
 |
 |
 |
 |
 |
Educação
|
|
| Cromatografia gasosa-espectrometria de massas e derivatização assistida por micro-ondas na identificação de isômeros de glicose: uma prática para o ensino avançado em análise e caracterização de compostos orgânicos Gas chromatography-mass spectrometry and microwave-assisted derivatization for identification of glucose isomers: experience for post-graduate teaching of analysis and characterization of organic compounds |
|
Célia Ferreira Frias; Sílvia Alexandre Gramacho; Marta Pineiro*
Departamento de Química, Universidade de Coimbra, 3004-535 Coimbra, Portugal Recebido em 07/05/2013 *e-mail: mpineiro@qui.uc.pt This work reports a practical case based on the use of microwave-assisted derivatization and GC-MS for the analysis of glucose. Using two different methods for derivatization, one reference compound and the calculated dipole moment, all the isomers of glucose were identified. Identification was corroborated for the assignment of structures using the mass spectra. With this work, students are expected to associate different types of information to solve the complex problem of the analysis of glucose. INTRODUÇÃO Os hidratos de carbono, sacarídeos, açúcares ou glicídios são compostos de fórmula empírica geral Cn(H2O)n. De origem natural, são sintetizados pelas plantas no processo da fotossíntese, encontram-se naturalmente em diversos alimentos tais como a fruta, o leite, o mel, o grão, diversos cereais e, evidentemente, na cana-de-açúcar. Devido a sua larga presença em produtos naturais, e sendo que fazem parte da cadeia nutritiva de animais e humanos, eles são incorporados através de diversos processos bioquímicos nos organismos superiores. Essas características fazem aumentar sua expressão e dispersão pelos mais variados contextos, aumentando a importância do desenvolvimento de métodos analíticos, que permitam determinar, identificar e quantificar os açúcares presentes numa determinada matriz.1 No que se refere à identificação e quantificação de compostos orgânicos, as técnicas cromatográficas gasosas hifenadas com a espectrometria de massas são ferramentas de eleição, mas, no caso particular da análise de glicídios, revelam-se pouco úteis. A análise desses compostos através dessas técnicas é dificultada porque, sendo os glicídios e os seus derivados compostos polihidroxilados, apresentam elevada polaridade, baixa volatilidade e ausência de grupos cromóforos. A baixa volatilidade impede a análise direta por cromatografia de gases (CG) e a ausência de cromóforos dificulta a análise recorrendo à cromatografia líquida (CL), já que esses cromatógrafos estão habitualmente equipados com detectores de UV-Vis. Para realizar a análise de açúcares utilizando cromatografia líquida poderia utilizar-se um detector de índice de refracção, detector amperométrico pulsado ou recorrer a espectrometria de massas.2,3 Assim, ou se recorre a equipamentos menos usuais, mais dispendiosos e com menor capacidade de resolução ou se modificam as propriedades dos glicídios de modo a poder realizar a análise com os sistemas cromatográficos mais comuns num laboratório de análise tal como a cromatografia de gás. A derivatização de açúcares permite a modificação dos grupos funcionais da molécula, aumentando a sua volatilidade, diminuindo a polaridade e melhorando a sua estabilidade. O aumento da volatilidade pela derivatização do analíto no cromatógrafo de gases melhora a resposta do detector e aumenta a eficiência na separação dos picos cromatográficos. Assim, uma apropriada derivatização permitirá a sua análise por CG-EM, uma das mais importantes técnicas analíticas disponíveis atualmente.4-6 As reações mais comumente utilizadas para