
 |
 |
 |
 |
 |
Revisão
|
|
| Distribuição, atividade biológica, síntese e métodos de purificação de podofilotoxina e seus derivados Distribution, biological activities, synthesis, and purification methods for podophyllotoxin and its derivatives |
|
Héctor Hugo S. MedradoI; Jorge M. DavidI,*; Juceni P. DavidII; Hugo N. BrandaoIII
IInstituto de Química, Universidade Federal da Bahia, 40170-280 Salvador - BA, Brasil Recebido em 15/06/2014 *e-mail: jmdavid@ufba.br Podophyllotoxin is the most studied lignan because of its use as an antimitotic agent and because it is a precursor of pharmacologically active derivatives. This review describes the anticancer activities of podophyllotoxin and the different processes that have been developed for its extraction and purification from Podophyllum spp. In addition, the synthesis routes of this compound and the development of three semi-synthetic procedures to obtain etoposide, teniposide, and Etopophos are detailed. INTRODUÇÃO Atualmente os anticancerígenos são responsáveis por cerca de 1/3 do mercado mundial de fármacos, sendo que os quimioterápicos derivados de taxanos e da camptotecina são responsáveis pelo faturamento de cerca de 3 bilhões de dólares.1 Somente o mercado norte americano apresenta aumento anual de 10% na comercialização de quimioterápicos.2 Desde o final da década de 1990, dentre os novos anticancerígenos efetivos obtidos a partir de produtos naturais, pode-se destacar o taxol (1) extraído de Taxus brevifolia (Taxaceae) e seu derivado, conhecido como docetaxel (2), (Figura 1).3 Além desses, podem ser citados vários compostos que ainda estão em fase de testes clínicos, incluindo os derivados da camptotecina e da epipodofilotoxina. Nos últimos anos, a combretastatina A4 isolada de Combretum caffrum (Combretaceae) passou também por processos de derivatização, fornecendo os derivados fosfatado 3 e AVE-8062 (4), que possuem atividade antineoplásica marcante. Homoharringtonina (5), um alcaloide isolado de Cephalotaxus harringtonia (Cephalotaxaceae) é um inibidor de síntese de proteínas e conhecido por sua atividade antineoplásica contra leucemia mieloide. O ingenol 3-O-angelato (6), um análogo do poli-hidroxi diterpeno ingenol (7), obtido a partir de Euphorbia peplus (Euphorbiaceae), é um agente quimioterápico de uso tópico contra câncer de pele. O fenoxodiol (8) é um análogo sintético da daidzeína, extraída da soja [Glycine max (Fabaceae)], está em fase de teste nos Estados Unidos e na Austrália para o tratamento de câncer cervical, de ovário, de próstata, de rins e vaginal. O protopanaxadiol (9), aglicona triterpênica derivada de várias saponinas do ginseng [Panax ginseng (Araliaceae)], exibe efeito apoptótico sobre células cancerígenas e tem se mostrado citotóxico a vários tumores resistentes a outros fármacos.4
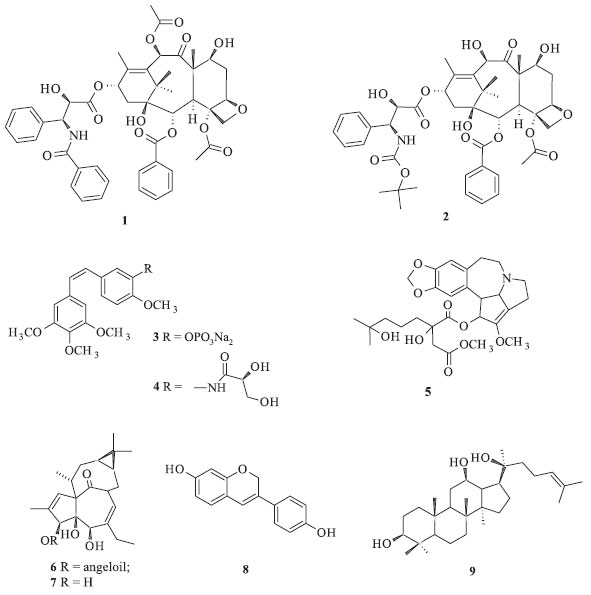 Figura 1. Anticancerígenos naturais obtidos de plantas e derivados
A podofilotoxina (10), desde o início do seu uso clínico, mostrou-se efetiva contra verrugas causadas por papilomas venéreos e também era empregada em tratamentos dermatológicos, além de possuir atividade antiviral. Desde a descoberta de suas propriedades anticancerígenas existe uma demanda comercial elevada por esta substância, principalmente para obtenção de vários derivados farmacologicamente ativos e usos clínicos que utilizam a podofilotoxina.5 Para suprir a demanda crescente dessa substância, vários processos de extração, purificação e síntese têm sido desenvolvidos. A presente revisão é um relato do estado da arte sobre a química e as atividades biológicas da podofilotoxina e seus derivados comerciais, bem como uma descrição dos diferentes processos de extração, purificação e isolamento dessa substância e das rotas sintéticas da produção do etoposide, Etopophos e teniposide. Para a presente revisão foram utilizadas as seguintes bases de dados: Scifinder, Web of Science, ScienceDirect, United States Patent e European Patent Office.
LIGNANAS O papel biológico das lignanas nos vegetais ainda não é completamente conhecido, mas acredita-se que esteja relacionado com a defesa dos mesmos e na regulação do seu crescimento. As diferentes estereoquímicas e padrões de substituição nos grupos arila contribuem na formação de diferentes estruturas que podem apresentar diversas atividades biológicas. Dentre as de maior interesse, destacam-se as atividades antiviral, antifúngica, imunossupressora, antiasmática, antioxidante e citotóxica.6 A atividade citotóxica indica o grau de morte celular induzida que um extrato, resina ou substância oferece a um determinado tipo de célula. Assim, os ensaios de citotoxicidade são uma ferramenta guia para o descobrimento de novos fármacos que podem ser usadas em tratamentos contra o câncer. Embora nem todos os compostos citotóxicos apresentem aplicação anticancerígena na terapêutica, todos os anticancerígenos mostram elevada citotoxicidade. Dentre as várias classes, destacam-se as lignanas ariltetralínicas, em especial, a podofilotoxina (10), devido ao seu efeito anticancerígeno comprovado. A podofilotoxina mostra-se um agente tanto com atividade citotóxica quanto anticancerígena e já foi utilizada por muito tempo para tratar condilomas venéreos. Histórico e ocorrência da podofilotoxina O termo podofilotoxina é derivado de espécies do gênero Podophyllum (Figura 2), como por exemplo, P. peltatum L. e P. hexandrum Royle (syn. P. emodi) (Berberidaceae) que produzem essa lignana e foram utilizadas por muito tempo como plantas medicinais com ações catárticas e colagogas.
 Figura 2. Espécimes floridas de Podophyllum peltatum L. e P. hexandrum Royle (syn. P. emodi) Fonte: http://eol.org/pages/61138/overview, acessado em 15 agosto de 2013 e http://en.wikipedia.org/wiki/Podophyllum_hexandrum acessado em 15 agosto de 2013
As raízes secas e os rizomas das mandrágoras (como essas espécies são conhecidas popularmente) fornecem uma resina obtida por extração com etanol, conhecida como podofilina. Esta resina foi a forma terapêutica mais utilizada no século XIX. Em 1872, já se havia observado que extratos das duas espécies de Podophyllum eram tóxicos a mucosas, e ficou cada vez mais evidente que a própria podofilotoxina também era tóxica, principalmente devido à ocorrência de sérias irritações gastrointestinais. Testes em sapos, gatos, cães e coelhos com podofilotoxina obtida a partir da resina de Podophyllum mostraram efeitos similares à colchicina, sendo que os efeitos foram mais potencializados nos carnívoros quando comparado aos herbívoros. Por essas razões, no século XIX, a podofilina não foi mais usada terapeuticamente como purgante ou como colagogo.7 Sem muitos dados sobre a toxicidade da podofilotoxina nas décadas seguintes, os extratos de Podophyllum foram retirados da Farmacopeia Americana em 1942. Neste mesmo ano, um estudo descreveu o uso bem sucedido da podofilina por meio do seu uso tópico para tratar condilomas venéreos. Esse resultado criou a possibilidade do uso da podofilina contra tecidos cancerosos.8 A partir dessa descoberta, a química e a atividade antineoplásica de muitos componentes da podofilina foram exaustivamente estudados. Destaca-se que o isolamento da podofilotoxina ocorreu em 1880, no entanto, sua determinação estrutural foi completada somente em 1951.9 Atividade citotóxica - mecanismo de ação A podofilotoxina e outras lignanas ariltetralínicas, bem como seus derivados apresentam atividades biológicas relevantes, principalmente como fortes agentes antineoplásicos e antivirais. Em 1946, estudos confirmaram a podofilina como potente agente antimitótico comparável à colchicina, sugerindo que seus mecanismos de ação são similares e envolvem a dinâmica de organização do fuso mitótico.10 Hartwell e Shear mostraram que a podofilina e em maior grau a podofilotoxina destruíam tumores em animais, enquanto a picropodofilotoxina (11) (Figura 3), não era ativa.11
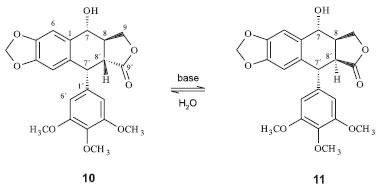 Figura 3. Epimerização da podofilotoxina (10) à picropodofilotoxina (11)
A podofilotoxina se liga à tubulina, subunidade monomérica fundamental dos microtúbulos, de forma tão forte quanto a colchicina.12 Os microtúbulos são longos filamentos em forma de tubo formados por polímeros de proteínas que são componentes chaves do citoesqueleto em células eucarióticas. São essenciais para o desenvolvimento e a forma das células, transportes de componentes, sinalização celular e no processo de mitose em que os cromossomos duplicados de uma célula são separados antes da clivagem. Esse processo requer o equilíbrio dinâmico entre organização e desorganização dos microtúbulos. Portanto, qualquer distúrbio nesse equilíbrio pode causar a interrupção da mitose e consequente morte da célula.13 A atividade citotóxica da podofilotoxina está diretamente relacionada com uma configuração única em C-7', C-8' e C-8, que é o sistema tensionado da trans-γ-lactona. Na presença de bases fracas, a podofilotoxina é epimerizada lentamente para o epímero em C-8', picropodofilotoxina (Figura 3), que é termodinamicamente mais estável devido à diminuição da tensão, mas que apresenta pouca ou nenhuma atividade citotóxica.9 Analisando as tensões presentes no anel B (átomos marcados de verde, Figura 4) observa-se que, na podofilotoxina, essas tensões provocadas pelo anel trans-lactônico levam à deformação deste anel B, forçando os átomos de carbono a permanecerem todos no mesmo plano do anel A. Na picropodofilotoxina, o anel cis-lactônico favorece a diminuição dessas torções, levando o anel B a adotar a conformação de barco, mais estável do que aquela presente na podofilotoxina.
