
 |
 |
 |
 |
 |
Artigo
| Interaction of naproxen with calcium carbonate: physicochemical characterization and in vitro drug release studies |
|
Shweta ParohaI; Ravindra Dhar DubeyII; Subrata MallickI, *
ISchool of Pharmaceutical Sciences, Siksha O Anushandhan University, Bhubneswar - 751003, Odisha, India Recebido em 23/05/2013 *e-mail: profsmallick@gmail.com Interaction and physicochemical characterization of dispersions of naproxen in calcium carbonate after freeze-drying the wet-state equilibrated mixture have been investigated by analytical methods. The FT-IR study revealed the acid-base reaction between naproxen and calcium carbonate. The DSC study indicated physical interaction and significantly diminished crystallinity of naproxen in the formulation containing higher quantities of calcium carbonate. Furthermore, the SEM study showed the reduced particle size and loss of crystalline morphology in the same sample. Drug release increased with the increase of calcium carbonate in the formulations. Formulation of naproxen with calcium carbonate in 1:2 ratio allowed its dissolution to the greatest extent (94.96%) while other compositions, 1:0.5 and 1:1, showed 80.86% and 78.30% release, respectively. INTRODUCTION Naproxen is a propanoic acid derivative with anti-inflammatory, antipyretic, and analgesic properties. Naproxen is used to treat pain or inflammation caused by conditions such as osteoarthritis, rheumatoid arthritis, spondylitis, tendinitis, and other such diseases. Naproxen is a choice of drug where nonsteroidal anti-inflammatory drug (NSAID) therapy is required for patients at risk of cardiovascular complications.1-3 Long term use of naproxen can cause serious side effects, such as ulceration, bleeding or perforation of the stomach. A large number of cases with these side effects have been reported.4-6 Although the incidences of frank hemorrhage with traditional NSAIDs vary, a real incidence of regular bleeding (melaena) may be in a higher level associated with naproxen. Administration of calcium carbonate also provides significant pain relief and delays the structural progression of osteoarthritis.7 Additionally, it gives relief from painful calcific periarthritis due to depression of serum calcium levels.8,9 Moreover, calcium carbonate is used as an antacid, a buffering agent, and as a dissolution aid in dispersible tablets. Calcium carbonate is also employed as a pharmaceutical excipient, mainly in solid-dosage forms as diluents.10-12 Doctor advises to use antacid concomitantly with NSAID. The antacid magnesium hydroxide enhances ibuprofen (another propanoic acid NSAID) solubility, dissolution and bioavailability; while another antacid aluminum hydroxide has a retarding effect.13 Calcium carbonate has no significant effects on famotidine (non-NSAID) pharmacokinetics when it is co-administered.14 Drug-drug or drug-excipient interaction can alter dissolution,15 stability and bioavailability of drugs, thereby, altering its safety and/or efficacy.16,17 Careful choice of the excipients is the prime factor in design and development of the solid dosage form; a prior preformulation study with such an excipient is a very useful guide in these investigations.18,19 We have undertaken the in vitro interaction study of naproxen and calcium carbonate to understand the biological effect of their co-administration. The interaction study was carried out by co-freeze-drying the mixture of naproxen and calcium carbonate after wet-state equilibration. Analytical techniques such as Fourier transform infrared spectroscopy (FT-IR), differential scanning calorimetry (DSC) and scanning electron microscope (SEM) studies were utilized for examination of the status of the drug in the freeze-dried powder. Afterward, in-vitro dissolution of freeze-dried powders was carried out to investigate the effect of wet state interaction on dissolution. To get a comparative view, acid-base interaction of naproxen and calcium carbonate was compared with non acid-base interaction of naproxen and silicified microcrystalline cellulose.
