
 |
 |
 |
 |
 |
Artigo
| Isolation and characterization of nanofibrillated cellulose from oat hulls |
|
Giovanni B. PaschoalI; Carmen M. O. MullerII; Gizilene M. CarvalhoIII; Cesar A. TischerI; Suzana MaliI,*
IDepartamento de Bioquímica e Biotecnologia, Centro de Ciências Exatas, Universidade Estadual de Londrina, CP 6001, 86051-990 Londrina - PR, Brasil Recebido em 18/06/2014 *e-mail: smali@uel.br The objectives of this work were to investigate the microstructure, crystallinity and thermal stability of nanofibrillated cellulose obtained from oat hulls using bleaching and acid hydrolysis at a mild temperature (45 ºC) followed by ultrasonication. The oat hulls were bleached with peracetic acid, and after bleaching, the compact structure around the cellulosic fibers was removed, and the bundles became individualized. The extraction time (30 or 60 min) did not affect the properties of the nanofibrillated cellulose, which presented a higher crystallinity index and thermal stability than the raw material (oat hulls). The nanocellulose formed interconnected webs of tiny fibers with diameters of 70-100 nm and lengths of several micrometers, producing nanofibers with a relatively high aspect ratio, thus indicating that these materials are suitable for polymer reinforcement. INTRODUCTION The use of biomass residues for production of high performance materials can result in a generation of value-added products from agro-industrial commodities.1-3 Several agro-based materials are waste products from various industrial processes.4,5 For example, oat hulls are a poorly utilized byproduct of oat groat milling and are discarded during processing, rendering them an environmental pollutant. Oat hulls are approximately 90% fiber, which is higher than wheat (47%) or corn bran (62%)6 and are an interesting raw material for use as a source of cellulose and nanocellulose. Over the last few years, some authors have reported advantages regarding the use of nanocellulose as filler in several biodegradable polymeric matrices, resulting in nanocomposites, which present improved properties related to nanocellulose reinforcement.7-9 Two different types of nanocellulose can be isolated from a cellulosic source: nanocrystals and nanofibrils. Nanocrystals have a perfect crystalline structure, and nanofibrils are fibrillar units containing both amorphous and crystalline regions and have the ability to create entangled networks.10 Abdul Khalil et al.10 reported that the term nanofibrillated cellulose is used to designate elongated rod-like nanoparticles with diameters less than 100 nm that consist of alternating crystalline and amorphous domains, and these materials have interesting properties such as high strength, flexibility and high aspect ratio, defined as the length to diameter ratio. Nanofibrillated cellulose derived from wood using a conventional high shear homogenizer without the use of chemical treatments was first reported more than two decades ago.11,12 Nanofibrillated cellulose has been prepared from a variety of sources using several mechanical processes, such as high pressure homogenization,13 grinding,14 microfluidization,15 cryocrushing16,17 and high intensity ultrasonication.18 Chemical or enzymatic treatments can be used before or after the mechanical process.19,20 It is worth noting that appropriate chemical pretreatments of cellulosic fibers promote the accessibility of hydroxyl groups, increase the inner surface, alter crystallinity, and break cellulose hydrogen bonds.21 Zimmerman et al.22 applied an acid hydrolysis step with sulfuric acid before pumping the sulfite pulp through the homogenizer to obtain nanofibrillated cellulose. The sulfuric acid treatment, combined with mechanical dispersion, resulted in finer fibril structures with diameters below 50 nm, with lengths in the micrometer range. Qua et al.17 reported the production of cellulose nanofibrils derived from microcrystalline cellulose and flax fibers using acid hydrolysis, combined with the application of ultrasound and high pressure microfluidization. The objectives of this work were investigate the microstructure, crystallinity and thermal stability of nanofibrillated cellulose obtained from oat hulls using bleaching and acid hydrolysis at a mild temperature (45 ºC) followed by ultrasonication.