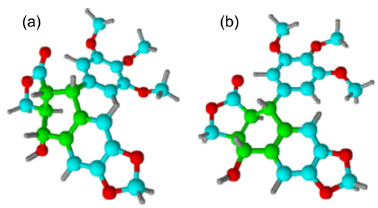 Figura 4. Modelos moleculares da podofilotoxina (a) e da picropodofiloxina (b)
A ação da podofilotoxina parece ilustrar o comportamento inibitório de microtúbulos fortemente dependente da concentração. Em concentrações suficientemente altas, a podofilotoxina provoca a desestruturação dos microtúbulos (o que a classifica no grupo de desestabilizadores de microtúbulos); em baixas concentrações de tubulina livre, há um efeito desproporcionalmente grande em inibir o crescimento e suprimir a dinâmica de microtúbulos, sem desmontar uma grande fração da população dos mesmos. Isto sugere um mecanismo de controle em potencial para a dinâmica e estabilidade dos microtúbulos, afetando a atividade celular, sem necessariamente causar desmontagem dos mesmos.14 A podofilotoxina não é o único fármaco que interrompe a estruturação e estagnação dos microtúbulos, outros tais como a colchicina, esteganancina, taxol e os alcaloides derivados da vinca são também exemplos de inibidores da polimerização/despolimerização de microtúbulos. Os mecanismos das interações com os microtúbulos da podofilotoxina e da colchicina são similares, mas não idênticos. Acredita-se que ambas não competem pelo mesmo sítio na tubulina, mas se ligam a grupos superpostos.15 Estudos de relação estrutura atividades sugerem que os anéis B e trans-lactônico estão envolvidos na interação com a tubulina. A configuração, volume e caráter hidrofóbico do substituinte em C-7 e impedimento estérico sobre o anel lactônico afetam significativamente a atividade dos derivados da podofilotoxina. Biossíntese da podofilotoxina Muitos estudos têm sido desenvolvidos para elucidação da rota biossintética da podofilotoxina que até o momento não é totalmente conhecida. De um modo geral, propõe-se que duas unidades de álcool coniferílico (12), derivado da via dos fenilpropanóides, promovam acoplamento oxidativo intermolecular 8-8' regioseletivo e estereosseletivo, via pinoresinol sintase, para formar o (+)-pinoresinol (13), com auxílio de uma proteína dirigente.16 As duas etapas seguintes têm atuação da enzima pinoresinol-lariciresinol redutase e levam a formação do (-)-secoisolariciresinol (15), passando antes pelo (+)-lariciresinol (14), por meio de reduções estereosseletivas sequenciais que resultam em inversão da configuração de C-7 e C-7' do pinoresinol. O secoisolariciresinol é oxidado pela enzima secoisolariciresinol desidrogenase, formando (-)-matairesinol (16). Este último é considerado o precursor comum das chamadas podofilotoxinas. Em trabalhos com marcação isotópica, pode-se verificar que quando matairesinol [14C] é administrado em Podophyllum, ocorre incorporação na podofilotoxina produzida. Estes experimentos demonstram que o (-)-matairesinol é provavelmente o precursor comum dessas substâncias via intermediários apropriados.2 Hoje também encontra-se estabelecido que o próximo intermediário formado na biossíntese é a yateína (17), seguido da desoxipodofilotoxina (18), que sob ação da enzima desoxipodofilotoxina 6-hidrolase leva a formação da β-peltatina (19) e com a ação da desoxipodofilotoxina 7-hidrolase, produz (-)-podofilotoxina (Figura 5).16
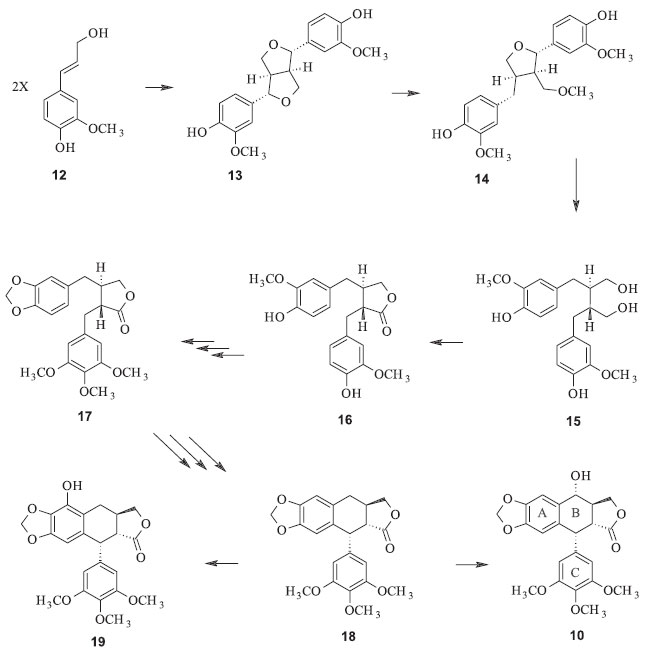 Figura 5. Proposta de biossíntese da podofilotoxina
ESTRATÉGIAS SINTÉTICAS PARA A PODOFILOTOXINA Os desafios para síntese da podofilotoxina são devidos à estereoquímica específica de quatro centros quirais, pela presença do anel trans-lactônico rígido e o substituinte aril em posição axial no anel B que oferece alto impedimento estérico. Esse desafio é acentuado pela epimerização termodicamente favorecida no C-8' para o isômero mais estável picropodofilotoxina, com um anel cis-lactônico mais flexível.17 Quatro rotas principais para a síntese da podofilotoxina e derivados foram desenvolvidas, embora diversas variações e inovações tenham sido introduzidas em cada um desses procedimentos: formação de um χ-oxoéster, lactonização de um ácido di-hidroxílico, ciclização de um produto de adição conjugada, e utilização de uma reação de Diels-Alder para sintetizar a unidade ariltetralínica (Figura 6).2,18
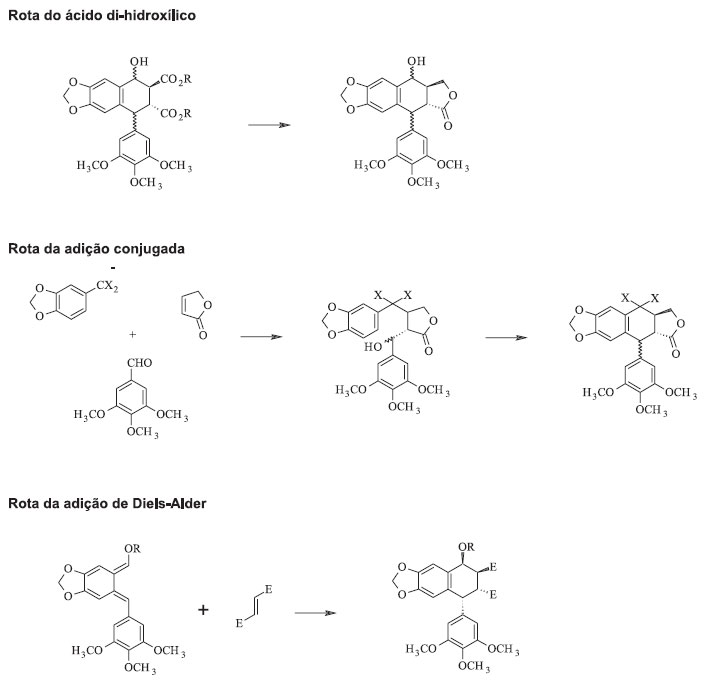 Figura 6. Quatro rotas principais para a síntese da podofilotoxina e derivados
A primeira síntese da podofilotoxina remete à década de 1960. A estratégia utilizada foi a epimerização da podofilotoxina (10) sob catálise básica para picropodofilotoxina (11) (Figura 3). A reação inversa também foi relatada, de modo que 11 foi convertida em equilíbrio para 10. A proporção de podofilotoxina na mistura em equilíbrio é muito pequena (cerca de 3%) para que este processo seja considerado uma forma prática de se obter a podofilotoxina. Acreditando que um enolato era o intermediário da isomerização, Gensler e colaboradores contornaram a limitação termodinâmica que impõe uma pequena proporção de 10 na mistura quando o equilíbrio é atingido. Para a formação do enolato, um átomo de hidrogênio na forma de próton de 11 é abstraído pelo catalisador de caráter básico. De acordo com esses autores, essa etapa ocorre em equilíbrio e a etapa seguinte, de abstração de prótons, é irreversível, na qual a mistura podofilotoxina/picropodofilotoxina não é controlada pelo equilíbrio e sim pelas velocidades de abstração dos prótons acima do plano do enolato (para formar a podofilotoxina) ou abaixo do plano, formando a picropodofilotoxina.19 A análise dos estados de transição para os dois modos de protonação pode dar uma ideia da utilidade deste fenômeno. Estudo empregando modelos construídos com átomos de preenchimento de espaço demonstrou que há espaço sensivelmente mais acima do que abaixo do ânion enolato. Assim sendo, a protonação pelo lado superior é cineticamenete mais favorecida que aquela pelo lado inferior.19 Outra estratégia sintética desenvolvida também fez uso das reações de epimerização. Uma delas mostra que o mesmo tratamento da isopodofilotoxina (20) também produziu a epimerização em C-8' para cis-8,8', fornecendo a estereoquímica da isopicropodofilotoxina (21) (Figura 7) onde os hidrogênios 8/8' e 8',7' encontram-se em cis. De modo a converter os produtos com estereoquímica cis-8,7' presente na isopodofilotoxina para a estereoquímica desejada, tal como na picropodofilotoxina (estereoquímica trans-8,7'), é necessária a epimerização em C-8 ou C -7' em vez da epimerização habitual em C-8'. Uma vez que não existem grupos funcionais próximos a C-7' que possam assistir a epimerização, o C-8 é o alvo da epimerização. A presença da função carbonila em C-7 foi apropriada para esta conversão, com a vantagem de que ela pode estar presente como um grupo funcional protegido (ditiana, ditiocetal ou cianidrina) nos intermediários não ciclizados e também nos produtos de ciclização.20
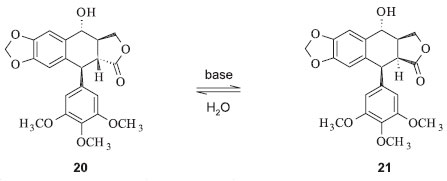 Figura 7. Epimerização da isopodofilotoxina (20) à isopicropodofilotoxina (21) catalisada por base