EXPERIMENTAL Materials Naproxen was collected from Ranbaxy laboratory, Gurgaon, India as a gift sample. Calcium Carbonate (assay: >98.5%), microcrystalline cellulose (pH value of 10% aqueous suspension: 4.5-7.5) and colloidal silicon dioxide (pH of 4% w/v aqueous dispersion: 3.5-4.4) were purchased from Merck, Mumbai, India. All other chemicals were obtained commercially as analytical grade reagents. Aqueous state equilibration and freeze-drying Physical blends of naproxen with calcium carbonate, microcrystalline cellulose and colloidal silicon dioxide were prepared as per the formulation code mentioned in Table 1. The drug-excipient slurry was prepared by adding small amount of water and was left overnight for a period of 12 h at ambient conditions as per Table 1. The wet mass was dried in a freeze-dryer (Lark, Penguin Classic Plus, India) for 10 hrs or until it was completely dry. The temperature and pressure during freeze-drying were maintained approximately at -40 ºC and 15-20 Pa, respectively. The freeze-dried sample was preserved in the desiccator till further analysis.
Fourier transform infrared spectroscopy FT-IR spectra were acquired from the KBr pellets of the sample. The sample containing approximately 1 mg of naproxen and 200 mg of dried KBr was ground into fine powder using an agate mortar and compressed into a translucent disk. Each KBr disk was scanned in FT-IR Spectrophotometer (Jusco-FTIR 4100 typeA, Japan) with 80 accumulations per spectrum and a resolution of 4 cm-1 and scanning speed of 2 mm s-1 over a wave number region of 500 cm-1 to 4000 cm-1. The characteristic peaks were recorded after smoothing of optimum points on all the spectra to remove spikes and negative peaks using Spectra Manager software. Differential scanning calorimetry DSC analysis was performed using Differential scanning calorimeter (model: DSC-60, Shimadzu, Japan.) instrument with nitrogen (30 mL/min) as the purge gas. The experiments were performed in hermetically sealed aluminum pans taking approximately either 16 mg of the pure drug or other samples containing 16 mg of drug. Temperature was increased from 40 ºC to 300 ºC at a rate of 10 ºC/min. The calorimeter was calibrated using pure Indium with a melting point of 156.60 ºC before the start of experiments. The thermograms of pure naproxen and formulated freeze-dried powder samples were reported. Scanning electron microscopy A scanning electron microscope (JSM, 6100, JEOL, Tokyo, Japan) was used for the morphological study of the pure crystalline drug, the physical mixture and the freeze-dried sample at 10 kV. After mounting the powder samples directly onto the aluminum sample stub, a thin (200 nm) coating of gold was applied under vacuum using SPI Sputter coating unit (SPI supplies, division of structure probe Inc., USA) to assess their morphology. In vitro dissolution Dissolutions studies (in triplicate) of pure naproxen and the freeze-dried powdered samples were carried out using USP XXIV type II dissolution apparatus (Electrolab, dissolution tester USP TDT 06L, India) with the agitation speed of 100 rpm maintained at 37 ± 0.2 ºC. Dissolutions studies were carried out after addition of 0.1% of sodium lauryl sulfate (SLS) in distilled water. This quantity of SLS was sufficient to improve the wettability of the powder samples while avoiding the undesirable strong hydrophobic nature and allowed for the maintenance of sink conditions. Under these conditions, it was possible to observe the differences among the dissolution profiles of the powder formulations. The final volume in all cases was 900 ml. At appropriate times, aliquots were withdrawn and replaced with a fresh dissolution medium. After proper rinsing of the cuvette and filtration of the aliquot through a 0.45 µm membrane filter, absorbance at 230.2 nm was recorded using the UV-Visible Spectrophotometer (JASCO V-630 spectrophotometer, Software: Spectra Manager). The respective concentration was calculated using the standard calibration curve. These data were utilized to calculate cumulative percent drug release from the pure drug and various freeze-dried formulations.
RESULTS AND DISCUSSION Aqueous mixtures of naproxen with calcium carbonate or silicified microcrystalline cellulose (SMCC) were prepared and left overnight for equilibration at ambient temperature (~25 ºC) and freeze-dried for wet-state interaction study. The physicochemical interaction of naproxen with calcium carbonate was studied using FTIR, DSC and SEM and is described below. FT-IR studies revealed the nature of the interaction between naproxen and calcium carbonate or SMCC. Results from DSC and SEM provide further support in the study of drug amorphization. Physicochemical analysis Intermolecular hydrogen bonding in crystalline naproxen and its solid dispersions with poly(vinylpyrrolidone) (PVP) has been well described in the literature.20-22 The crystal structure of naproxen reveals that it exists as a trimolecular catemer.23 The catemer of naproxen is mainly characterized by the vibrations at 1725 cm-1 and 1684 cm-1 in the fingerprint region attributed to non-hydrogen-bonded -C=O stretching and hydrogen-bonded - C=O stretching, respectively. The FTIR spectra of the pure naproxen, the physical mixture, and other freeze-dried materials are shown in Figure 1.