EXPERIMENTAL SECTION Materials Unpurified oat hulls (OH), containing approximately 48.0, 25.5 and 14.5% of cellulose, hemicelluloses and lignin, respectively, were kindly supplied by SL-Alimentos (Mauá da Serra-PR, Brazil). Bleaching of oat hulls Approximately 20 g of raw oat hulls were dispersed in 250 mL of a peracetic acid solution (50% acetic acid (Synth - Brazil), 38% hydrogen peroxide (Synth - Brazil) and 12% distilled water) at 60 ºC and vigorously stirred for 24 h. After this treatment, the fibers were vacuum filtered, washed with distilled water until the pH value was between 6 and 7 and dried at 35 ºC for 12 - 24 h in an air-circulating oven (Tecnal - São Paulo-Brazil). The bleached oat hull was labeled as OHB. Preparation of the nanofibrillated cellulose from oat hulls Approximately 10 g of bleached oat hulls were dispersed in 100 mL of 63.7% (w/v) sulfuric acid (Synth - Brazil) at 45 ºC and vigorously stirred for 30 or 60 min. Then, 200 mL of cold distilled water was added to stop the reaction. The sulfuric acid was partially removed from the resulting suspension through centrifugation at 10,000 rpm for 10 min. The non-reactive sulfate groups were removed using centrifugation followed by dialysis in tap water with a cellulose membrane (Sigma-Aldrich: D9402) until the pH value was between 6 and 7. Dialysis was performed in running distilled water during 3-4 days. The neutral suspension was ultrasonicated (Ultrasonic Processor - Fisher Scientific - USA) for 15 min and stored in a refrigerator after adding two drops of chloroform. The nanofibrillated cellulose samples were labeled OHNF30 or OHNF60, depending on the extraction time. For X-ray diffraction, Fourier transform-infrared spectroscopy, thermogravimetric and nuclear magnetic resonance spectroscopy, a 50 mL aliquot was dried at 35 ºC for 12 h in an air-circulating oven (Tecnal - São Paulo-Brazil). The yield of the acid hydrolysis was determined by weighing an aliquot of 10 mL of the supernatant of the suspension after being allowed to stand overnight to dry. The yield (%) was calculated from the difference between the initial and final weight. The lignin content of OHB, OHNF30 and OHNF60 was determined according to the standard method of the Technical Association of Pulp and Paper Industry TAPPI T222 om-88.23 Nanofibrillated cellulose characterization Colorimetry The sample colors were determined using a colorimeter (CR 10, Minolta Chroma Co., Osaka, Japan). The color parameters range from L = 0 (black) to L = 100 (white), -a (greenness) to +a (redness), and -b (blueness) to +b (yellowness). The instrument was calibrated using a set of three Minolta calibration plates. The reported values are the averages from five measurements of each sample. Scanning electron microscopy (SEM) SEM analyses were performed with a FEI Quanta 200 microscope (Oregon, USA) to observe the morphology of OH and OHB. The dried samples were mounted for visualization on bronze stubs using double-sided tape. The surfaces were then coated with a thin gold layer (40-50 nm). All samples were examined using an accelerating voltage of 30 kV. Transmission electron microscopy (TEM) The suspensions of OHNF30 and OHNF60 were observed by TEM using a FEI-TECNAI 12 transmission electron microscope (Oregon - USA) with an acceleration voltage of 80 kV. A droplet of diluted suspension was deposited on a carbon coated grid and allowed to dry. The grid was stained with a 1.5% solution of uranyl acetate and dried at room temperature. The diameters of the nanofibrillated cellulose from oat hulls were determined using an image analysis program (ImageJ 1.37v®), and a minimum of 30 measurements were performed for this determination. X-ray diffraction The crystallinity of each sample was investigated using X-ray diffraction (XRD). The samples were finely powdered (particles < 0.149 mm), and the analysis was performed using a PANalytical X´Pert PRO MPD diffractometer (Netherlands) with copper Kα radiation (λ = 1.5418 Å) under the operational conditions of 40 kV and 30 mA. All assays were performed with a ramp rate of 1º/min. The relative crystallinity index (CI) was calculated using the Ruland method24 as follows: CI = ((Ac)/(Ac+Aa)), where Ac is the crystalline area and Aa is the amorphous area. Fourier Transform-Infrared Spectroscopy (FT-IR) The pulverized and dried samples were then mixed with potassium bromide and compressed into tablets. The FT-IR analyses were carried out with a Shimadzu FT-IR - 8300 (Japan), which has a spectral resolution of 4 cm-1 and a spectral range of 4000 - 500 cm-1. Thermogravimetric analysis (TGA) Thermogravimetric analysis (TGA 50 - Shimadzu - Japan) was carried out under a nitrogen atmosphere (50 mL min-1), and the samples (approximately 10 mg) were heated from 0 to 600 ºC at a heating rate of 10 ºC/min. The weight loss (%) was evaluated by measuring the residual weight at 600 ºC. Solid state NMR spectroscopy (13 C CP MAS NMR) Solid state 13C spectra were recorded on a Bruker (Avance III) instrument operating at 400 MHz. A multinuclear probe (4 mm) and a zirconium oxide rotor were used with a Cross Polarized - Magic Angle Spinning (CP-MAS) unit. The relative crystallinity index (CI) was measured as the ratio between the amorphous and crystalline C4 regions, which was obtained through integration.