A reação com geração de espécies de carbo-hidroxipaládio produz simultaneamente e de forma diastereosseletiva ligações C-C e C-O para formar um anel furano. A analogia com o anel lactônico da podofilotoxina, incluindo o grupo hidroxílico em C-7, levou a se considerar que essa reação era uma nova abordagem sintética da lignana. Dessa forma, foram introduzidas funções no grupo tetralínico de análogos da podofilotoxina a partir de duas reações empregando paládio como catalisador. Essa reação levou a uma ciclização diastereosseletiva concomitante à hidroxilação para formar o anel furano funcionalizado. A segunda reação, uma nova ciclização também catalisada por paládio, apresentou rendimentos expressivamente diferentes com a variação do substrato, da base e do solvente utilizado. Condições otimizadas foram obtidas com o uso do ligante bidentado difenilfosfinoferroceno (dppf) e do p-metoxifenil (PMP) e tálio como bases, além de aditivos em dioxano a 110 ºC. A introdução do grupo benzil em C-7' foi realizada por meio de uma adição de Michael intermolecular. Esta síntese se constituiu num procedimento rápido com rendimento de 30% na preparação de um precursor da podofilotoxina e de seus análogos (Figura 8).21
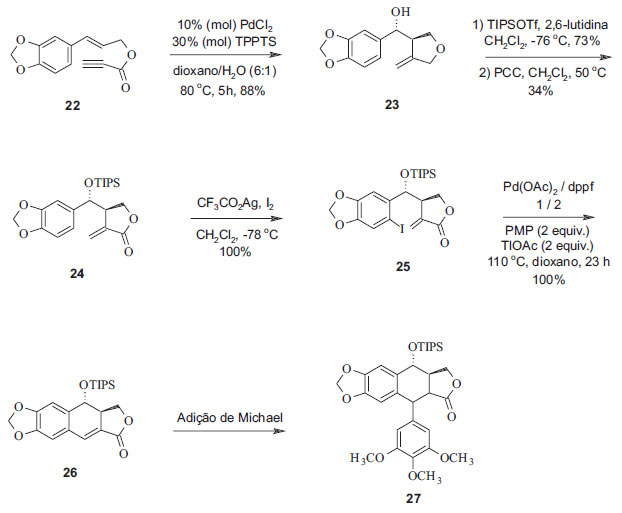 Figura 8. Síntese de análogos da podofilotoxina catalisada por paládio
Outra estratégia retrossintética importante para a obtenção dos derivados da podofilotoxina partiu da isopicropodofilona (28) e encontra-se representada na Figura 9. Esta transformação exige um desvio incomum do curso tradicional da reação de Barton-McCombie. Restrições geométricas sobre a formação das duas ligações intramoleculares C-C controlam a estereoquímica nos dois novos estereocentros; a regioquímica de substituição no grupo migratório aromático é garantida por uma etapa de substituição ipso-aromática 32 → 33 → 34.22
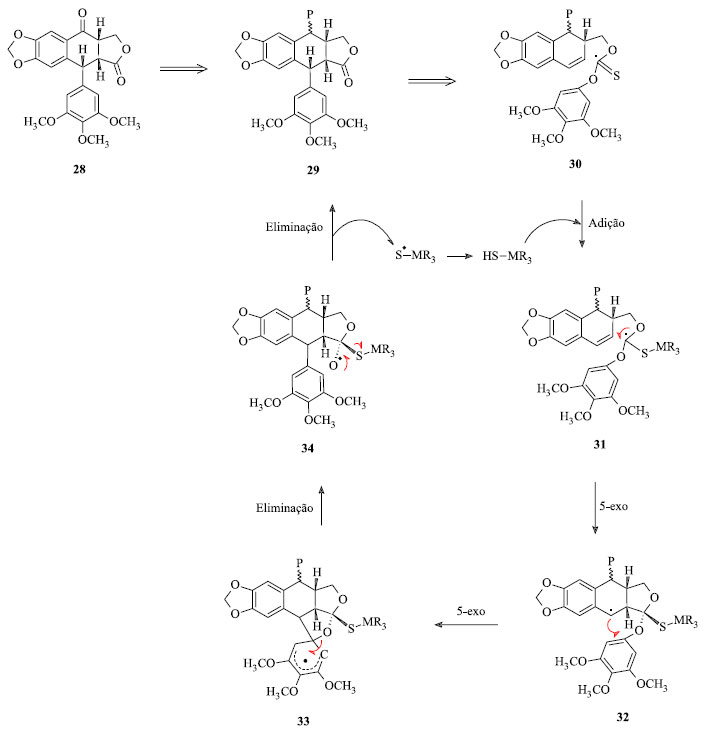 Figura 9. Estratégia retrossintética para síntese da podofilotoxina usando isopicropodofilona como precursor
A isopicropodofilona (28) pode ser obtida da reação de desaromatização naftalênica de Meyer. A adição nucleofílica estereosseletiva de dimetilfenilsililítio ao derivado oxazolínico obtido entre a valina e o derivado naftila (35), seguida por adição ao aza-enolato pelo cloroformiato de alila, forneceu o aduto 38 como um único diastereoisômero. Esta reação foi inesperadamente acompanhada pela formação do produto 36 de uma SNAr. A oxazolina 38 foi convertida no aldeído 39, que por sua vez gerou o álcool 40. A conversão seguinte ao tionocarbonato 41 e a exposição a tris-(trimetilsilil)-silano levou à lactona ariltetra-hidronaftalênica 42 (40% de rendimento). A oxidação de Fleming-Tamao do silano benzílico levou à formação do álcool correspondente, que foi imediatamente oxidado à cetona 43. Finalmente, a desalilação-descarboxilação mediada por paládio forneceu a isopicropodofilona (28), com rendimento total de 11% (Figura 10).22
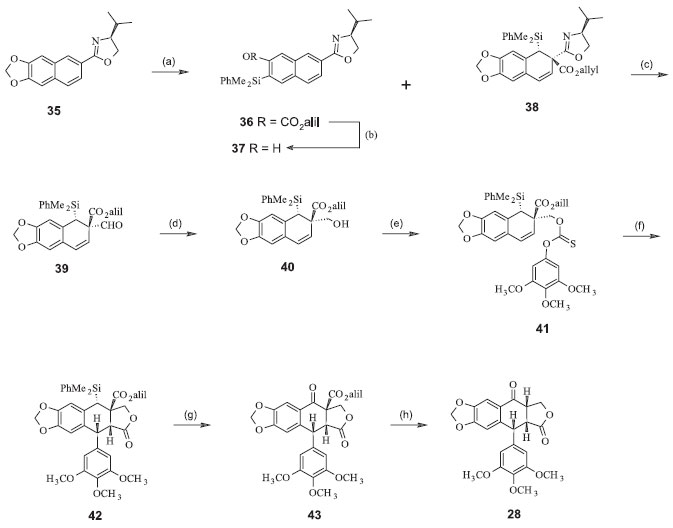 Figura 10. Síntese da isopicropodofilona para posterior conversão à podofilotoxina. (a) PhMe2SiLi (5,0 equiv.), THF, -78ºC, 3 h, seguido de cloroformiato de alila (7,5 equiv.), aquecimento de -78 ºC a 25 ºC; (b) KHCO3 (0,7 equiv.), K2CO3 (2,5 equiv.), MeOH-H2O, 25ºC, 55 min., 30% de (31), 57% de (30) nas duas etapas; (c) MeOTf (2,0 equiv.), CH2Cl2, 25 ºC, 2h, seguido de NaBH4 (4,0 equiv.) THF-MeOH, 25 ºC, 30 min., seguido de (COOH)2.2H2O (5,1 equiv.), THF-H2O, 25 ºC, 16 h, 100% nas três etapas; (d) Bu3SnH (2,0 equiv.), SiO2 , PhMe, 80ºC, 10 h, 79%; (e) 3,4,5-trimetoxifenol (1,1 equiv.), piridina (2,0 equiv.), CH2Cl2 , 25 ºC, 2 h, 99%; (f) (Me3Si)3SiH (1,1 equiv.), AIBN (0,6 equiv. adicionado em 14 h), PhH, 80 ºC, 14 h, 40%; (g) BF3.2AcOH (9,0 equiv.), CH2Cl2 , frasco selado, 50ºC, 27 h, seguido de m-CPBA (6,9 equiv.), KF (1,2 equiv.), DMF, 25 ºC, 1 h, seguido de periodinano de Dess-Martin (1,8 equiv.), CH2Cl2 , 25 ºC, 30 min., 60% baseado no (36) recuperado nas três etapas; (h) Pd(OAc)2 (4 equiv.), PPh3 (8,0 equiv.), HCO2H (40 equiv.), Et3N (50 equiv.), THF, 25 ºC, 43 min., 100%
PROCESSOS DE EXTRAÇÃO E PURIFICAÇÃO DA PODOFILOTOXINA A síntese total de podofilotoxina, molécula que possui quatro centros quirais, requer um processo sintético de custo elevado.23 Existe uma demanda muito grande por podofilotoxina, para conversão desta aos vários derivados farmacologicamente ativos e para os usos clínicos que utilizam a podofilotoxina pura.5 De modo a suprir a demanda crescente dessa substância em quantidades comerciais, inúmeros processos de extração e purificação têm sido desenvolvidos em todo o mundo. Em meados do século XX, a podofilotoxina era isolada a partir da podofilina por meio de processos trabalhosos que incluíam, por exemplo, extração com solvente seguida de outras etapas de purificação.24 Assim, a cromatografia se tornava cada vez mais essencial para a purificação desse composto, apesar da presença de seus congêneres naturais em pequenas quantidades. A purificação por cromatografia necessária para se obter a podofilotoxina com grau de pureza aceitável utilizava benzeno, apesar de se ter conhecimento dos perigos oferecidos por este solvente. Dessa forma, a podofilotoxina pura se tornava rara, com alto custo e difícil de obter comercialmente.5 O primeiro processo patenteado para a obtenção de podofilotoxina buscando alta eficiência e baixo custo data do ano de 1985.5 A estratégia baseou-se na preparação da podofilotoxina, com alto grau de pureza, a partir de uma solução da resina obtida de espécies de Podophyllum em um solvente orgânico, e, em seguida, formação de um complexo sólido da podofilotoxina com solventes aromáticos e/ou heteroaromáticos, separando o complexo da solução por meio de cristalização (Figura 11). O processo pode ser feito empregando-se vidrarias comuns de laboratório com capacidade acima das corriqueiras. Este é um processo simples que compreende duas etapas de recristalização. Uma desvantagem do processo é o emprego de substâncias aromáticas tóxicas, sendo contraditório extrair uma substância utilizada em diversas quimioterapias com materiais que podem causar neoplasias.
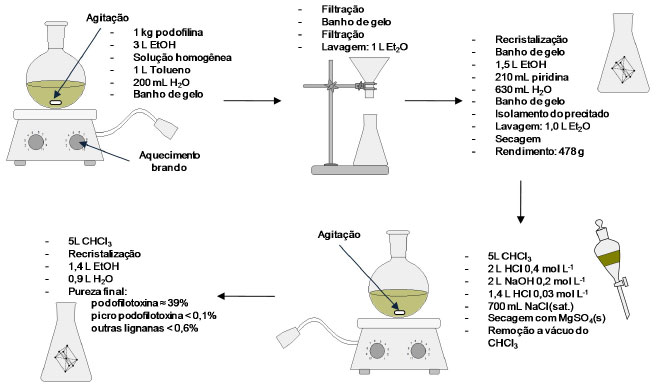 Figura 11. Esquema do processo de purificação da podofilotoxina a partir da podofilina desenvolvido por Buchardt e colaboradores
Esse processo extrativo faz uso de solução básica na segunda recristalização, sendo possível assumir que o produto final pode estar contaminado, como já discutido, com o epímero em C-8', a picropodofilotoxina. Entretanto, as análises do produto final surpreendem pelo grau de pureza da podofilotoxina obtida e a baixa contaminação com a picropodofilotoxina. Posteriormente, Jennings e colaboradores25 propuseram outro processo para a purificação da podofilotoxina a partir da mesma resina, empregando duas recristalizações intercaladas por adição de um adsorvente, preferencialmente em alumina, para a remoção de impurezas da resina e principalmente da demetilpodofilotoxina. Todavia, a partir da primeira recristalização seguida por processo de adsorção é possível obter um material rico em podofilotoxina, mas que pode ter seu grau de pureza aumentado por uma segunda recristalização, como é proposto pelos autores (Figura 12).25 O contato entre a solução do produto de primeiro estágio e uma fase sólida inorgânica e adsorvente com grande área superficial, favorecendo a adsorção de impurezas da solução de primeiro estágio (principalmente a demetilpodofilotoxina), fornece o produto de segundo estágio. A patente depositada com esse processo fornece um exemplo com quantidades reais de cada um dos reagentes, soluções e resina. O controle da pureza durante o processo é feito por CLAE e o rendimento final da extração é de 14,2% em massa de podofilotoxina em relação à resina (podofilina).25
