentes poses foram obtidas e agrupadas em diferentes clusters, definidos por proximidade energética e valores de RMS ("Root Mean Square deviation"), de acordo com o default do AutoDock.26 A validação da metodologia utilizada foi feita através da técnica de reancoramento (redocking), utilizando outro PDB do mesmo alvo proteico contendo um ligante complexado ("nativo"): o fármaco bromocriptina [PDB ID: 3UA1].27 Todos os cálculos foram efetuados em desktop Intel Xeon, 16 GB DDR3 RAM com Ubuntu linux 10.04. As análises moleculares e representações dos complexos foram obtidas com o pacote UCSF Chimera28 e a representação das interações hidrofóbicas com o software Ligplot.29
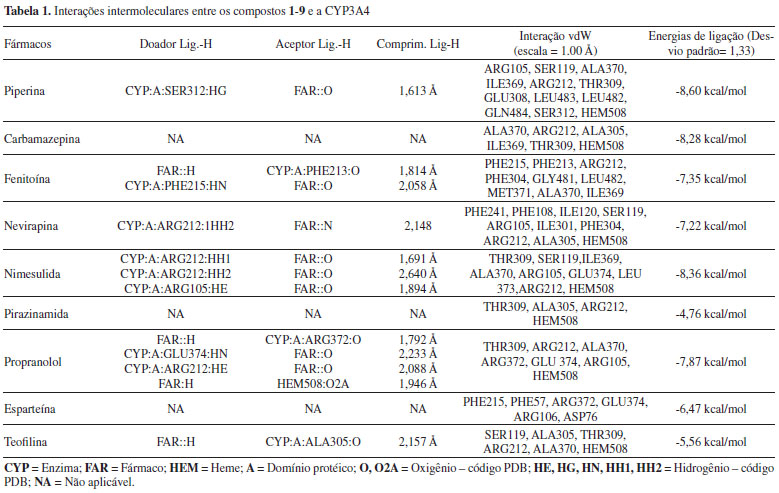
RESULTADOS E DISCUSSÃO Os compostos 1-9 (Figura 1) foram estudados via ancoramento molecular no sítio de ativo do CYP3A4 para comparar seus padrões de interação com este alvo, avaliando as suas energias de ligação em comparação com a piperina. A conformação farmacofórica para cada molécula (energia mais baixa) foi determinada a partir do cluster mais populoso. Os complexos podem ser visualizados na Figura 2, e na Figura 3 podem-se observar os diversos ligantes sobrepostos no sítio ativo. Na Tabela 1 estão listados os fármacos e tipos de interação com os resíduos envolvidos. Na coluna dos doadores e aceptores de ligação de hidrogênio, o padrão utilizado para representação foi: molécula (CYP3A4 ou fármaco): domínio protéico: número do resíduo: tipo de átomo que participa da ligação. As energias de ligação (afinidade) para cada ligante estão apresentadas na última coluna.
