
 |
 |
 |
 |
 |
Artigo
|
|
| Método alternativo para a síntese e mecanismo de 2-(1,3-ditiano-2-ilideno)-acetonitrila Alternative method for synthesis and mechanism of 2-(1,3-dithian-2-ylidene)-acetonitrile |
|
Marcelle S. Ferreira; José D. Figueroa-Villar*
Departamento de Química, Instituto Militar de Engenharia, Praça General Tiburcio 80, 22290-270 Rio de Janeiro - RJ, Brasil Recebido em 18/08/2014 *e-mail: jdfv2009@gmail.com We report an alternative method for the synthesis of 2-(1,3-dithian-2-ylidene)-acetonitrile using 3-(4-chlorophenyl)-3-oxopropanenitrile and carbon disulfide as starting materials. The methanolysis of the intermediate 3-(4-chlorophenyl)-2-(1,3-dithian-2-ylidene)-3-oxopropanenitrile occurs via three possible intermediates, leading to the formation of the product at a 75% overall yield. Molecular modeling simulation of the reaction pathway using B3LYP 6-311G++(2df,2p) justified the proposed reaction mechanism. INTRODUÇÃO Ceteno ditioacetais são intermediários versáteis na síntese orgânica,1 que têm sido utilizados com sucesso na construção de compostos heterocíclicos e aromáticos e na preparação de intermediários orgânicos.2,3 O uso mais comum dos ceteno ditioacetais é na síntese de pirimidinas,3 tiofenos,4-6 tioacetamidas,5 piridinas,7-9 piridonas,10 furanos,11 β-lactamas,12 quinolonas13 e compostos heterocíclicos indeno fundidos.14 De fato, ceteno ditioacetais têm sido usados no desenvolvimento de novas drogas, como na introdução de grupos CO2H em novos inibidores de neuraminidases15 e na síntese de produtos naturais bioativos.16 Um interessante ceteno ditioacetal para a síntese de novos compostos heterocíclicos é o 2-(1,3-ditiano-2-ilideno)-acetonitrila (1), que pode ser submetido à alquilação no carbono-α17 ou redução seletiva do grupo ciano,18 como no Esquema 1.
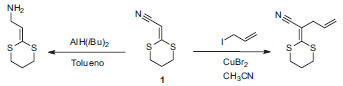 Esquema 1. Alquilação e redução seletiva do 2-(1,3-ditiano-2-ilideno)-acetonitrila (1)17,18
A síntese do composto 1 foi reportada na literatura utilizando somente dois procedimentos diferentes, como mostrado no Esquema 2.1,19 A primeira síntese do 1 foi reportada por Creemer e colaboradores usando a reação do enolato de lítio da acetonitrila com dissulfeto de carbono, seguida por alquilação com 1,3-dibromopropano,19 como mostrado no Esquema 2. Neste primeiro procedimento, o composto 1 foi obtido com 40% de rendimento. Em 2007, Yin e colaboradores desenvolveram uma rota sintética mais eficiente para a preparação deste composto por meio de uma deacilação do α-acetil-α-(aminocarbonil)ceteno ditioacetal, seguido por desidratação do α-(aminocarbonil) ceteno ditioacetal com POCl3.1 Neste caso, o rendimento total do composto 1 foi de 71%.
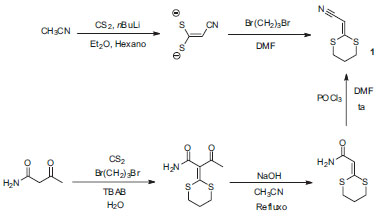 Esquema 2. Síntese de 2-(1,3-ditiano-2-ilideno)-acetonitrila (1) por Creemer et col. a partir de acetonitrila19 e por Yin et col. a partir de α-acetil-α-(aminocarbonil) ceteno ditioacetal1
Em 2009, Wang e colaboradores utilizaram a 2-(1,3-ditiano-2-ilideno)-acetonitrila (1) e análogos como intermediários para a síntese de cumarinas por meio de reação com salicialdeído, utilizando CuBr2 como catalisador.20 Devido ao pequeno número de artigos publicados sobre a síntese de 2-(1,3-ditiano-2-ilideno)-acetonitrila (1) e o nosso interesse no seu uso para a preparação de novos compostos heterocíclicos com potencial para o tratamento de malária e hanseníase, o objetivo deste trabalho foi o desenvolvimento de um método alternativo mais simples e eficiente para a síntese de 2-(1,3-ditiano-2-ilideno)-acetonitrila (1).