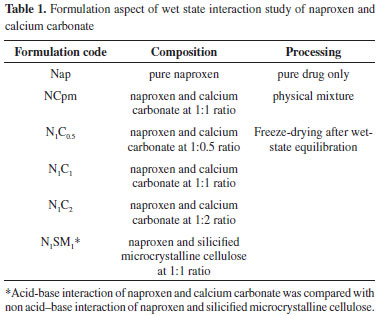
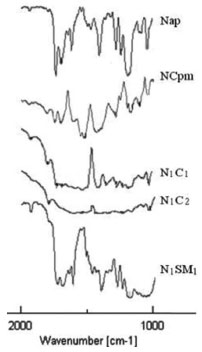 Figure 1. FTIR spectra of (i)Nap; (ii) NCpm; (iii) N1C1; (iv) N1C2; (v) N1SM1
The interaction between naproxen and calcium carbonate affected the more sensitive carbonyl stretching region. A broad shoulder-like carboxylate peak in the range 1599-1540 cm-1 has appeared in the freeze-dried powders (N1C1 and N1C2). Even the physical mixture NCpm has shown a slight shoulder-like absorption band in the same region. However, the carboxylic acid peaks (1725 and 1684 cm-1) were very weak in N1C1 and N1C2. Considering the acidic nature of carboxylic acid group of naproxen, the possibility of an acid-base interaction between the drug and calcium carbonate was investigated. An acid-base reaction between the carboxylic acid-containing drug and calcium carbonate does explain the changes observed in the FTIR spectra of freeze-dried powders, N1C1 and N1C2. Similar acid-base reactions have been reported in previous interaction studies.24-26 In the case of N1SM1, prepared by freeze-drying after the wet-state equilibration of drug and SMCC in 1:1 ratio, the vibrations at 1727 cm-1 and 1685 cm-1 appeared slightly shifted and broadened and with decreased intensity when compared to that of the pure drug. In addition, the absence of the carboxylate peak in the spectra indicates partial amorphization of the drug and absence of any acid-base interaction. The appearance of a weak carboxylate ion peak in the spectra of NCpm indicated that a small fraction of naproxen is involved in acid-base interaction with calcium carbonate in the solid state. Even minute quantities of moisture in the drug and calcium carbonate have brought about the acid-base interaction in the solid state. The intensity of carboxylate peak is higher in N1C2 when compared to that of N1C1. It is likely that the higher content of calcium carbonate in N1C2 has reacted with a larger amount of the carboxylic acid. This acid-base reaction can be represented unambiguously (Figure 2) when considering the chemical structures involved.
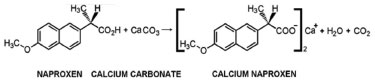 Figure 2. Acid-base reaction of naproxen and calcium carbonate, containing the chemical structures involved
Sharp and intense endothermic peak at 157 ºC accompanied by melting was noticed in the DSC scan of pure naproxen (Figure 3). The DSC scan of N1SM1 showed a melting endotherm at 156 ºC with a decreased heat of melting. On the other hand, the heat of melting at 152 ºC is barely noticeable in the endotherm of N1C2. Heat required for the melting of naproxen in the samples are N1C2 (-7.27 J/g), N1SM1 (-29.30 J/g), and Nap (-42.04 J/g). From DSC study showed similar endothermic nature to that of pure naproxen, which indicates no major interaction.27,28 Only physical interaction diminished the crystallinity of naproxen significantly as observed in the case of NCpm. In N1C2, naproxen is present in almost a molecularly dispersed state, whereas in N1SM1, it has undergone partial amorphization.