derivatizar açúcares e seus derivados são as reações de acetilação e de sililação. A acetilação em meio básico é normalmente realizada utilizando piridina ou acetato de sódio como catalisador básico. No primeiro caso, obtêm-se o derivado penta-acetilado com a mesma proporção de anômeros α e β que na amostra não derivatizada. Utilizando acetato de sódio como catalisador obtem-se majoritariamente o anómero β.7 Em 1957, Clarence Chang estabeleceu um método para a determinação de sacarose por cromatografia de gás. A derivatização do glícido foi realizada por reação com tetrametilclorosilano (TMCS) em piridina a 85 ºC durante 2 h.8 Desde então, vários métodos de derivatização de açúcares para análise por CG-EM têm sido publicados. Tipicamente, as reações de acetilação podem ser realizadas à temperatura ambiente com tempos de reação que variam entre aproximadamente 10 min9 e 24 h.10 Métodos que utilizam temperaturas superiores à temperatura ambiente resultam geralmente em tempos de reação mais curtos, caracteristicamente de 1 h.11,12 O processo de derivatização acelerada por micro-ondas, DAM, (MAD - do inglês Microwave Assisted Derivatization) tem ganhado popularidade significativa em diversas áreas de aplicação da química analítica.13,14 A utilização de irradiação de micro-ondas em substituição ao aquecimento convencional permite reduzir significativamente o tempo de reação, reduzir a quantidade de solvente - permitindo inclusive a realização de reações em fase sólida - e ainda, em alguns casos, aumentar o rendimento da reação e a sua seletividade. Essas características aproximam esse método dos princípios da química verde, encontrando-se muitas vezes associadas, o que tem levado a uma crescente utilização em síntese química e diversas aplicações no ensino.15-18 As vantagens que daí advêm para a implementação de um método analítico que inclua reações de derivatização são evidentes, sendo a mais importante a diminuição do tempo necessário para a realização da análise. A aplicação de DAM à análise de açúcares é um bom exemplo em que a utilização dessa técnica permite uma análise rápida e eficiente por CG-EM de analitos que, sem a aplicação dessa metodologia, não são analisáveis rapidamente. Muitos outros analitos podem ser analisados utilizando-se DAM. Alguns exemplos, dentre os compostos de origem natural, são os aminoácidos e os ácidos graxos que são habitualmente transformados nos derivados metilados para serem analisados por CG-EM.19,20 A glicose foi escolhida para a realização deste trabalho de laboratório porque 1) é um composto muito acessível; 2) a sua análise requer derivatização prévia, o que permite ilustrar as novas metodologias de derivatização de compostos orgânicos; 3) apresenta-se em diversas formas isómericas (Figura 1), ilustrando muito bem a capacidade deste método de separação de compostos muito semelhantes e 4) identificar isômeros através da interpretação dos espectros de massas é um exercício muito interessante para a iniciação no estudo de espectrometria de massas em compostos orgânicos.
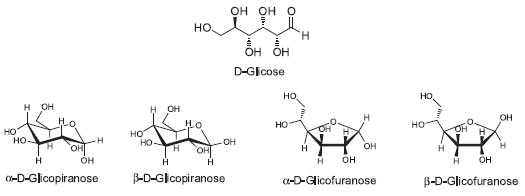 Figura 1. Isômeros que resultam do equilíbrio entre a estrutura com grupo funcional aldeído e acetal, no caso da glicose