EXPERIMENTAL Os pontos de fusão foram determinados em um aparelho Fisher-Johns. Os espectros de RMN foram determinados em um espectrômetro Varian UNITY-300 (300 MHz para 1H e 75 MHz para 13C), usando CDCl3 como solvente e TMS como padrão interno. Os espectros de infravermelho (IV) foram medidos usando um espectrômetro Shimadzu 21, com amostras preparadas com pastilhas de brometo de potássio anidro (KBr). As análises por cromatografia em camada fina foram realizadas utilizando cromatofolhas de alumínio 60 F254 da Merck e por cromatografia em coluna utilizando como fase estacionária sílica gel de 40-63 µm (230 - 400 mesh) da Vetec e como fase móvel uma mistura de hexano e acetato de etila (7:3). Síntese da 3-(4-clorofenil)-2-(1,3-ditiano-2-ilideno)-3-oxopropanonitrila (3) Em um balão de fundo redondo de 100 mL foram adicionados 1,80 g (10 mmol) de 3-(4-clorofenil)-3-oxopropanonitrila (2), 4,14 g de K2CO3 dissolvidos em 20 mL de DMF. A mistura reacional foi mantida sob agitação magnética por 30 min à 25 ºC. Em seguida, foi adicionado 0,90 mL (15 mmol) de dissulfeto de carbono em um banho de gelo sob agitação. Após 30 min de reação, foi adicionado lentamente 1,22 mL (12 mmol) de 1,3-dibromopropano à 25 ºC e a reação foi mantida sob agitação por 24 h. Depois foi adicionada à reação água fria (20 mL) e deixou sob agitação por mais 20 min. Após o término da reação, o produto foi extraído com diclorometano (3x 20 mL) e a fase orgânica seca com sulfato de sódio anidro, filtrada e concentrada a vácuo. O produto bruto, um sólido laranja, foi purificado por recristalização em água destilada. Síntese da 2-(1,3-ditiano-2-ilideno)-acetonitrila (1) Em um balão de fundo redondo de 100 mL foram adicionados 0,400 g (1,4 mmol) de 3-(4-clorofenil)-2-(1,3-ditiano-2-ilideno)-3-oxopropanonitrila (2) dissolvidos em 15 mL de THF seco, 0,140 g (20 mmol) de sódio e 15 mL de metanol seco sob atmosfera de nitrogênio. A mistura reacional foi mantida sob agitação à 25 ºC por 48 h. Em seguida, a mistura reacional foi dissolvida em 30 mL de água destilada e extraída com acetato de etila (3 x 20 mL). A fase orgânica foi seca em sulfato de sódio anidro, filtrada e concentrada a vácuo para se obter o produto bruto, que foi purificado por cromatografia em coluna (silica gel e hexano:acetato de etila 7:3). 3-(4-clorofenil)-2-(1,3-ditiano-2-ilideno)-3-oxopropanonitrila (3): Cristal amarelo. Rendimento: 95%, 2,80 g, pf 158-160 ºC, lit.21 159-160 ºC; IV (KBr, cm -1): 2198 (CN), 1612 (C=O), 1585, 1560 (aromático), 678 cm -1 (C-S); 1H RMN (300 MHz, CDCl3) δ 2,38 (m, J 6,9, 2H, CH2); 3,01 (t, J 6,6, 2H, SCH2); 3,17 (t, J 7,2 , 2H, SCH2); 7,43 (d, J 8,5, 2H); 7,83 (d, J 8,5, 2H); 13C RMN (75 MHz, CDCl3) δ 23,9 (CH2), 30,4 (SCH2), 104,2 (CCO), 117,5 (CN), 128,9, 130,5, 135,6, 139,2 (aromático), 185,2 (C=CS), 185,4 (CO). 2-(1,3-ditiano-2-ilideno)-acetonitrila (1): Cristal branco. Rendimento: 75%, 165 mg, pf. 60-63 ºC, lit1 60-62 ºC; 1H RMN (300 MHz, CDCl3) δ 2,23 (m, J 6,8, 2H, CH2); 3,01 (t, J 7,5, 2H, SCH2); 3,06 (t, J 6,9, 2H, SCH2), 5,39 (s, 1H, CH); 13C RMN (75 MHz, CDCl3) δ 22,9 (CH2), 28,7 (SCH2), 28,8 (SCH2), 90,4 (CHCN), 116,3 (CN), 163,8 (C=CS). Modelagem molecular Os cálculos de modelagem molecular de todas as moléculas, intermediários (INT) e estados de transição (ET) foram realizados na forma isolada (vácuo) utilizando o pacote de programas Spartan 06 (Windows).22 Os materiais de partida, intermediários, estados de transição e a geometria e energia dos produtos foram calculados utilizando o método B3LYP com o conjunto de bases 6-311++G(2df,2p). O caminho reacional foi confirmado utilizando o método de coordenada de reação intrínseca (IRC).