 Figure 3. DSC thermograms of (i) Nap; (ii) N1SM1; (iii) N1C2
Scanning electron micrographs of naproxen crystals, NCpm, and freeze-dried powders are shown in Fig. 4. SEM study of the pure drug showed distinctive morphological features of geometric shape of naproxen crystals (Fig. 4, A). Naproxen crystal can also be clearly identified in the physical mixture of drug and calcium carbonate in 1:1 ratio (Figure 4, B). Nearly complete loss of crystallinity and a reduction in particle size was particularly observed in the freeze-dried sample of wet equilibrated mixture of drug and calcium carbonate (Figure 4, C). Co-lyophilized powder after aqueous state equilibration of naproxen with SMCC at 1:1 ratio is characterized by submicron crystals in clustered form as observed in Figure 4, D. Taken together, the results from SEM study have been well supported by FT-IR and DSC data discussed above.
 Figure 4. Scanning electron micrograph of samples of (A) nap; (B) NCpm; (C) N1C2; (D) N1SM1. All magnifications are thousand fold
The dissolution profiles of crystalline naproxen and freeze-dried powders have been evaluated over a period of 90 minutes (Figure 5). Because of its poor aqueous solubility, crystalline naproxen (Nap) exhibited a slow rate of dissolution even when the medium contained 0.1% SLS (i.e., 66.27% released at the end of the 90 min period). Drug dissolution was improved with the increase in the amount of calcium carbonate in the freeze-dried formulations. The freeze-dried powder, N1C2, exhibited the greatest percentage of dissolution (94.96%) while N1C0.5 and N1C1 showed 80.86 and 78.30% release, respectively. A small improvement in dissolution of naproxen was observed in N1SM1 (74.82%).
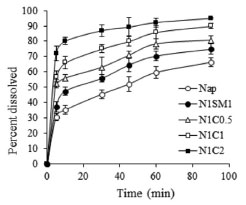 Figure 5. Cumulative percentage of naproxen released in in vitro dissolution studies from samples of pure naproxen and powders obtained by co-lyophilization after aqueous state equilibration with calcium carbonate and silicified microcrystalline cellulose. Each point represents mean ± SD, n=3
The acid-base reaction between the carboxylic acid-containing drug naproxen and calcium carbonate can also explain the observed improvement in dissolution of naproxen from the freeze-dried powders after their wet-state equilibration. The extent of dissolution was inversely related to a function of the drug-calcium carbonate ratio. Improvement of drug dissolution in case of N1SM1 is due to the decreased particle size and partial amorphization of the drug. Moreover, the presence of silicone dioxide increased the hydrophilicity and facilitated dissolution to some extent. Decreased particle size, increased wettability and dispersibility, and partial amorphization have been reported to bring about improved dissolution of ketoprofen (another propionic acid based NSAID) present in a solid dispersion composed of PVP K30 and D-mannitol.29
CONCLUSION Freeze-drying after wet-state equilibration of carboxylic acid-containing drug naproxen with calcium carbonate can improve drug dissolution. The acid-base reaction between naproxen and calcium carbonate was investigated in N1C0.5, N1C1 and N1C2 by FT-IR. The FT-IR spectra of the SMCC containing N1SM1 did not display the carboxylate peak, but showed a slightly shifted and broadened weaker carbonyl stretching peak when compared to that displayed by the pure drug. This indicated partial amorphization of drug and an absence of acid-base interaction. DSC thermogram indicated a significantly diminished crystallinity of naproxen in N1C2 when compared to the other formulations. Scanning electron micrograph of samples exhibited a reduction of particle size and loss of crystalline morphology in the freeze-dried sample of N1C2. In vitro drug release studies reveal a trend in drug dissolution: Nap < N1SM1 < N1C0.5 < N1C1 < N1C2. These studies indicate that the onset of pain relief can be shortened in patients taking naproxen and calcium carbonate together. This also provides the additional benefit of a delayed structural progression of osteoarthritis. Further studies in clinical settings will be required for evaluating in vivo efficacy of these formulations.
ACKNOWLEDGEMENT The authors would like to thank Prof. M. R. Nayak, President, Siksha O Anusandhan University for his encouragement and facilities.