PARTE EXPERIMENTAL Instrumentação As reações de derivatização foram realizadas recorrendo ao reator de micro-ondas para síntese Discover S-Class do fabricante CEM com controle de temperatura, pressão e potência. As análises de CG-EM foram realizadas num cromatógrafo a gás Agilent 7820A com detector de espectrometria de massas quadrupolar serie MSD Agilent 5975. A coluna cromatrográfica utilizada foi uma HP1-MS (30 m × 250 µm × 0,25 µm) da Agilent. Padrões, solventes e reagentes D(+)-glicose monohidratada (Merck, C6H12O6.H2O), α-D-glicopiranose penta-acetilada (Aldrich), piridina (Panreac, 99,5%), anidrido acético (Panreac, 99%), acetato de sódio (Panreac, PA-ACS-ISO) diclorometano (Alcrich, 99,5%) seco por refluxo com cloreto de cálcio (Merck, ACS-ISO) durante três horas, seguido de destilação à pressão atmosférica. He (Air Liquide, 99,999%) e N2 (Air Liquide, 99,99%). Derivatização assistida por micro-ondas Método A: utilização de piridina como catalisador básico Colocam-se 200 mg de glicose num tubo de micro-ondas e adiciona-se 1 mL de piridina e 2 mL de anidrido acético. A solução é aquecida em micro-ondas durante 1 minuto a 80 ºC com potência máxima de 100 watts. Após arrefecimento, evapora-se até a secura com fluxo de nitrogênio. Retiram-se 2 mg, adiciona-se 1 mL de diclorometano e analisa-se por CG-EM. Método B: utilização de acetato de sódio como catalisador básico Colocam-se 200 mg de glicose num tubo de micro-ondas e adiciona-se 1 g de acetato de sódio e 1,5 mL de anidrido acético. A solução é aquecida em micro-ondas durante 1 minuto a 110 ºC com potência máxima de 100 watts. Após arrefecimento, evapora-se até a secura com fluxo de nitrogênio. Retiram-se 2 mg adiciona-se 1 mL de diclorometano e analisa-se por CG-EM. Método cromatográfico Registaram-se os espectros de massas com ionização por impacto electrónico (70 eV) e com aquisição de dados no modo de varredura linear com um intervalo de massa entre 50 e 550 Daltons. Um volume de 1 µL de amostra foi injetada no modo com divisão de fluxo ("Split") com taxa de 100:1 a uma temperatura de 270 ºC. Os analitos foram separados numa coluna capilar de sílica fundida com fase estacionária HP-1MS. A coluna foi mantida a uma temperatura de 70 ºC durante 4 min e depois a temperatura foi incrementada de 70 ºC até 250 ºC com uma velocidade de 6 ºC/min. O tempo total de execução foi de 40 min. O gás de arraste utilizado foi o hélio. A temperatura da interface e da fonte iónica foram 260 ºC e 250 ºC, respetivamente.
RESULTADOS E DISCUSSÃO Os alunos realizaram as reações de derivatização, evaporação e recuperação da amostra, bem como a análise por CG-EM. Após completarem o procedimento experimental, os alunos realizaram a análise de resultados utilizando o seguinte conjunto de dados: 1) cromatograma da solução padrão de glicose penta-acetilada obtida pelo método A (Figura 2); 2) cromatograma da solução padrão de glicose penta-acetilada obtida pelo método B (Figura 1S, Material Suplementar); 3) os espectros de massas correspondentes a cada um dos picos que se encontram no cromatograma da Figura 2 (Figuras 3 e 4 correspondentes à α-D-glicopiranose e α-D-glicofuranose e Figuras 2S e 3S do Material Suplementar das correspondentes formas β); 4) os espectros de massas correspondentes a cada um dos picos do cromatograma da Figura 1S, não apresentados por serem idênticos aos das Figuras 3, 4, 2S e 3S; 5) o cromatograma e respectivo espectro de massas da α-D-glicopiranose penta-acetilada utilizada como padrão de referência (Figura 5 e Figura 4S do Material Suplementar) e os momentos dipolares dos anômeros alfa e beta de vários derivados da glicose (Tabela 1).
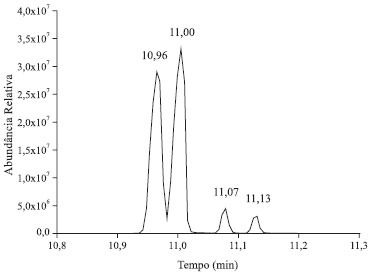 Figura 2. Cromatograma da glicose penta-acetilada obtida por derivatizaçao assistida por micro-ondas utilizando piridina (Método A)
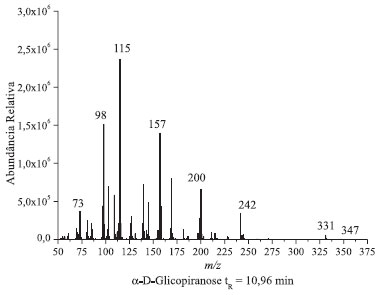 Figura 3. Espectro de massas correspondentes ao pico cromatográfico da glicose penta-acetilada com tempo de retençao 10,96 min
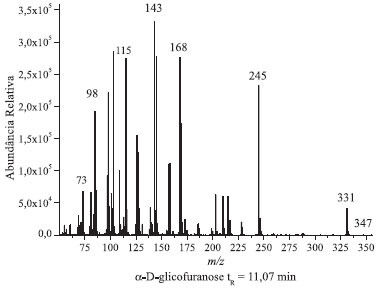 Figura 4. Espectro de massas correspondentes aos picos cromatográficos da glicose penta-acetilada com tempo de retençao 11,07 min