RESULTADOS E DISCUSSÃO O método desenvolvido neste trabalho para a preparação da 2-(1,3-ditiano-2-ilideno)-acetonitrila (1) consiste em duas etapas, como mostrado no Esquema 3. Foi utilizado como material de partida a 3-(4-clorofenil)-3-oxopropanonitrila (2) com CS2 dissolvido em DMF, seguido por alquilação com 1,3-dibromopropano, obtendo a 3-(4-clorofenil)-2-(1,3-ditiano-2-ilideno)-3-oxopropanonitrila (3) em excelente rendimento (95%). Este composto é muito similar aos diversos cetenos ditioacetais preparadas por Mellor e colaboradores com rendimentos mais baixos.23 Interessantemente, os procedimentos reportados por Mellor para a redução seletiva da dupla ligação carbono-carbono de vários destes ceteno ditioacetais utilizando Zn em ácido acético ou Mg em metanol23 não tiveram sucesso com o composto 3.
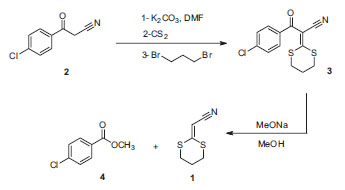 Esquema 3. Nova síntese da 2-(1,3-ditiano-2-ilideno)-acetonitrila (1)
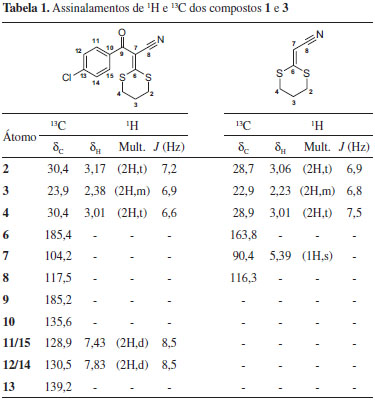
Para a preparação da 2-(1,3-ditiano-2-ilideno)-acetonitrila (1) foi realizada uma reação de metanólise. A 3-(4-clorofenil)-2-(1,3-ditiano-2-ilideno)-3-oxopropanonitrila (3) foi tratada com metóxido de sódio em metanol à 25 ºC por 48 h, levando à formação do 1 com 75% de rendimento e produzindo o 4-clorobenzoato de metila (4) como produto secundário. Nessa reação a 3-(4-clorofenil)-3-oxopropanonitrila foi selecionada por ser ativa para ataque nucleofílico de metóxido. Os compostos 1 e 3 foram caracterizados utilizando técnicas espectroscópicas de infravermelho e RMN de 1H e 13C. Os dados dos assinalamentos de RMN de 1H e 13C são mostrados na Tabela 1.
O mecanismo proposto para a formação da 2-(1,3-ditiano-2-ilideno)-acetonitrila (1) por meio de uma reação do composto 3 com MeONa é mostrado no Esquema 4. A primeira etapa da metanólise ocorre a partir do ataque nucleofílico do ânion metóxido para a carbonila do ceteno, formando o primeiro intermediário (INT1) através do primeiro estado de transição (ET1). Então, o INT1 sofre degradação e ocorre a perda do éster (4). Esta etapa pode conduzir a formação de três intermediários diferentes; INT2a, INT2b e INT2c. Assim, os três intermediários poderiam participar no processo da reação, mas seguindo vias de finalização diferentes. Uma simples protonação do intermediário INT2a leva ao produto final (1), enquanto o intermediário INT2b deve executar a protonação com movimento eletrônico a partir do nitrogênio, mas possivelmente com a mesma energia que INT2a. Por outro lado, a transformação do intermediário INT2c para o produto 1 inclui um ataque nucleofílico do RS- ao carbono, seguido da protonação. De acordo com a energia relativa de cada um dos intermediários, o processo mais favorável pode ser por meio do intermediário INT2a, no qual ocorre sua protonação de forma direta, mas os intermediários INT2b e INT2c, cuja protonação inclui mais etapas, como possuem energia igual, certamente têm a possibilidade de participarem no mecanismo, pelo menos na estabilização da carga negativa do intermediário.