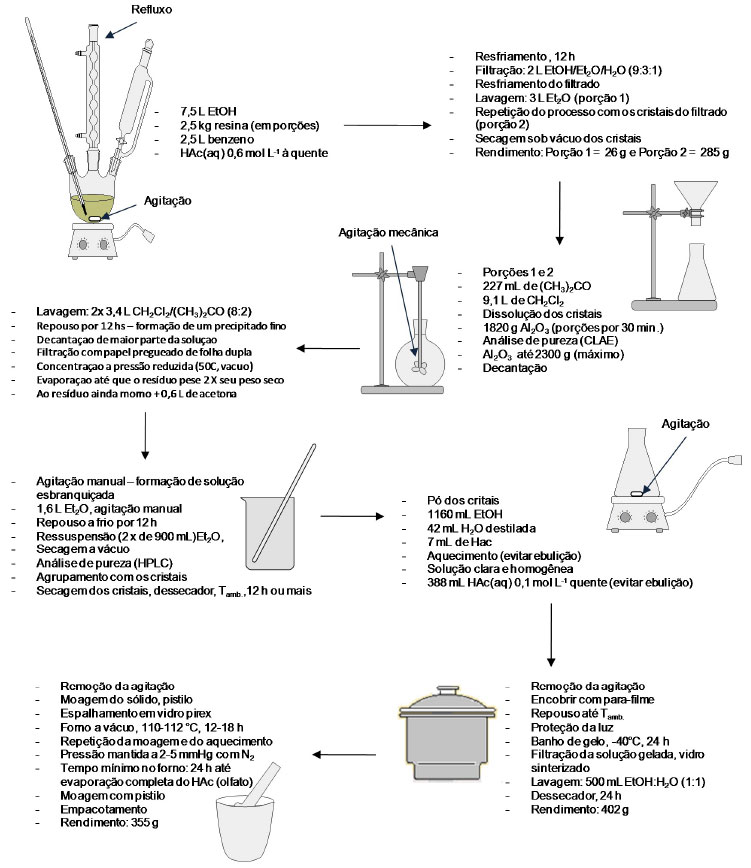 Figura 12. Esquema do processo de purificação da podofilotoxina a partir de resinas de Podophyllum desenvolvido por Jennings e colaboradores
A comparação dos processos desenvolvidos por Buchardt e Jennings mostra que o rendimento do primeiro é mais que o dobro do segundo. Enquanto que a metodologia proposta por Buchardt faz uso de solventes aromáticos, a de Jennings utiliza solventes clorados, de modo que em ambas existe a exposição acentuada dos operadores a solventes tóxicos. Uma alternativa ao uso de solventes capaz de purificar podofilotoxina cristalina anidra partindo-se da podofilotoxina obtida em estado bruto (baixa pureza) foi proposta a partir da dissolução em um solvente cujo ponto de ebulição deve ser menor que 130 ºC e teor de água em torno de 1%. A solução é resfriada até a precipitação da podofilotoxina, fornecendo os cristais que são secos a temperatura ambiente. Durante a secagem a temperatura é elevada até que ocorra fusão dos cristais, na faixa de 183-184 ºC. Desse modo, o resíduo de solvente fica em torno de 500 ppm. Na presença de solvente orgânico adsorvido no material, a adição de outra substância leva à formação de azeótropo para a remoção desse solvente. Além disso, é observado aumento do rendimento quando água é adicionada à mistura na última etapa de cristalização (Figura 13).26
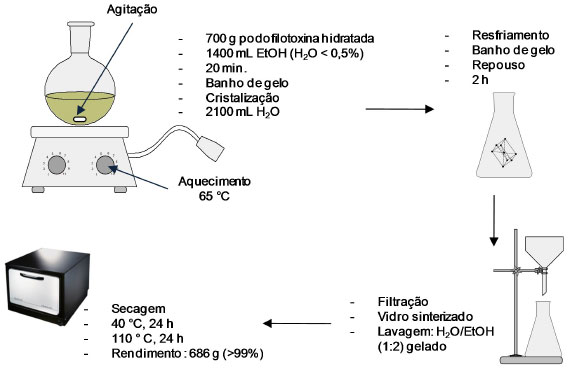 Figura 13. Esquema do processo de purificação da podofilotoxina utilizando solventes não aromáticos ou halogenados desenvolvido por Roedovre e Vaerloese
Existem metodologias mais recentes que também estão patenteadas, mas ainda não estão disponibilizadas na íntegra para acesso ao público. Assim, em um processo de 1997 é proposto a purificação da podofilotoxina a partir de P. emodi em apenas uma etapa.27 Outro método foi patenteado para obter podofilotoxina de alta pureza a partir da raiz de espécies de Podophyllum da China. A metodologia consiste da etapa de extrações com alcoóis para a obtenção do extrato inicial, seguida de outra extração para obter a podofilotoxina bruta; a segunda etapa consiste no isolamento da podofilotoxina em coluna de cromatografia de alta pressão, fornecendo um produto com pureza > 99 %.28
FONTES ALTERNATIVAS DE PODOFILOTOXINA A forma de obtenção de podofilotoxina utilizada atualmente consiste no isolamento a partir dos rizomas de P. emodii e P. peltatum. Porém, esse método não representa um sistema de produção ideal, visto que, apesar do rizomas de P. emodii possuírem cerca de 4% de podofilotoxina em peso seco da droga vegetal (P. peltatum, esse valor é pouco menor)29, o provimento dessa substância torna-se cada vez mais limitado em função da intensiva colheita dessas espécies e a falta de cultivo das mesmas. Essas espécies fazem parte da CITES, que lista não necessariamente as espécies em extinção, mas que podem vir a ser, caso não haja um controle.30 Estudos recentes mostraram que algumas espécies de Lamiaceae podem ser consideradas fontes alternativas de podofilotoxina e compostos relacionados, sendo utilizadas suas partes aéreas. Espécies de Hyptis e Eriope apresentaram teor total de lignanas do grupo das podofilotoxinas próximo a 4% em relação ao peso seco do vegetal, um valor comparável àquele exibido pelas espécies de Podophyllum utilizadas como fontes comerciais de podofilotoxina. A possível obtenção comercial desses componentes das partes aéreas dos vegetais representa uma grande vantagem por serem fontes renováveis. Uma indicação para prospecção de novas fontes de podofilotoxina em Lamiaceae foi dada pela triagem de extratos apresentando atividade citotóxica. Assim, a partir do estudo dos extratos citotóxicos das folhas de Eriope blanchetii e E. latifolia (Lamiaceae) foi possível obter podofilotoxina e seu precursor yateína, dentre outras lignanas.32 Na literatura também estão descritas outras fontes vegetais de diferentes famílias das quais a podofitoxina e seus derivados foram isolados, sendo registrado que essa substância foi isolada de Juniperus chinensis e J. scopulorum (Cupressaceae),33 Dyosma versipellis (Berberidaceae)34 e Bursera fagaroides var. fagaroides (Burseraceae).35 As informações contidas nesse estudo servem de subsídio para trabalhos que visem exploração comercial e sustentável da podofilotoxina a partir de fontes alternativas. Uma abordagem alternativa para o provimento de podofilotoxina tem sido a produção por via biotecnológica. A seleção da melhor linhagem de células, a sua manutenção e estabilização são necessárias para a produção em biorreatores e posterior aumento de escala do processo de cultivo ao nível industrial. A escala de crescimento e de rendimento do produto depende de uma multiplicidade de fatores, como meio de cultura, condições físico-químicas de inóculo da semente, tipo de reator e as condições de processamento. A composição do meio de cultura, elicitores e precursores também influenciam sensivelmente a produção.36 Uma proposta de cultivo submerso de P. hexandrum (P. emodi) para a produção de podofilotoxina foi realizada em biorreator sob agitação, equipado com um impulsor de baixo cisalhamento. Os requisitos específicos do meio, como fonte de carbono (açúcar) e de luz foram estabelecidos para o aumento da produção de podofilotoxina por P. hexandrum em culturas em suspensão. A substituição da sacarose por glicose resultou em maior crescimento e produção de podofilotoxina. A biossíntese da podofilotoxina foi favorecida quando as células das plantas foram cultivadas no escuro. A velocidade de agitação de 100 rpm foi suficiente para homogeneização no biorreator sem causar significativos danos às células.37 Na busca de uma fonte alternativa viável dos compostos bioativos de espécies de Podophyllum foram avaliados espécimes de P. hexandrum, das altitudes do Himalaya, que proporcionam um terreno ideal para a formação de relacionamentos simbióticos entre microorganismos e vegetais. Neste estudo, foi isolado de P. hexandrum o fungo endofítico Trametes hirsuta, que produz especificamente a podofilotoxina. O crescimento e a produção sob condições de fermentação foram padronizadas. O novo endófito pode ser utilizado para fermentação, com potencial biotecnológico capaz de torná-lo uma alternativa à produção de moléculas bioativas. Os endófitos isolados podem ser facilmente cultivados in vitro, o acúmulo de biomassa é rápido, com extrapolação factível e o aumento de metabólitos secundários é independente das condições ambientais (ao contrário de plantas). Além desse, foi descoberto que o fungo endofítico, morfologicamente e filogeneticamente similar a Mucor fragilis, isolado de Sinopodophyllum hexandrum (Royle) Ying (Berberidaceae) produz kaempferol e podofilotoxina.38 Paralelamente, uma substância estruturalmente relacionada com a podofilotoxina, conhecida como DPT (deoxipodofilotoxina ou anthricina) tem sido bem estudada. Esse derivado, sem o grupo hidroxílico em C-4, é também um potente agente antitumoral e anti-inflamatório. É o principal constituinte de Anthriscus sylvestris L. Hoffm. (Apiacae) e que pode também ser utilizado como precursor dos fármacos comerciais. Todavia, ainda não é possível a sua comercialização a partir de fontes naturais devido a sua baixa concentração na planta.39