REFERENCES 1. Antman, E. M.; Bennett, J. S.; Daugherty, A.; Furberg, C.; Roberts, H.; Taubert, K. A.; Circulation 2007, 115, 1634. 2. Lanza, F. L.; Chan, F. K.; Quigley, E. M. M.; Am. J. Gastroenterol. 2009, 104, 728. 3. Scheiman, J. M.; Fendrick, A. M.; Lancet 2007, 369, 1580. 4. Katona, G.;. Drugs Exp. Clin. Res. 1977, 2, 57. 5. Melton, J. W.; Lussier, A.; Ward, J. R.; Neustadt, D.; Multz, C.; J. Rheumatol. 1978, 5, 338. 6. Rainsford, K. D.; Rheumatol. Int. 1982, 2, 1. 7. Reginster, J. Y.; Neuprez, A.; Lecart, M. P.; Sarlet, N.; Bruyere, O.; Rheumatol. Int. 2012, 32, 2959. 8. Bilge, S. Y.; Korkmaz, C.; Rheumatol. Int. 2012, 32, 1503. 9. Walton, K.; Swinson, D. R.; Br. J. Rheumatol. 1983, 22, 179. 10. Allen, L. V.; Int. J. Pharm. Compd. 2000, 4, 306. 11. Mattsson, S.; Nystrom, C.; Eur. J. Pharm. Sci. 2000, 10, 53. 12. Serra, M. D.; Robles, L. V.; Int. J. Pharm. 2003, 258, 153. 13. Parojcić, J.; Corrigan, O. I.; Eur. J. Pharm. Biopharm. 2008, 69, 640. 14. Zhai, Q.; Fu, J.; Huang, X.; Xu, B.; Yuan, Y.-Z.; Jiang, T.; Rong, Z.-X.; Chen, H.-Z.; Arzneim.-Forsch. 2008, 58, 581. 15. Takahashi, A. I.; Lourenço, F. B.; Duque, M. D.; Consiglieri, V. O.; Ferraz, H. G.; Braz. Arch. Biol. Technol. 2012, 55, 477. 16. Bozdag-Pehlivan, S.; Subasi, B.; Vural, I.; Unlu, N.; Capan, Y.; Acta Pol. Pharm. 2011, 68, 423. 17. Verma, R. K.; Garg, S.; J. Pharm. Biomed. Anal. 2005, 38, 633. 18. Batista De Carvalho, L. A.; Marques, M. P. M.; Tomkinson, J.; Biopolymers 2006, 82, 420. 19. Vueba, M. L.; Batista De Carvalho, L. A.; Veiga, F.; Sousa, J. J.; Pina, M. E.; Drug Dev. Ind. Pharm. 2005, 31, 653. 20. Nair, R.; Nyamweya, N.; Gönen, S.; Martinez-Miranda, L. J.; Hoag, S. W.; Int. J. Pharm. 2001, 225, 83. 21. Tomasko, D. L.; Timko, M. T.; J. Cryst. Growth 1999, 205, 233. 22. Velazquez, M. M.; Valero, M.; Rodriguez, L. J.; Costa, S.; Santos, M. A.; J. Photochem. Photobiol., B 1995, 29, 23. 23. Bogdanova, S.; Pajeva, I.; Nikolova, P.; Tsakovska, I.; Müller, B.; Pharm. Res. 2005, 22, 806. 24. Maclean, J.; Medina, C.; Daurio, D.; Alvarez-Nunez, F.; Jona, J.; Munson, E.; Nagapudi, K.; J. Pharm. Sci. 2011, 100, 3332. 25. Mallick, S.; Pattnaik, S.; Swain, K.; De, P. K.; Saha, A.; Ghoshal, G.; Mondal, A.; Eur. J. Pharm. Biopharm. 2008, 68, 346. 26. Mallick, S.; Pattnaik, S.; Swain, K.; De, P. K.; Saha, A.; Mazumdar, P.; Ghoshal, G.; Drug Dev. Ind. Pharm. 2008, 34, 726. 27. Kundu, S. P.; Amjad, F. M.; Sultana, S.; Sultan, M. Z.; Hossain, M. A; Amran, M. S.; Bangladesh Pharm. J. 2012, 15, 7. 28. Rus, L. M.; Tomuta, I.; Iuga, C.; Maier, C.; Kacso, I.; Borodi, G.; Bratu, I.; Bojita, M.; Farmacia 2012, 60, 92. 29. Yadav, P. S.; Kumar, V.; Singh, U. P; Bhat, H. R.; Mazumder, B.; Saudi Pharm. J. 2013, 21, 77. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access