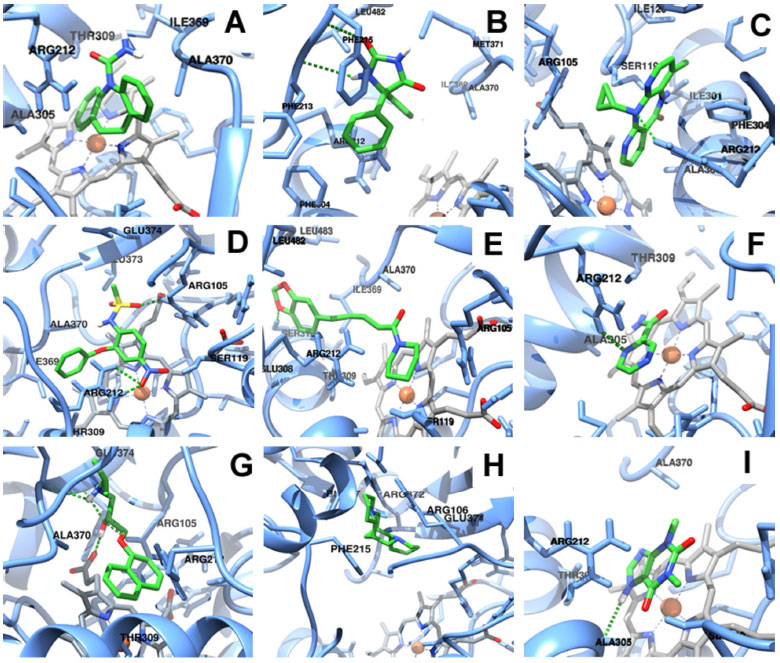 Figura 2. Ilustraçoes dos complexos do CYP3A4 com: (A) carbamazepina, (B) fenitoína, (C) nevirapina, (D) nimesulida, (E) piperina, (F) pirazinamida, (G) propranolol, (H) esparteína e (I) teofilina. Cadeia proteica: Bastoes em azul. Ligantes: Bastoes em verde com heteroátomos coloridos (oxigênios em vermelho, nitrogênios em azul escuro e hidrogênios em branco). Grupo heme: Bastoes em cinza. Ligaçoes de hidrogênios: Tracejados verdes
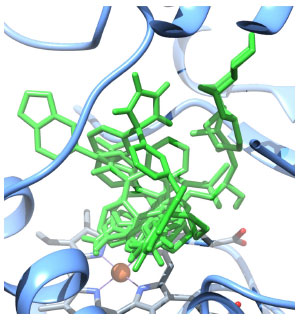 Figura 3. Sobreposiçao das moléculas ligantes no sítio ativo do CYP3A4
O reancoramento apresentou valor de RMS = 1,63 Å e energia de ligação = -13,69 kcal/mol, considerando a pose mais estável do cluster mais populoso. Esse procedimento é considerado satisfatório quando o RMS (o qual mede o desvio) entre a melhor pose e o ligante cristalográfico complexado é menor que 2,0 Å.30 Deste modo, o valor apresentado nesta etapa valida as condições utilizadas para os ancoramentos investigados. A Figura 4 apresenta a pose cristalográfica original (azul) e após o reancoramento (verde) do fármaco bromocriptina no sítio e ligação.
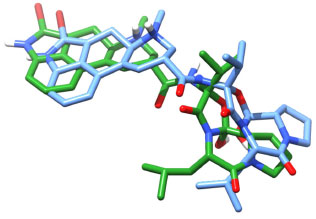 Figura 4. Sobreposiçao entre a melhor pose (verde) e o ligante cristalográfico (azul)
Dentre todas as moléculas submetidas ao cálculo de ancoramento molecular comparativo, o complexo formado entre piperina e CYP3A4 foi o mais estável, com a energia de ligação de -8,60 kcal/mol. Isso indica uma seletividade enzima-piperina em relação aos fármacos avaliados, o que corrobora a hipótese de inibição competitiva. Para o cálculo desse parâmetro (energia de Gibbs de ligação, ΔG) o Autodock utiliza um campo de força específico, parametrizado para reproduzir energias de Gibbs derivadas de constantes de equilíbrio experimentais, o qual inclui parâmetros típicos de dispersão/repulsão, ligações de hidrogênios, fatores eletrostáticos, dessolvatação e entropia torsional.26 Considerando aspectos moleculares intrínsecos, a estrutura da piperina pode ser dividida em três fragmentos: o anel metilenodioxifenila (MDP), a cadeia lateral conjugada, e o grupamento carbonila acoplado ao anel piperidínico, Figura 5.