DERIVADOS SEMI-SINTÉTICOS DA PODOFILOTOXINA O etoposide (44) (4'-demetil-7-[4,6-O-etilideno-β-D-glicopiranosil epipodofilotoxina]) e o teniposide (45) (4'-demetil-7-[4,6-O-tenilideno-β-D-glicopiranosil epipodofilotoxina]) são derivados semissintéticos da podofilotoxina, desenvolvidos pela Sandoz (Figura 14) na tentativa de preparar derivados desta substância que não possuam elevado efeito tóxico gastrointestinal. Nos quinze anos seguintes, o etoposide e o teniposide foram introduzidos no tratamento clínico e estes fármacos têm tido uma participação relevante no tratamento quimioterápico do câncer. Em 1996, um análogo do etoposide na forma de fosfato recebeu a aprovação do FDA para ser comercializado com o nome de Etopophos (46). A grande vantagem do Etopophos é sua solubilidade em água, o que facilita bastante a administração.40 Este representa um exemplo de pró-fármaco, pois no corpo humano o Etopophos é hidrolizado e ocorre a liberação da espécie bioativa, que é o etoposide. As indicações desse fármaco incluem câncer de pulmão, linfomas Não Hodgkin, sarcoma de Kaposi, sarcomas de tecidos moles e neuroblastoma.41
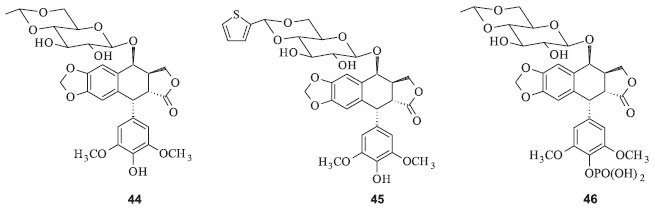 Figura 14. Derivados semissintéticos da podofilotoxina
O teniposide, análogo do etoposide, teve seu uso aprovado em 1993 nos Estados Unidos. Este fármaco é utilizado principalmente no tratamento de linfomas infantis e leucemias. In vitro o teniposide é dez vezes mais potente que o etoposide, provavelmente por ser absorvido mais facilmente pelas células. Porém, in vivo ambos possuem efeitos semelhantes. O teniposide possui menor depuração renal e maior afinidade por proteínas plasmáticas, sendo indicado para casos de leucemia linfoblástica aguda infantil, e para os adultos em casos de câncer de bexiga, sarcoma de Kaposi, leucemias e linfomas.41 Nas duas últimas décadas vários outros derivados químicos biologicamente ativos da podofilotoxina e da α-peltatina foram obtidos. Principalmente uma série de derivados esterificados e éteres sintetizados a partir de modificação do grupo OH nos anéis B e C destas lignanas. Também foi obtida uma série de derivados no anel E da podofilotoxina e substâncias não naturais estruturalmente relacionadas à podofilotoxina por meio de processos sintéticos. Muitos deles apresentam atividades inibitórias da Topo-II promissoras e foram alvos de estudos relacionados à estrutura/atividade com o objetivo de se obter substâncias mais ativas do que os fármacos comerciais derivados da podofilotoxina. A literatura já registra revisões devotadas a uma análise crítica desses novos desenvolvimentos, embora nenhuma das substâncias sintetizadas já esteja sendo comercializada.42 Mecanismo de ação do etoposide e seus análogos Anticancerígenos podem atuar diretamente no DNA das células ou indiretamente por inibição de funções metabólicas dessa macromolécula. O DNA possui uma tensão causada durante a replicação, transcrição e remodelagem da cromatina. A enzima DNA topoisomerase II (TOP2) induz a quebra das duas fitas do DNA, reduzindo a sua tensão torcional provocada pela replicação e condensação dos cromossomos nos núcleos durante a divisão da célula. A quebra é momentânea e o reparo do DNA é feito pela mesma TOP2. A função de religação da TOP2 pode ser bloqueada por inibidores dessa enzima, como por exemplo o etoposide e o teniposide.43 A vantagem desses dois derivados semissintéticos da podofilotoxina são seus efeitos colaterais menos acentuados do que os do precursor. Ambos conseguem diferenciar células cancerígenas de células normais devido às duas formas da TOP2, α e β, que são muito similares, mas geneticamente distintas. TOP2α é expressa em células proliferativas no final de uma das fases da divisão celular. Em células normais, a TOP2α ocorre em uma etapa diferente. Os derivados semissintéticos da podofilotoxina induzem o bloqueio pré-mitótico devido à ligação destes com a TOP2α, impedindo que essa enzima complete sua função e repare as duas fitas de DNA que foram previamente quebradas.13 Ao passar pela dupla fita de DNA, a TOP2 regula por ambos os lados o enrolamento da dupla hélice e desenrola os emaranhados de ácidos nucléicos. O etoposide e outros inibidores da TOP2 envenenam as enzimas aumentando a concentração no estado estacionário do complexo covalente enzima/inibidor, uma toxina fisiológica.44 O potencial de letalidade desse complexo aumenta drasticamente quando se iniciam os mecanismos de replicação ou as tentativas das helicases em desobstruir a interrupção criada pelo complexo enzima/inibidor na fita de DNA. Isto converte as quebras transitórias das fitas em permanentes, que já não são mantidas juntas por pontes proteicas. Estas pausas prolongadas geram uma série de eventos, tais como a produção de aberrações cromossômicas. Quando esses fragmentos permanentes de DNA estão presentes em concentração suficiente, inicia-se uma série de eventos que culminam na morte celular por apoptose (Figura 15).44,45
 Figura 15. Células normais e células tratadas com etoposide - cromossomos extensivamente fragmentados no tratamento com etoposide45
Estratégias sintéticas e produção industrial As modificações estruturais que convertem a podofilotoxina, uma entidade que interage com a tubulina, em outra que interage com a TOP2 são: demetilação em C-4', epimerização e glicosilação em C-7 e acetilação dos grupos 4-hidróxi e 6-hidróxi das unidades glicopiranosídicas via transacetalização. A configuração 7',8'-cis 8,8'-trans é essencial para atividade biológica e a estereoquímica de C-7 é menos importante visto que a podofilotoxina e a epipodofilotoxina são facilmente interconvertidas.2 Experimentos de RMN de 1H e de Raio-X indicaram que o anel trans-lactônico força uma conformação muito tensionada nos anéis A e B, fazendo-os permanecer no mesmo plano, enquanto o anel C encontra-se praticamente perpendicular a este plano. Esses dados ainda indicaram que o grupo açúcar encontra-se sobre o anel trans-lactônico. Como este anel é o sítio pelo qual a podofilotoxina interage com a tubulina, e neste caso ele está protegido, o etoposide e seus derivados não apresentam atividade citotóxica relacionada com a dinâmica dos microtúbulos.46 A formação estereoseletiva dos O-glicosídeos derivados da podofilotoxina é regida pela química dos carboidratos. A ligação O-glicosídica nesses compostos pode ser obtida por meio de duas estratégias sintéticas (Figura 16).