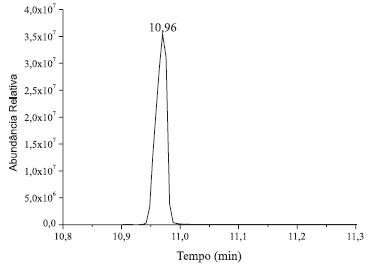 Figura 5. Cromatograma da α-D-glicose penta-acetilada de referência

Através da comparação dos cromatogramas das análises do padrão derivatizado de glicose (Figuras 2 e 1S) com o cromatograma do padrão de referência de α-D-glicopiranose penta-acetilada (Figura 5) realiza-se facilmente a atribuição do primeiro sinal cromatográfico com tempo de retenção (tR) de 10,96 minutos à α-glicopiranose penta-acetilada. Utilizando esse dado como ponto de partida, realiza-se a análise do espectro de massas correspondente ao mesmo. Recomenda-se que seja fornecido aos alunos a publicação de Ricardo Vessecchi e colaboradores de nomenclaturas de espectrometria de massas em língua portuguesa.22 Em primeiro lugar, é evidente a similitude entre as amostras, a da referência e a do primeiro pico do cromatograma da glicose. Ambas apresentam sinais com m/z 73, 98, 115, 157, 200, 242 e 331, sendo que este último é o de maior massa apreciável (não se observa o pico de massa correspondente à massa molar da glicose penta-acetilada), e o sinal em m/z 115 é o de maior abundância relativa (pico base). Estendendo a análise de comparação dos íons moleculares dos 4 picos cromatográficos da solução padrão de glicose derivatizada, o que pode ser realizado através da Tabela 2, apreciam-se as diferenças nos espectros de massas dos diferentes picos cromatográficos. Os sinais com tR's de 10,96 e 11,00 minutos apresentam picos de massa característicos em m/z 157, 200 e 242, enquanto que nos picos cromatográficos com tR's 11,07 e 11,13 estes sinais estão ausentes e observam-se picos de massa com m/z 143, 168 e 245.
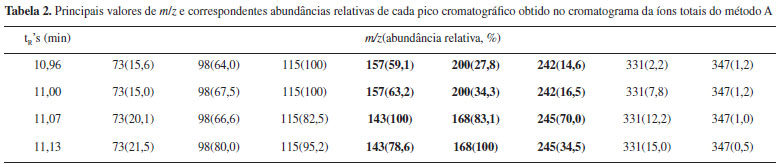
Esta análise permite agrupar os picos cromatográficos aos pares. Os dois primeiros correspondem às formas isoméricas da glicopiranose e os dois últimos aos isômeros da glicofuranose. Sendo que, se o primeiro pico cromatográfico tinha sido identificado por comparação com o isômero puro como α-D-glicopiranose penta-acetilada, o segundo pode ser atribuído à β-D-glicopiranose penta-acetilada. Essa atribuição é corroborada pelo momento dipolar dos isômeros apresentados na Tabela 1. Por analogia com os picos da glicopiranose, o segundo par de picos cromatográficos será atribuído aos isômeros α-D-glicofuranose e β-D-glicofuranose a 11,07 e 11,13 minutos respectivamente. Feita esta atribuição de picos cromatográficos, os alunos realizarão a análise dos picos de massa, com especial incidência na atribuição de uma estrutura aos picos diferenciadores dos isômeros. A diferenciação de isómeros tem representado um grande desafio químico requerendo em muitos casos o uso de diferentes técnicas analíticas.23 Na Figura 6, apresenta-se uma proposta de fragmentação para os espectros de massas da α-D-glicopiranose e α-D-glicofuranose baseada nas propostas de trabalhos anteriores.24,25 Para os anômeros β observa-se o mesmo tipo de fragmentação, e por uma questão de simplicidade não são apresentados na figura. Na fragmentação dos açúcares piranose e furanose apreciasse um pico com m/z 331 que se corresponde em ambos casos a fragmentação de um grupo acetoxilo (CH3COO) que se exemplifica como a fragmentação designada por 1 na Figura 6. Podem-se apreciar como elementos altamente diferenciadores a estrutura com o anel de furanose correspondente ao sinal com m/z 245, resultante da fragmentação da cadeia linear de dois carbonos, assinalada como 2 na Figura 6. Estes fragmentos não tem correspondência nos isômeros piranose que apresentam como pico diferenciador o sinal com m/z 242 e m/z 200 atribuíveis ao dieno triacetilado formado pela fragmentação identificada por 2 na Figura 6, e eliminação consecutiva de ceteno (CH2CO, 42 Daltons).