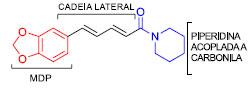 Figura 5. Representaçao esquemática dos fragmentos da piperina
Como podemos observar nos resultados, a porção da molécula correspondente ao anel metilenodioxifenila apresentou uma ligação de hidrogênio com a Serina-312, com comprimento de ligação de 1,613 Å, bem como interações de Van der Walls importantes com os resíduos dos aminoácidos LEU-482, LEU-483, GLN-484 e GLU-308. Já a cadeia lateral conjugada apresenta interações lipofílicas com os resíduos de ILE-369, ARG-212, ALA-370 e THR-309. Para o anel piperidínico foram observadas interações lipofílicas envolvendo o arcabouço pirrólico que compõe o grupo heme, bem como os resíduos SER-119 e ARG-105. Essas interações estão representadas na Figura 6.
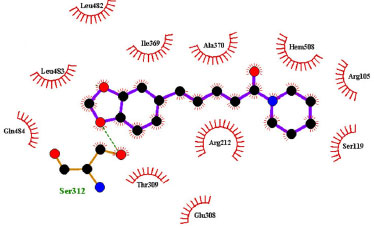 Figura 6. Interaçoes lipofílicas e ligaçoes de hidrogênio entre o complexo CYP3A4 e piperina
A inibição do metabolismo de fármacos pode ser um processo desejável, visto que promove aumento da biodisponibilidade de compostos susceptíveis às isoformas de CYP do sistema microssomal hepático e/ou intestino. Contudo, pode trazer severa toxicidade considerando fármacos que possuem uma pequena janela terapêutica (razão entre a dose tóxica e a dose eficaz). Em adição, efeitos colaterais de fármacos comuns podem ser aumentados, devido a uma maior quantidade de fármaco na circulação. Os resultados de ancoramento molecular apresentados neste trabalho (energia de ligação e modos de interação) estão de acordo com a capacidade da piperina inibir a CYP, isoforma 3A4,31 corroborando os dados da literatura sobre o aumento da biodisponibilidade de fármacos co-administrados com este produto natural.
CONCLUSÃO Os resultados teóricos de ancoramento molecular desenvolvidos neste trabalho permitiram uma melhor visão, a nível molecular, das interações entre diversos fármacos bem como da piperina na enzima CYP3A4. Foi possível observar que a piperina forma um complexo mais estável no sítio catalítico do CYP3A4 em comparação com os fármacos estudados. Estes resultados sugerem a ocorrência de um mecanismo de interação mais seletiva na CYP3A4, explicando o aumento observado da biodisponibilidade dos fármacos 2-9 quando co-administrados com piperina. A abordagem aqui considerada encontra-se em consonância com as interações erva-fármaco descritas na literatura. Sendo a piperina uma molécula de fácil obtenção e baixo custo, pode ser vista como uma alternativa para desenvolvimento de inibidores das enzimas do citocromo, objetivando modular a biodisponibilidade de fármacos na terapêutica.
AGRADECIMENTOS CAPES e CNPq pelo apoio financeiro e pelas bolsas de IC e de produtividade.