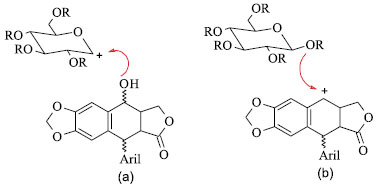 Figura 16. Formação da ligação O-glicosídica - acoplamentos de Koenigs-Knorr (a) e Kuhn (b).45
A primeira delas é semelhante ao acoplamento de Koenigs-Knorr (Figura 16a) de um derivado da glicose, que necessita de um bom grupo de saída ligado ao carbono anomérico. Esta reação envolve o ataque do grupo hidroxila da aglicona ao íon carboxônio formado no carbono anomérico da unidade glicosídica. Assim, a configuração no carbono anomérico do O-glicosídeo formado é dependente apenas do grupo de proteção em C-2 do anel glicopiranose em favorecer a formação do íon glicopiranoxônio. A estereoquímica em C-7 da aglicona é a mesma da lignana de partida.47 A segunda estratégia proposta (Figura 17b) envolve um ataque da hidroxila do carbono anomérico do derivado da glicopiranose pela face menos impedida do cátion benzílico gerado em C-7 da aglicona. Nesta situação, a estereoquímica da ligação glicosídica é altamente dependente da configuração no carbono anomérico do grupo glicopiranose e a estereoquímica de C-7 do produto final é independente da lignana precursora. Esta última estratégia é mais útil na síntese do etoposide e seus congêneres, uma vez que a configuração β está presente tanto no carbono anomérico quanto no carbono benzílico (C-7).47
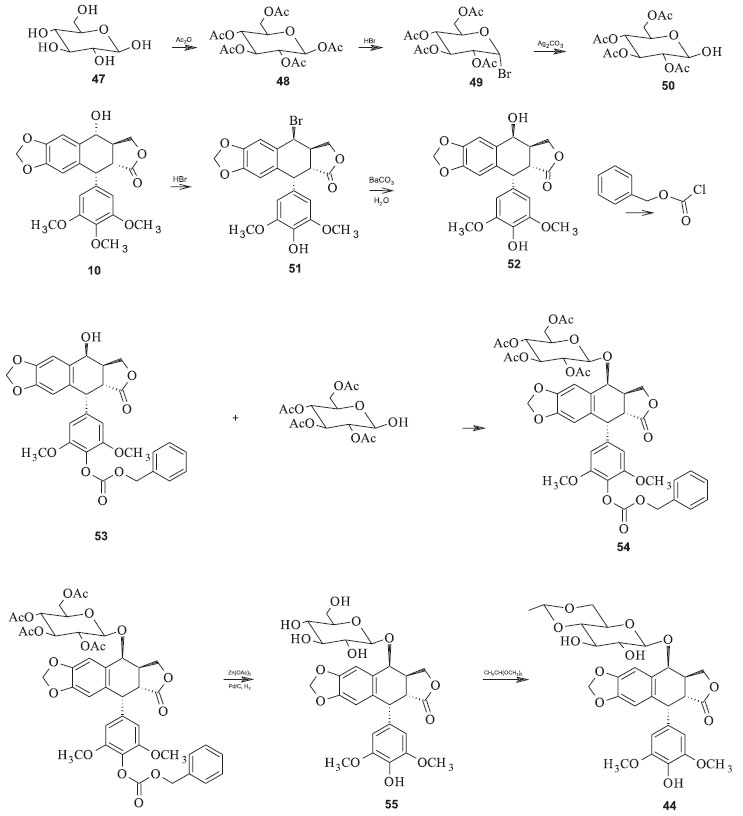 Figura 17. Síntese convencional do etoposide
Os processos convencionais para a produção dessas substâncias são complicados e onerosos. O etoposide tem sido preparado pela reação da 7-carbobenzoxi-4'-demetilepipodofilotoxina (53) e 2,3,4,6-tetra-O-acetil-4,6-O-etileno-D-glicopiranose (50) (Figura 17). O derivado carbobenzoxi é preparado a partir da podofilotoxina. Depois da reação de condensação, os grupos protetores carbobenzoxi e acetoxi devem ser removidos separadamente para se obter o glicosídeo resultante para então se isolar o etoposide. Para se obter o teniposide se adiciona o grupo tienilideno. O rendimento é de apenas 18% baseado na 4'-demetilepipodofilotoxina.48 A sequência de reações possui vários inconvenientes, além de ser muito difícil de reproduzir em escala industrial. Entre as desvantagens desse processo pode-se listar o emprego de reagentes onerosos e perigosos, como o cloroformiato de benzila, e condições reacionais muito precisas para a proteção seletiva da hidroxila fenólica da epipodofilotoxina; necessidade de duas etapas reacionais para a remoção dos dois grupos de proteção a partir do produto de condensação; exigência de intermediários glicosídicos para um processo complexo de várias etapas, com controle rígido das condições reacionais. Além disso, os açúcares possuem grupos hidroxila em átomos de carbono anoméricos que dificultam o controle das estereoquímicas.48,49 Utilizando essa estratégia com algumas modificações, Sterling e colaboradores propuseram a síntese de derivados semissintéticos da podofilotoxina a partir do intermediário 56, no qual W = H ou 2,3-di-O-acetil-4,6-O-etileno-D-glicopiranosila (Figura 18). Esses intermediários são facilmente convertidos no etoposide e no teniposide, dentre outros derivados. A principal modificação apresentada por esse processo é o uso de trialquilestanho para a desproteção da hidroxila anomérica no açúcar. Os rendimentos variaram de valores baixos (44 %) até 100 % dependendo dos substratos utilizados.48
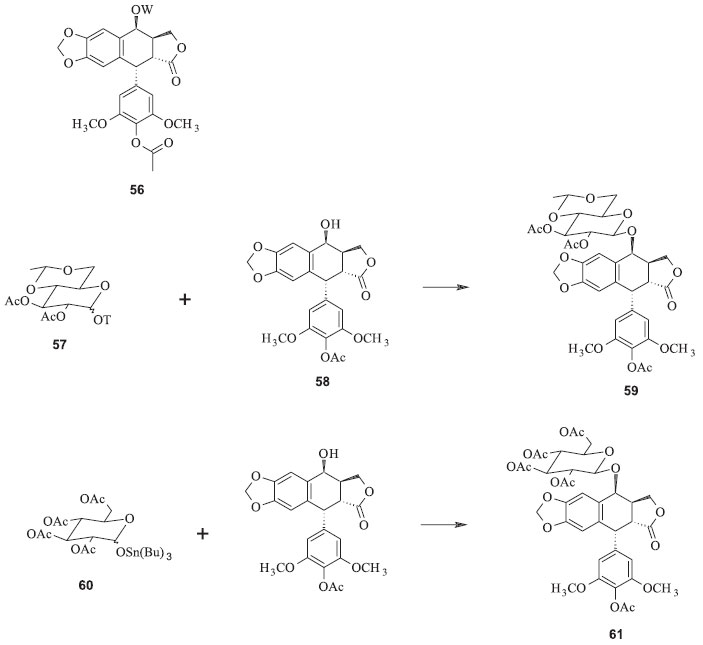 Figura 18. Intermediários para conversão ao etoposide e teniposide pelo processo de Sterling e colaboradores (OT = grupo ligado ao carbono anomérico do derivado do açúcar)48
Utilizando o derivado 57 para reagir com 58, sob catálise de ácidos de Lewis como o trifluoreto de boro, há a formação do intermediário 59, que pode ser rapidamente convertido ao etoposide pela remoção dos grupos protetores acetila com acetato de zinco em solvente alcoólico. O teniposide pode ser preparado de forma análoga utilizando o intermediário 62, a partir do derivado da glicose 60 e da aglicona 58 (Figura 18). Num processo de semissíntese mais recente foi desenvolvida uma metodologia na qual a modificação da estratégia sintética foi o reagente utilizado para proteger a hidroxila anomérica da glicose. Além disso, foi utilizado o mesmo grupo protetor tanto para as demais hidroxilas do grupo açúcar quanto para a hidroxila de C-4' da aglicona. Essas modificações permitiram a diminuição do número de etapas e o rendimento aumentou, em relação ao procedimento anterior, para 18% a partir de 52. A inconveniência dessa rota ainda reside na necessidade de condições rigidamente controladas para a monoacetilação da 4'-demetilepipodofilotoxina (52) na hidroxila fenólica (Figura 19).49