RESULTS AND DISCUSSION The original lignin content of OH (14.5%) decreased in all samples, the lignin contents of OHB, OHNF30 and OHNF75 were 3.50 ± 0.80, 1.12 ± 0.50 and 1.10 ± 0.32%, respectively, showing that the bleaching and acid hydrolysis were effective for cellulose purification. The yield of acid hydrolysis was 32 ± 5% (by dry mass); each 100 g of raw oat hulls resulted in 32 g of nanofibrillated cellulose. Figure 1 shows the aspect of the oat hulls (OH) both before and after bleaching with peracetic acid. After bleaching, the fiber was discolored and turned into a white color, indicating that most of the initial non-cellulosic components were removed by this purification process and that bleached oat hulls have higher cellulose content than raw oat hulls. This result agreed with the color parameter results (Table 1). The luminosity (L* parameter) was indicative of the whiteness of these samples, and in the bleached oat hull (OHB), this parameter significantly (p < 0.05) increased compared to the raw oat hulls (OH), confirming the tendency of bleached samples to have a white color.
 Figure 1. Appearance of the raw oat hulls (A) and bleached oat hulls (B)
The increase in the a* and b* color parameters were related to the redness and yellowness of the samples, respectively, and in this work, these color parameters (Table 1) significantly (p < 0.05) decreased in the OHB samples compared to those. The hydrolysates (OHNF30 and OHNF60) presented similar results for the color parameters (Table 1) to those of the OHB samples. Morphological analyses (SEM and TEM) The morphology of the longitudinal surface of the fiber before and after bleaching is shown in Figure 2. The original fiber forms a compact structure (Figure 2a) with the nonfibrous components (hemicellulose and lignin). After bleaching, some of the components around the fiber bundles were removed and these bundles become individualized (Figure 2b), and the microfibrils could be visualized. In plant cells, lignin and hemicelluloses are deposited between the cellulosic microfibrils, which results in an interrupted lamellar structure,20 and the removal of these non-cellulosic components could be accomplished using bleaching agents.
 Figure 2. Micrographs obtained using SEM: raw oat hulls (a) and bleached oat hulls (b)
The suspensions that result from the acid hydrolysis were very stable, and there was no sedimentation when they were stored at room temperature for a long time. TEM micrographs of the nanofibrillated oat hulls are shown in Figure 3 and display the homogeneity and nanometric dimensions of these materials.
 Figure 3. Micrographs obtained using TEM of the nanofibrillated cellulose: extracted at 30 min (a) and 60 min (b)
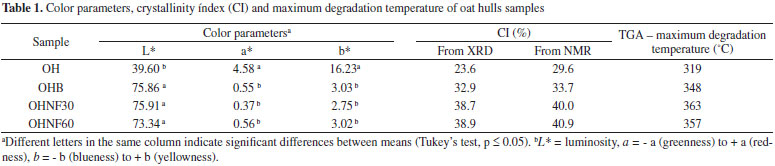
The hydrolysis time did not influence the morphological properties of the nanofibrillated oat hulls (Figure 3). The nanocellulose obtained in our work showed an aspect of interconnected webs of tiny nanofibers with diameters of 70-100 nm and lengths of several micrometers, which results in nanofibers with relatively high aspect ratio. Zhou et al.8 reported that these types of materials are suitable for polymer reinforcement. X-ray Diffraction The X-ray diffraction patterns of OH, OHB, OHNF30 and OHNF60 are shown in Figure 4. These patterns are typical of semicrystalline materials with an amorphous broad hump and crystalline peaks. The OH sample showed a lower crystallinity index (CI) than the bleached (OHB) and hydrolyzed (OHNF30 and OHNF60) samples (Table 1). This lower CI of OH sample occurred because of the reduction and removal of amorphous, non-cellulosic compounds, which was induced by bleaching and acid hydrolysis. According to Abraham et al.,20 lignin removal results in an increase of crystallinity index.