REFERÊNCIAS 1. Ferreira, W. S.; Franklin, T. N.; Lopes, N. D.; de Lima, M. E. F.; Rev. Virtual Quim. 2012, 4, 208. 2. Hanch, C.; Sammes P. G.; Anand, N.; Contribution of Ayurvedic medicine to medicinal chemistry. In: Comprehensive Medicinal Chemistry, 1st ed, Pergamon Press: Oxford, 1990. 3. Reddy. S. V.; Srinivas, P. V.; Praveen, B.; Kishore, K. H.; Raju, B. C.; Murthy, U. S.; Rao, J. M.; Phytomedicine 2004, 11, 697. 4. Pradeep, C. R.; Kuttan, G.; Int. Immunopharmacol. 2004, 4, 1795. 5. Ribeiro, T. S.; Lima L, F.; Previato, L. M.; Previato, J. O.; Heise, N.; Lima, M. E. F.; Bioorg. Med. Chem. Letters 2004, 14, 3555. 6. Ferreira, C.; Soares, D. C.; Barreto-Junior, C. B.; Lima, L. F.; Delorenzi, J. C.; Lima, M. E. F.; Atella, G. C.; Folly, E.; Carvalho, T. M. U.; Saraiva, B. E. M.; Pinto-da-Silva, L. H.; Phytochemistry 2011, 72, 2155. 7. Bano, G.; Raina, R. K.; Zutsi, U.; Bedi, K. L.; Johri, R. K.; Sharma, S. C.; Eur. J. Clin. Pharmacol. 1991, 41, 615. 8. Bano, G.; Amla, V.; Raina, R. K.; Zutshi, U.; Chopra, C. L.; Planta Med. 1987, 53, 569. 9. Kasibhatta, R.; Naidu, M. U. R.; Drugs in R&D 2007, 8, 383. 10. Atal, C. K.; Zutshi, U.; Rao, P .G.; J. Ethnopharmacol. 1981, 4, 229. 11. Gupta, S. K.; Bansal, P.; Bhardwaj, R. K.; Velpandian, T.; Pharmacological Research 2000, 41, 657. 12. Pattanaik, S.; Hota, D; Prabhakar, S.; Kharbanda, P.; Pandhi, P.; Phytotherapy Research 2009, 23, 1281. 13. Singh, J.; Dubey, R. K.; Atal, C. K.; Love, J.; J. Pharmacol Exp Ther Suppl. 1 1986, 236 S21, 488. 14. Reen, R. K.; Roesch, S. F.; Kiefer, F.; Wiebel, F. J.; Singh, J.; Biochem. Biophys. Res. Commun. 1996, 218, 562. 15. Atal, C. K.; Dubey, R. K.; Singh, J.; J. Pharmacol. Exp. Ther. 1985, 232, 258. 16. Jayakanthan, M.; Chandrasekar, S.; Muthukumaran, J.; Mathur. P. P.; J. Mol. Graphics Modell. 2010, 28, 455. 17. Baily, D. G.; Malcolm, J.; Arnold, O.; Br. J. Clin. Pharmacol. 1998, 46, 101. 18. Kane, G. C.; Lipsky, J. J.; Mayo Clin. Proc. 2000, 75, 933. 19. Byrne, B. E.; Endodontic Topics 2003, 4, 9. 20. Kitchen, D. B.; Decornez, H.; Furr, J. R.; Bajorath, J.; Nat. Rev. Drug Discovery 2004, 3, 935. 21. Gohlke, H.; Klebe, G.; Angew. Chem. Int. Ed. 2002, 41, 2644. 22. Meng, X. Y.; Zhang, H. X.; Mezei, M.; Cui, M.; Curr. Comput. Aided Drug Des. 2011, 7, 146. 23. Berman, H. M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T. N.; Weissig, H.; Shindyalov, I. N.; Bourne, P. E.; Nucleic Acids Res. 2000, 28, 235. 24. Yano, J. K.; Wester, M. R.; Schoch, G. A.; Griffin, K. J.; Stout, C. D.; Johnson, E. F.; J. Biol. Chem. 2004, 279, 38091. 25. Hanwell, M. D.; Curtis, D. E.; Lonie, D. C.; Vandermeersch, T.; Zurek, E.; Hutchison, G. R.; J. Cheminformatics 2012, 4, 17. 26. Morris, G. M.; Huey, R.; Lindstrom, W.; Sanner, M. F.; Belew, R. K.; Goodsell, D. S.; Olson, A. J.; J. Comput. Chem. 2009, 30, 2785. 27. Sevrioukova, I. F.; Poulos, T. L.; J. Biol. Chem. 2011, 287, 3510. 28. Pettersen, E. F.; Goddard, T. D.; Huang, C. C.; Couch, G. S.; Greenblatt, D. M.; Meng, E. C.; Ferrin, T. E.; J. Comput. Chem. 2004, 25, 1605. 29. Wallace, A. C.; Laskowski, R. A., Thornton, J. M.; Protein Eng. 1995, 8, 127. 30. Thomsen., R.; Christensen, M. H.; J. Med. Chem. 2006, 49, 3315. 31. Laurie, P.; Volak, S. G.; John, R. C.; Court, M. H.; Drug Metab. Dispos. 2008, 36, 1594. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access