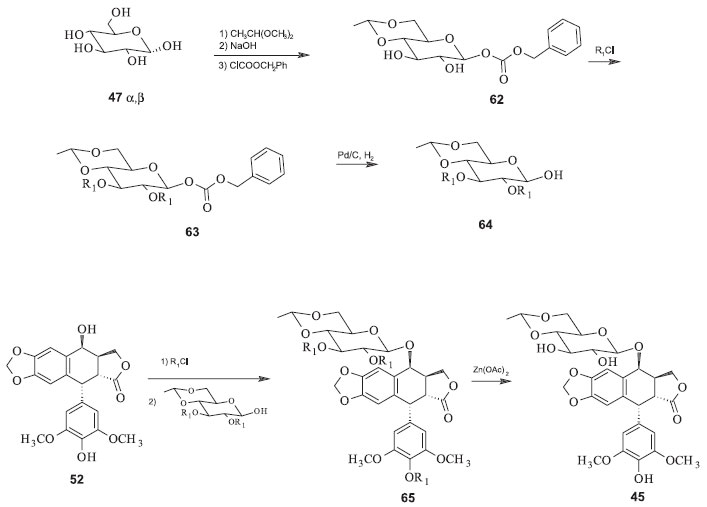 Figura 19. Síntese do etoposide pelo processo de Kurabayashi e Kalshiko50
Uma melhora desse último processo foi desenvolvida e patenteada em 1991 por Wang e colaboradores. A metodologia consta de uma rota sintética curta, de procedimento fácil, alto rendimento e baixo custo, facilitando a manufatura deste agente quimioterápico. Neste processo, o etoposide é produzido a partir da condensação direta de 52 e 64 em temperaturas de 0º a 30 ºC, na presença do trifluoreterato de boro como catalisador. Após a remoção dos grupos protetores cloroacetila (R) de 52 com acetato de zinco em metanol em temperatura a 80º a 85 ºC por duas horas, o etoposide (38) é obtido com rendimento total de 54% em relação à 46 (Figura 20).49
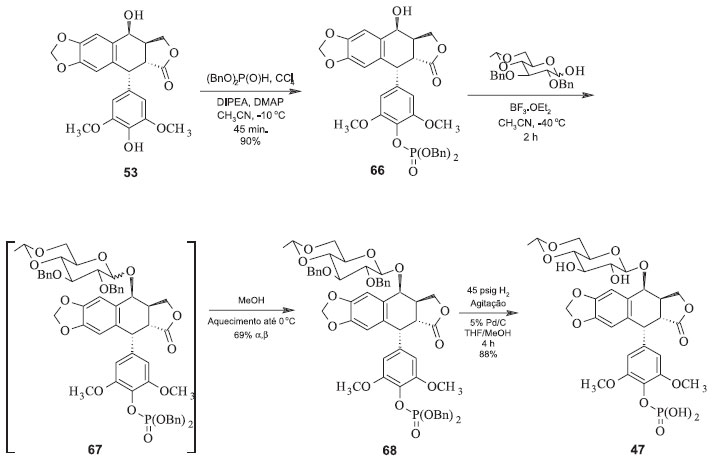 Figura 20. Síntese do Etopophos pelo processo de Silverberg e colaboradores52
Nesta rota, a proteção do grupo hidroxílico em C-4' é evitada e todas as operações do processo são relativamente mais simples que as anteriores, o rendimento é relativamente alto e o monitoramento e o controle das condições para a glicosilação são mais fáceis. Ao final do processo o etoposide é separado dos demais produtos da reação por meio de cromatografia líquida tradicional. Mais recentemente foi proposta uma metodologia de síntese do etoposide a partir da junção das estratégias de Kurabayashi e Kalshiko com a de Wang. A partir da condensação de 52 com o derivado 64, na presença de solvente orgânico e temperatura de -30 ºC, na presença de triflato de trimetilsilila como catalisador. Em seguida, o tratamento de 65 com acetato de zinco em metanol fornece o etoposide (44), com rendimento total em relação a 52 de 65%.50 As sínteses do Etopophos são análogas aquelas do etoposide. A diferença reside somente no grupo fosfato adicionado à hidroxila fenólica em C-4'. Em 1994, Miyazawa e colaboradores propuseram a síntese do Etopophos a partir da proteção dos grupos hidroxila da porção glicosídica do etoposide com grupos haloacetila. Em seguida, procedeu-se com a fosforilação da hidroxila em C-4' e posterior remoção dos grupos haloacetila na presença de uma amina. O processo fornece o Etopophos com rendimento de 62% em relação a 52.51 Mais recentemente, Silverberg e colaboradores sintetizaram o Etopophos usando uma rota sintética de três etapas com rendimento de 55% em relação à 4'-demetilepipodofilotoxina (52). A estratégia sintética está na proteção tanto do grupo fosfato quanto das hidroxilas da parte glicosídica com éteres benzílicos, facilitando a remoção dos mesmos de uma só vez. Duas características dessa síntese são a anomerização térmica e completamente diastereosseletiva do grupo glicosídeo e a recristalização conjunta dos anômeros α e β. Aumentando-se a temperatura da mistura até 0 ºC, o anômero β recristaliza preferencialmente em relação ao anômero α, fornecendo então 55% do Etopophos (Figura 20) em relação à 52.52
CONCLUSÕES Dentre as lignanas, a podofilotoxina é a substância que apresenta o maior número de estudos desde o seu isolamento. As atividades citotóxica e antiviral dessa substância, além do fato de ser precursora semissintética de quimioterápicos utilizados na atualidade, fazem dela uma substância ímpar. Assim, inúmeros processos de extração e purificação dessa substância têm sido desenvolvidos, tentando utilizar solventes menos tóxicos e agressivos. Entretanto, a falta de plantio e o uso extrativista das suas principais fontes têm levado à sua escassez. Esse fato vem aumentando o interesse por novas fontes alternativas de podofilotoxina, principalmente de fontes renováveis. A extração a partir de fontes naturais ainda é preferível sobre sua produção sintética. As rotas de síntese vêm sendo aprimoradas desde que esses medicamentos foram inicialmente produzidos. Os maiores problemas dos processos sintéticos são os usos de grupos protetores, reagentes caros e tóxicos, além da etapa de hidrogenação, desaconselhada em plantas industriais. Das estratégias sintéticas abordadas, a de Naidu parece ser a mais simples e com resultados satisfatórios.