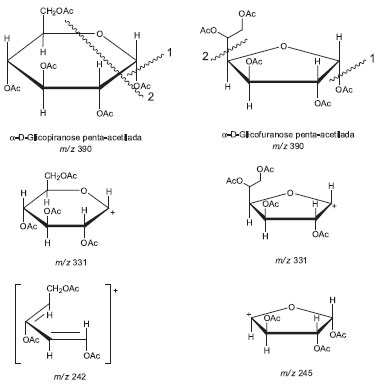 Figura 6. Proposta de fragmentaçao da α-D-glicopiranose e α-D-glicofuranose
Por último, os alunos deverão interpretar os resultados obtidos na análise da amostra obtida em relação aos diferentes métodos de derivatização utilizados. No cromatograma da amostra derivatizada utilizando piridina como catalisador (Método A) observa-se uma relação de α-D-gluocopiranose/β-D-gluocopiranose de aproximadamente 1:1, que será representativa da mistura na amostra original. No cromatograma da amostra derivatizada utilizando acetato de sódio como base (Método B) a relação entre isômeros altera-se obtendo-se majoritariamente o isômero β da forma piranose. Esta rápida anomerização é devida à preferencial acetilação do anômero mais reativo. A deprotonação da glicose dá lugar à formação de um β-oxi-ânion, que interage desfavoravelmente com o par de elétrons do oxigênio vizinho, esta espécie é estabilizada pela rápida acetilação do ânion (Figura 7).
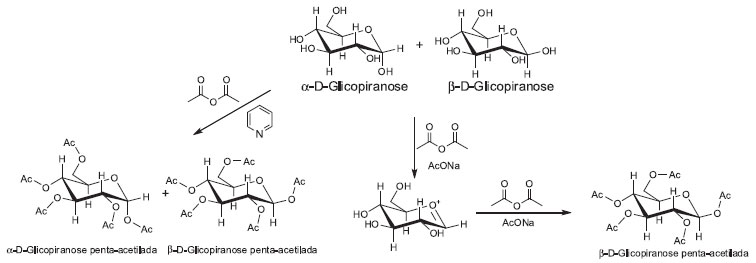 Figura 7. Formaçao dos isômeros α- e β- da glicose acetilada
Os alunos poderão concluir que a escolha do método de derivatização não deve ser aleatória e deve considerar o tipo de análise que se pretende fazer, isto é, para determinar a quantidade total de glicose numa amostra seria mais conveniente o Método B, em que a maioria da glicose é transformada praticamente num único isômero facilitando a identificação e a quantificação na amostra, mas, para a determinação da presença de diferentes anômeros este método é inadequado, devendo ser escolhido, nesses casos, o Método A, que mantém inalterada a proporção entre anômeros. Ainda, os alunos poderão refletir quanto às vantagens e desvantagens da utilização da derivatização assistida por micro-ondas, quando comparada com a derivatização por métodos convencionais26 ou com métodos mais recentes, como a utilização de líquidos iônicos,27 para o que poderão ser fornecidos aos alunos, como material suplementar, os artigos científicos aqui referidos.