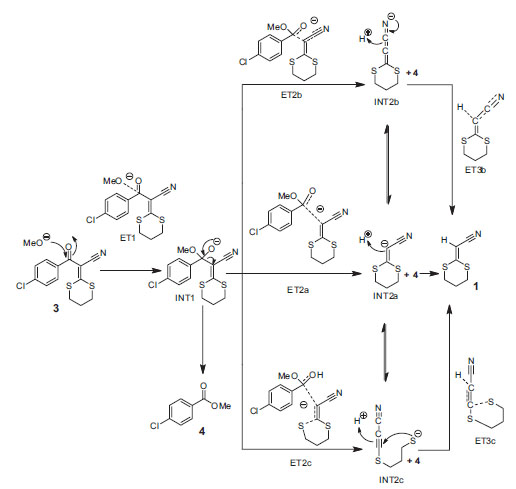 Esquema 4. Proposta mecanística para a formação do composto 1
Para confirmar esse mecanismo da última etapa reacional (metanólise) foi simulado por modelagem molecular usando B3LYP e o conjunto de bases 6-311++G(2df,2p) com Spartan'06.22 A energia relativa calculada dos intermediários propostos INT2a, INT2b e INT2c foi a mesma (1,128 kcal mol -1), confirmando que os três intermediários podem participar do processo reacional. Os componentes das vias da reação são mostrados na Figura 1 e sua participação no mecanismo proposto da reação foi confirmada pelo cálculo de coordenada de reação intrínseca (IRC).24-26
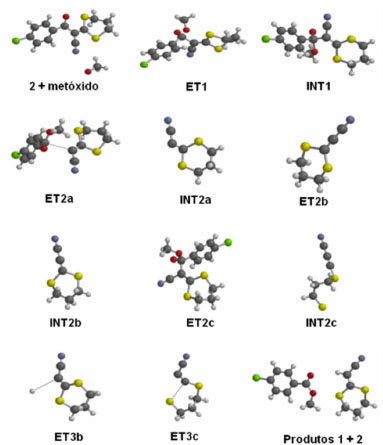 Figura 1. Cálculo dos componentes do mecanismo proposto para a reação de conversão do 3-(4-clorofenil)-2-(1,3-ditiano-2-ilideno)-3-oxopropanonitrila (2) para 2-(1,3-ditiano-2-ilideno)-acetonitrila (1) por metanólise
Os intermediários da segunda etapa do processo de metanólise, os ânions INT2a, INT2b e INT2c têm uma energia calculada muito semelhante (-6,79 × 105 para -6,77 × 105 kcal mol -1), com INT2a e INT2c tendo a mais baixa energia. Esses resultados indicam que os três intermediários são capazes de participar do processo de metanólise, mas com ênfase nos intermediários INT2a e INT2c. A variação da energia relativa dos diferentes intermediários e estados de transição da reação para a formação do composto 1 se encontra na Figura 2, mostrando que a proposta mecanística da reação está correta.
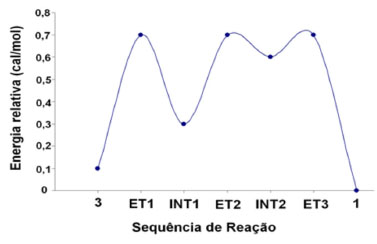 Figura 2. Energia relativa e sequência de reação para a formação do composto 1
Em termos gerais, a metodologia utilizada na preparação da 2-(1,3-ditiano-2-ilideno)-acetonitrila (1) e os resultados de modelagem molecular confirmam o proposto mecanismo da reação, inclusive indicando que basicamente todos os estados de transição (ET) e intermediários (INT), por possuírem energias basicamente similares, com a mesma estabilidade, contribuem com a estabilização do ânion, participando no processo da síntese, como mostra a Figura 2.