REFERÊNCIAS 1. Oberlies, N. H; Kroll, D. J.; J. Nat. Prod.2004, 67, 129; Thayer, A.; Chem. Eng. News2003, 81, 6. DOI: http://dx.doi.org/10.1021/np030498t PMID: 14987046 2. Canel, C.; Moraes, R. M.; Dayan, F. E.; Ferreira, D.; Phytochemistry2000, 54, 115. DOI: http://dx.doi.org/10.1016/S0031-9422(00)00094-7 PMID: 10872202 3. Harvey, A.; Drug Discovery Today2000, 5, 294. DOI: http://dx.doi.org/10.1016/S1359-6446(00)01511-7 PMID: 10856912 4. Chin, Y.W.; Balunas, M. J.; Chai, H. B.; Kinghorn, D.; AAPS J. 2006, 8, 239. DOI: http://dx.doi.org/10.1208/aapsj080228 5. Buchardt, O.; US pat.4,680,3991985. 6. Kitamura, M.; Hirokawa Y.; Maezaki, N.; Chem. Eur. J.2009, 15, 9911; Ríos, J. L.; Giner, R. M.; Prieto, J. M.; Stud. Nat. Prod. Chem.2002, 26, 183; Willfor, S. M.; Smeds, A. I.; Holmbom, B. R.; J. Chromatogr. A. 2006, 1112, 64. DOI: http://dx.doi.org/10.1002/chem.200901212 7. Neuberger, J.; Arch. Exp. Pathol. Pharmakol.1891, 28, 32. DOI: http://dx.doi.org/10.1007/BF01949656 8. Imbert, F.; Biochimie1998, 80, 207. DOI: http://dx.doi.org/10.1016/S0300-9084(98)80004-7 PMID: 9615861 9. Botta, B.; Monache, G. D.; Misiti, D.; Vitali, A.; Zappia, G.; Curr. Med. Chem. 2001, 8, 1363. DOI: http://dx.doi.org/10.2174/0929867013372292 PMID: 11562272 10. King, I. S.; Sullivan, M.; Sci.1946, 104, 244 DOI: http://dx.doi.org/10.1126/science.104.2698.244 11. Hartwell, J. L.; Shear, M. J.; Cancer Res.1947, 7, 716. 12. Wilson, I.; Friedkin, M.; Biochem.1967, 6, 3126. DOI: http://dx., doi.org/10.1021/bi00862a021 13. Brandao, H. N.; David, J. P.; Couto, R. D.; Nascimento, J. A. P.; David, J. M.; Quim. Nova2010, 33, 1359. DOI: http://dx.doi.org/10.1590/S0100-40422010000600026 14. Schiltra, M. J.; Martin, S. R.; Bayley, P. M.; J. Biol. Chem.1989, 264, 8827. 15. Bhattacharyya, B.; Wolff, J.; Proc. Natl. Acad. Sci. U. S. A.1974, 71, 2627. DOI: http://dx.doi.org/10.1073/pnas.71.7.2627 PMID: 4527949 16. Fuss, E.; Phytochem. Rev.2003, 2, 307; Kuhlmann, S.; Kranz, K.; Lücking, B.; Alfermann, A. W.; Petersen, M.; Phytochem. Rev. 2002, 1, 37; Seidel, V.; Windhövel, J.; Eaton, G.; Alfermann, A. W.; Arroo, R. R. J.; Medarde, M.; Petersen, M.; Woolley, J. G.; Planta2002, 215, 1031; Xu, H.; Lv, M.; Tian, X. A.; Curr. Med. Chem. 2009, 16, 327. DOI: http://dx.doi.org/10.1023/B:PHYT.0000045500.56476.f5 17. Forsey, S. P; Rajapaksa, D.; Taylor, N. J.; Rodrigo, R.; J. Org. Chem.1989, 54, 4280. DOI: http://dx.doi.org/10.1021/jo00279a011 18. Hadimani, S. B.; Tanpure, R. P.; Bhat. S. V.; Tetrahedron Lett. 1996, 37, 4791. DOI: http://dx.doi.org/10.1016/0040-4039(96)00937-9 19. Gensler, W. J.; Gatsonis, C. D.; J. Org. Chem.1966, 31, 4004. DOI: http://dx.doi.org/10.1021/jo01350a028 PMID: 5978075 20. Medarde, M.; Ramos, A. C.; Caballero, E.; López, J. L.; Tetrahedron Lett.1996, 37, 2663. DOI: http://dx.doi.org/10.1016/0040-4039(96)00355-3 21. Charruault, L.; Michelet, V.; Genêt, J-P.; Tetrahedron Lett. 2002, 43, 4757. DOI: http://dx.doi.org/10.1016/S0040-4039(02)00908-5 22. Reynolds, J. A.; Scott, A. J.; Turner, C. I.; Sherburn, M. S.; J. Am. Chem. Soc. 2003, 125, 12108. DOI: http://dx.doi.org/10.1021/ja0376588 PMID: 14518993 23. Gordaliza, M.; García, P. A.; Corral, J. M. M. C.; Castro, M. A.; Zurita, G.; Toxicon2004, 44, 441. DOI: http://dx.doi.org/10.1016/j.toxicon.2004.05.008 PMID: 15302526 24. Hartwell, J. L.; Detty, W. E.; J. Am. Chem. Soc.1950, 72, 246; Hearon, W. M.; Macgregor, W. S.; Chem. Rev.1955, 55, 957. DOI: http://dx.doi.org/10.1021/ja01157a068 25. Jennings, R. A.; Stearns, J. F.; US pat.5,057,6161989. 26. Roedovre, H. F. H; Vaerloese, K. K.; US pat.005,315,0161992. 27. Mohan, A. S.; Mohan, J. S.; Singh, K. R.; Chander, P. S.; IN178,8741997. 28. Zhang, W.; Cai, Z.; Tong, Z.; Wang, Q.; Zhang, A.; Gao, S.; CN101,503,4102009. 29. Choudhary, D. K.; Kaul, B. L.; Khan, S.; J. Med. Aromat. Plant Sci.1998, 20, 1071; Rai, L. K., Prasad, P.; Sharma, E.; Biol. Cons.2000, 93, 27. 30. World Conservation Monitoring Centre. Checklist of CITES species: a reference to the appendices to the Convention on international trade in endangered species of wild fauna and flora, CITES Secretariat/World Conservation Monitoring Centre, Chatelâine-Genève, 2001. Disponível em: http://www.unep-wcmc.org/species/sca/scs.htm, acessada em abril 2014. 31. David, J. P.; Silva, E. F.; Moura, D. L.; Guedes, M. L. S.; Assunçao, R. J.; David, J. M.; Quim. Nova2001, 24, 730. DOI: http://dx.doi.org/10.1590/S0100-40422001000600004 32. Santos, E. O.; Lima, L. S.; David, J. M.; Martins, L. C.; Guedes, M. L. S.; David, J. P.; Nat. Prod. Res. 2011, 25, 1450. DOI: http://dx.doi.org/10.1080/14786410902809500 PMID: 20234970 33. Miyata, M.; Itoh, K.; Tachibana, S.; J. Wood Sci.200644, 397; Schroeder, K. A.; French, A. N.; Jeliazkov, V.; ACS National Meeting & Exposition, April 7-11, 2013, New Orleans, LA. DOI: http://dx.doi.org/10.1007/BF01130454 34. Yang, Z.; Liu, X.; Wang, K.; Cao, X.; Wu, S.; J. Sep. Sci. 2013, 36, 1022. DOI: http://dx.doi.org/10.1002/jssc.201200618 PMID: 23418155 35. Rojas-Sepulveda, A. M.; Mendieta-Serrano, M.; Mojica, M. Y. A.; Salas-Vidal, E.; Marquina, S.; Villarreal, M. L.; Puebla, A. M.; Delgado, J. I.; Alvarez, L.; Molecules2012, 17, 9506. DOI: http://dx.doi.org/10.3390/molecules17089506 PMID: 22878225 36. Farkya, S.; Bisaria, V. S.; Srivastava, A. K.; Appl. Microbiol. Biotechnol.2004, 65, 504. PMID: 15378293 37. Chattopadhyay, S.; Srivastava, A. K.; Bhojwani, S. S.; Bisaria, V. S.; J. Biosci. Bioeng.2002, 93, 215. DOI: http://dx.doi.org/10.1016/S1389-1723(02)80017-2 PMID: 16233190 38. Puri, S. C.; Nazir, A.; Chawla, R.; Arora, R.; Hasan, S. R.; Amna, T.; Ahmed, B.; Verma, V.; Singh, S.; Sagar, R.; Sharma, A.; Kumar, R.; Sharma, R. K.; Qazi, G. N.; J. Biotechnol.2006, 122, 494; Huang, J-X.; Zhang, J.; Zhang, X-R.; Zhang, K.; Zhang, X.; He, X-R.; Pharm. Biol. 2014, 52, 1237. DOI: http://dx.doi.org/10.1016/j.jbiotec.2005.10.015 PMID: 16375985 39. Khaled, M.; Jiang, Z. Z.; Zhang, L. Y.; J. Ethnopharmacol. 2013, 149, 24. DOI: http://dx.doi.org/10.1016/j.jep.2013.06.021 PMID: 23792585 40. Witterland, A. H.; Koks, C. H.; Beijnen, J. H.; Pharm. World Sci.1996, 18, 163. DOI: http://dx.doi.org/10.1007/BF00820727 PMID: 8933576 41. Hande, K. R.; Update on Cancer Therapeutics2006, 1, 3. DOI: http://dx.doi.org/10.1016/j.uct.2006.04.001 42. Damayanthi, Y.; Lown, J. W.; Curr. Med. Chem. 1998, 5, 205; You, Y.; Curr. Pharm. Des.2005, 13, 1695. PMID: 9562603 43. Mann, J.; Nat. Rev. Cancer. 2002, 2, 143. DOI: http://dx.doi.org/10.1038/nrc723 PMID: 12635177 44. Hande, K. R.; Eur. J. Cancer. 1998, 34, 1514. DOI: http://dx.doi.org/10.1016/S0959-8049(98)00228-7 PMID: 9893622 45. Larsen, K. A.; Escargueil, A. E.; Skladanowski, A.; Pharmacol. Ther. 2003, 99, 167. DOI: http://dx.doi.org/10.1016/S0163-7258(03)00058-5 PMID: 12888111 46. Bewer, C. F.; Loike, J. D.; Horwitz, S. B.; Sternlicht, H.; Gensler, W. J.; J. Med. Chem. 1979, 22, 215; Issel B. F., Muggia F. M.; Carter S. K. In Etoposide (VP-16) - current status and new developments; Issel, B. F.; Muggia, F. M.; Carter, S. K., eds.; Academic Press: Orlando, 1984. DOI: http://dx.doi.org/10.1021/jm00189a001 47. Allevi, P.; Anastasia, M.; Ciuffreda, P.; Bigatti, E.; Macdonald, P.; J. Org. Chem.1993, 58, 4175. DOI: http://dx.doi.org/10.1021/jo00067a071 48. Sterling, J.; Nudelman, A.; Herzig, J.; Keinan, E.; Weiner, B. Z.; US.4,900,8141986. 49. Wang, Z.; Ma, W.; Zhang, C.; US pat.5,206,3501991. 50. Naidu, R.; US pat.6,384,2012000. 51. Miyazawa, Y.; Sato, H.; Yoshikawa, H.; Ohkawa, K.; Menos, N. T. US5,637,6801995. 52. Silverberg, L .J.; Dillon, J. L.; Vemishetii, P.; Sleezer, P. D.; Discordia, R. P.; Hartung, K. B.; Gao, Q.; Org. Process Res. Dev. 2000, 3, 34. DOI: http://dx.doi.org/10.1021/op990193e |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access