MATERIAL SUPLEMENTAR No material suplementar (disponível em http/quimicanova.sbq.org.br com acesso livre, em arquivo pdf) pode-se encontrar as seguintes figuras: Figura 1S, cromatograma da solução padrão de glicose penta-acetilada obtida utilizando o método B, isto é, utilizando acetato de sódio como catalisador básico; Figura 2S e 3S, espectros de massas correspondentes aos picos cromatográficos com tempos de retenção de 11,00 e 11,13 min da β-D-glicopiranose e β-D-glicofuranose, respectivamente; Figura 4S, espectro de massas da α-D-gluocopiranose penta-acetilada utilizada como padrão de referência.
REFERÊNCIAS 1. da Cunha, P. L. R.; de Paula, R. C. M.; Feitosa, J. P. A.; Quim. Nova 2009, 32, 649. 2. http://www.dionex.com/en-us/webdocs/61831-Bro_Carbohydrates_Food_Beverage_29Aug2007_LPN1971-R2.pdf, acessada em Julho 2013. 3. http://www.dionex.com/en-us/webdocs/113460-Pittcon12_2140-1_BABailey_PN70026_Carbos.pdf, acessada em Julho 2013. 4. Ruiz-Matute, A. I.; Hernández-Hernández, O.; Rodríguez-Sánchez, S.; Sanz, M. L.; Martínez-Castro, M.; J. Chromatogr. B 2011, 879, 1226. 5. Penteado, J. C. P.; Magalhães, D.; Masini, J. C.; Quim. Nova 2008, 31, 2190. 6. Montero, C. M.; Dodero, M. C.; Sanchez, D. A. G.; Barroso, C. G.; Chromatographia 2004, 59, 15. 7. Robyt, J. F.; Essentials of carbohydrate chemistry; Springer: New York, 1998, p 77. 8. Chang, C. D.; Hass, H. B.; J. Org. Chem. 1958, 23, 773. 9. Silva, F. O.; Clin. Chem. 2006, 2, 334. 10. Simkovic, I.; Šurinab, I.; Vričan, M.; J. Anal. Appl. Pyrolysis 2003, 70, 493. 11. Price, N. P. J.; Anal. Chem. 2004, 76, 6566. 12. Di Gioia, M.; Leggio, A.; Le Pera, A.; Liguoria, A.; Napoli, A.; Siciliano, C.; Sindona, G.; J. Chromatogr. B 2004, 801, 355. 13. Söderholm, S. L.; Damm, M.; Kappe, C. O.; Mol. Divers. 2010, 14, 869. 14. Arruda, M. A. Z.; Santelli, R. E.; Quim. Nova 1997, 20, 638. 15. de Souza, R. O. M. A.; Miranda, L. S. M.; Quim. Nova 2011, 34, 497. 16. Konrath, E. L.; Piedade, M.; Eifler-Lima, V. L.; Quim. Nova 2012, 35, 1887. 17. Navarro, M.; de Sena., V. L. M.; Srivastava, R. M.; Navarro, D. M. A. F.; Quim. Nova 2005, 28, 1111. 18. Machado, A. A. S. C.; Quim. Nova 2011, 34, 535. 19. Library, T. A. L.; http://lipidlibrary.aocs.org, acessada em Julho 2013. 20. Silva, F. O.; Ferraz, V. Food Chem. 2004, 88, 609. 21. Ramakrishna, V.; Kolloid Zeitschrift 1957, 154, 152. 22. Vessecchi, R.; Lopes, N. P.; Gozzo, F. C.; Dörr, F. A.; Murgu, M.; Lebre, D. T.; Abreu, R.; Bustillos, O. V.; Riveros, J. M.; Quim. Nova 2011, 34, 1875. 23. Eberlin, M. N.; Benassi, M.; Quim. Nova 2012, 35, 17. 24. Simkovic, I.; Surina, I.; Vrican, M.; J. Anal. Appl. Pyrolysis 2003, 70, 493. 25. Jerkovic, I.; Maastelic, J.; Croat. Chem. Acta 2004, 3, 529. 26. Ahluwalia, V. K.; Aggarwai, R.; Comprehensive Practical Organic Chemistry. Preparation and Quantitative Analysis; Universities Press (India) Private Limited 2000: Hyderguda, 2000. 27. Forsyth, S. A.; MacFarlane, D. R.; Thomson, R. J.; von Itzstein, M.; Chem. Commun. 2002, 714. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access