CONCLUSÃO Neste trabalho foi desenvolvido um método alternativo para a preparação de 2-(1,3-ditiano-2-ilideno)-acetonitrila (1) que consiste em duas etapas. Este método sintético é mais simples e eficiente e isto facilita o uso do composto 1 como intermediário para a síntese de novos compostos heterocíclicos. Para o completo entendimento desta reação foi proposto um mecanismo de reação de metanólise da 3-(4-clorofenil)-2-(1,3-ditiano-2-ilideno)-3-oxopropanonitrila (2), que foi reforçado por cálculos de modelagem molecular da reação usando o método IRC com DFT por B3LYP. Esta última etapa do processo sintético ocorre em duas fases, iniciando com a formação do primeiro intermediário (INT1) por meio de um único estado de transição (ET1). A transformação do INT1 para os compostos 1 e 2 pode ocorrer por três vias diferentes. A primeira ocorre por meio da formação do segundo estado de transição ET2a, o que leva à formação do intermediário INT2a e o composto 4 como produto secundário. A simples protonação do INT2a leva diretamente ao produto 1. A segunda via ocorre a partir da formação de um diferente estado de transição ET2b, o qual é convertido no respectivo intermediário INT2b. A conversão do INT2b para o produto 1 deve ocorrer por meio de um terceiro estado de transição (ET3b). A terceira via alternativa é a conversão do INT1 ao intermediário INT2c através do respectivo estado de transição (ET2c), seguido para à conversão do produto 1 via outro estado de transição (ET3c). Os resultados do IRC indicam que a via preferencial para o processo de metanólise para produzir o composto 1 ocorre pela protonação do INT2a. As vias, com participação dos outros dois intermediários (INT2b e INT2c) são também possíveis, mas porque incluem um maior nível de energia e o seu potencial do terceiro estado de transição é menor. Em termos gerais, esses resultados mostram que este método sintético é eficiente e possui bom rendimento, e o mecanismo proposto é confirmado pelos cálculos de modelagem molecular. Essa metodologia vai ser futuramente aplicada na preparação de novos fármacos.
MATERIAL SUPLEMENTAR O material suplementar, disponível em http://quimicanova.sbq.org.br, na forma de arquivo PDF, com acesso livre, apresenta os espectros de RMN de 1H e 13C e infravermelho dos compostos sintetizados.
AGRADECIMENTOS Nós agradecemos ao INBEB, CNPq, CAPES e FAPERJ pelo apoio financeiro.
REFERÊNCIAS 1. Yin, Y.; Zangh, Q.; Liu, Q.; Liu, Y.; Sun, S.; Synth. Commun.2007, 37, 703. 2. Xu, X-X; Wang, M.; Liu, Q.; Pan, L.; Zhao, Y-L.; Chin. J. Chem2006, 24, 1431 DOI: http://dx.doi.org/10.1002/cjoc.200690004 3. Metwally, M. A.; Abdel-Latif, E.; J. Sulfur Chem.2004, 25, 359. DOI: http://dx.doi.org/10.1080/17415990412331300025 4. Wang, Y.; Dong, D.; Yang, Y.; Huang, J.; Ouyang, Y.; Liu, Q.; Tetrahedron2007, 63, 2724. DOI: http://dx.doi.org/10.1016/j.tet.2006.10.044 5. Liang, F.; Li, D.; Zhang, L.; Gao, J.; Liu. Q.; Org. Lett.2007, 9, 4845. DOI: http://dx.doi.org/10.1021/ol7021752 PMID: 17929827 6. Thomae, D.; Perspicace, E.; Henryon, D.; Xu, Z.; Schneider, S.; Hesse, S.; Kirsch, G.; Seck, P. Tetrahedron2009, 65, 10453. DOI: http://dx.doi.org/10.1016/j.tet.2009.10.021 7. Hu, J.; Zhang, Q.; Yuan, H.; Liu, Q.; J. Org. Chem. 2008, 73, 2442. DOI: http://dx.doi.org/10.1021/jo702586p PMID: 18290663 8. Hagimori, M.; Mizuyama, N.; Hisadome, Y.; Nagaoka, J.; Uedab, K.; Tominagab, Y.; Tetrahedron2007, 63, 2511. DOI: http://dx.doi.org/10.1016/j.tet.2006.12.031 9. Thomas A. D.; Asokan, C. V.; Tetrahedron Lett.2002, 43, 2273. DOI: http://dx.doi.org/10.1016/S0040-4039(02)00174-0 10. Tominaga, Y.; Matsuda, Y.; J. Heterocycl. Chem. 1985, 22, 937. DOI: http://dx.doi.org/10.1002/jhet.5570220401 11. Padwa, A.; Eidell, C. K.; Ginn, J. D.; McClure, M. S.; J. Org. Chem. 2002, 67, 1595. DOI: http://dx.doi.org/10.1021/jo010986a PMID: 11871892 12. Yang, Y.; Xiang, D.; Zhao, X.; Liang, Y.; Huang, J.; Dong, D.; Tetrahedron2008, 64, 4959. DOI: http://dx.doi.org/10.1016/j.tet.2008.03.093 13. Cheng, D.; Zhou, J.; Saiah, E.; Beaton, G.; Org. Lett.2002, 4, 4411. DOI: http://dx.doi.org/10.1021/ol020179d PMID: 12465900 14. Verma, R. K.; Ila, H.; Singh, M. S.; Tetrahedron2010, 66, 7389. DOI: http://dx.doi.org/10.1016/j.tet.2010.07.031 15. Chand, P.; Babu, Y. S.; Bantia, S.; Rowland, S.; Dehghani, A.; Kotian, P. L.; Hutchison, T. L.; Ali, S.; Brouillette, W.; El-Kattan, W.; Lin, T-S.; J. Med. Chem.2004, 47, 1919. DOI: http://dx.doi.org/10.1021/jm0303406 PMID: 15055992 16. Miguel, Y.; Carmen, N.; Francisco F.; Tetrahedron2003, 59, 6147. DOI: http://dx.doi.org/10.1016/S0040-4020(03)00955-4 17. Yuan, H-J.; Wang, M.; Liu, Y-J.; Liua, Q.; Adv. Synth. Catal. 2009, 351, 112 . DOI: http://dx.doi.org/10.1002/adsc.200800584 18. Bargar, T. M.; Broersma, R. J.; Creemer, L. C.; McCarthy, J. R.; Hornsperger, J. M.; Attwood, P.V.; Jung, M. J.; J. Am. Chem. Soc.1988, 110, 2975. DOI: http://dx.doi.org/10.1021/ja00217a050 19. Creemer, L. C.; Bargar, T. M.; Wagner, E. R.; Synth. Commun. 1988, 18, 1103. DOI: http://dx.doi.org/10.1080/00397918808060896 20. Wang, M.; Liu, Q.; Yuan, H-Y.; Liu, Y-L.; Adv. Synth. Catal.2009, 351, 112. DOI: http://dx.doi.org/10.1002/adsc.200800584 21. Rudorf, W. D.; Augustin, M.; Phosphorus Sulfur Relat. Elem.1981, 9, 329. DOI: http://dx.doi.org/10.1080/03086648108078258 22. Shao, Y.; Molnar, L. F.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S. T.; Gilbert, A. T. B.; Slipchenko, L. V.; Levchenko, S. V.; O'Neill, D. P.; DiStasio J., R. A.; Lochan, R. C.; Wang, T.; Beran, G. J. O.; Besley, N. A.; Herbert, J. M.; Lin, C. Y.; Van Voorhis, T.; Chien, S. H.; Sodt, A.; Steele, R. P.; Rassolov, V. A.; Maslen, P. E.; Korambath, P. P.; Adamson, R. D.; Austin, B.; Baker, J.; Byrd, E. F. C.; Dachsel, H.; Doerksen, R. J.; Dreuw, A.; Dunietz, B. D.; Dutoi, A. D.; Furlani, T. R.; Gwaltney, S. R.; Heyden, A.; Hirata, S.; Hsu, C-P.; Kedziora, G.; Khalliulin, R. Z.; Klunzinger, P.; Lee, A. M.; Lee, M. S.; Liang, W. Z.; Lotan, I.; Nair, N.; Peters, B.; Proynov, E. I.; Pieniazek, P. A.; Rhee, Y. M.; Ritchie, J.; Rosta, E.; Sherrill, C. D.; Simmonett, A. C.; Subotnik, J. E.; Woodcock III, H. L.; Zhang, W.; Bell, A. T.; Chakraborty, A. K.; Chipman, D. M.; Keil, F. J.; Warshel, A.; Hehre, W. J.; Schaefer, H. F.; Kong, J.; Krylov, A. I.; Gill, P. M. W.; Head-Gordon, M.; Phys. Chem. Chem. Phys. 2006, 8, 3172. DOI: http://dx.doi.org/10.1039/b517914a PMID: 16902710 23. Mellor, J. M.; Schofield, S. R.; Tetrahedron1997, 53, 17151. DOI: http://dx.doi.org/10.1016/S0040-4020(97)10136-3 24. Fukui, K.; Acc. Chem. Res.1981, 14, 363. DOI: http://dx.doi.org/10.1021/ar00072a001 25. Deng, L.; Ziegler, T.; J. Chem. Phys.1993, 99, 3823. DOI: http://dx.doi.org/10.1063/1.466129 26. Deng, L.; Ziegler, T.; Int. J. Quantum Chem.1994, 52, 731. DOI: http://dx.doi.org/10.1002/qua.560520406 |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access