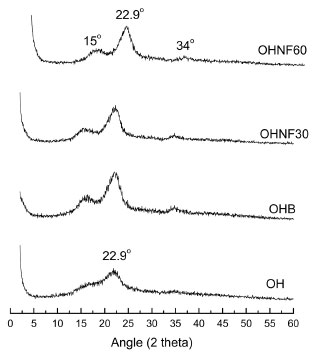 Figure 4. X-ray diffractograms of OH, OHB, OHNF30 and OHNF60
The CI values of hydrolyzed samples (OHNF30 and OHNF60) were similar between each other (Table 1), and these results were consistent with TEM analysis, which showed the same morphology for OHNF30 and OHNF60. Possible, the time range was not sufficient to result in higher removal of amorphous cellulose at this temperature. The CI values obtained in this work are lower than the values reported by other authors for nanofibrillated cellulose obtained by high pressure defibrillation of 2,2,6,6,-tetramethylpiperidine-1-oxyl radical (TEMPO) oxidized eucalyptus fibers,25 which ranged from 49 to 86%. They are also lower than the values reported by other authors18 for nanofibrillated cellulose from flax fibers obtained by acid hydrolysis combined with ultrasound and high pressure microfluidisation, which ranged from 70 to 77%. According to Križman et al.,26 peracetic acid is an oxidant agent used for fiber bleaching and the main advantage of bleaching with peracetic acid is that a satisfactory degree of whiteness can be obtained at 60 ºC in 40 min at neutral pH without the addition of auxiliary agents, and this implies in lower energy and water consumption during bleaching resulting in much less damage to the fiber. Probably, the bleaching step in our work resulted in less damage of the oat hull fiber, generating materials with low CI. In the OH pattern one peak was observed at 22.9o, and in the other diffractogram profiles (OHB, OHNF30 and OHNF60), peaks were observed at 15o, 22.9o and 34o, which are characteristic of type I cellulose (Figure 4), thus compared with the original fibers, both of the bleached and hydrolyzed samples did not exhibit any variation in their polymorph type. Fourier Transform-Infrared Spectroscopy (FT-IR) Figure 5 shows the FT-IR spectra of OHB, OHNF30 and OHNF60. All of the spectra showed a wide absorption band corresponding to O-H stretching at approximately 3400 cm-1, which could indicate the occurrence of H-bonding interactions in these materials. The peaks observed at 2900 cm-1 correspond to -C-H stretching; H-C-H and -C-O-H conjugated bending vibrations appeared in all spectra (Figure 5).
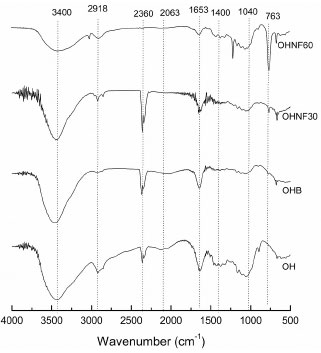 Figure 5. FT-IR spectra of OH, OHB, OHNF30 and OHNF60
For all samples, bands were observed at approximately 1650 cm-1, which are associated with the angular O-H bending of water molecules.27 According to Abraham et al.,20 the water adsorbed in the cellulose molecules is very difficult to extract due to the cellulose-water interaction. Sun et al.28 reported that the band at approximately 1650 cm-1 may result from water, but could also be attributed to the aromatic C=C stretch of the aromatic ring in the lignin. The contribution from the absorbed water predominates in the case of OHB, OHNF30 and OHNF60, but in the raw oat hull (OH), this band could be attributed to the lignin. The bands related to the aromatic C=C stretching of lignin (1513 - 1433 cm1)28 disappeared in the bleached (OHB) and hydrolyzed samples (OHNF30 and OHNF60), indicating that lignin was removed from these samples. Raw oat hulls (OH) showed a band at 1043 cm-1 that corresponds to the linear and branched (1→4)-β-xylans,29 which are present in hemicelluloses. A tiny band appeared in the OHB sample, and this band disappeared in the hydrolysates (Figure 5), which confirms the removal of these non-cellulosic compounds. The differences between the spectra of OH and the other samples (OHB, OHNF30 and OHNF60) were a clear indicative that these samples have higher cellulose content than the raw sample and suggest that they are almost pure cellulose. Thermogravimetric analysis (TGA) The curves obtained from TGA analysis were used to examine the changes in thermal stability caused by the bleaching and acid hydrolysis (Figure 6). A small weight loss (< 10%) was found in the range from 50-100 oC due to the evaporation of water or other low molecular weight compounds from the materials.
 Figure 6. TGA curves of OH, OHB, OHNF30 and OHNF60
The maximum degradation temperature (Table 1) was higher for the bleached (OHB) and hydrolyzed samples (OHNF30 and OHNF60) than the raw material (OH), indicating that the extracted cellulose and nanocellulose had increased thermal stability. These results were related to the different cellulose, hemicellulose and lignin compositions of the samples. Great differences are found in the thermal degradations of cellulose, hemicelluloses and lignin. Hemicelluloses start its decomposition easily, with the weight loss primarily occurring between 220-315 ºC. Cellulose decomposition occurs at a higher temperature range (315-400 ºC). Lignin decomposition happens slowly over a large temperature range from ambient conditions to 900 ºC.30 Our results agreed with Abraham et al.,20 who produced nanofibrillated cellulose from banana, pineapple leaf and jute fibers using alkaline treatment followed by steam explosion and acid hydrolysis. According to these authors, nanocellulose obtained from natural fibers shows higher thermal stability than the cellulose present in untreated lignocellulosic fibers. This occurs probably because the bleached (OHB) and hydrolyzed samples (OHNF30 and OHNF60) were more crystalline than the raw material (OH), and a greater crystalline structure required a higher degradation temperature.31 Solid state NMR spectroscopy (13C CP MAS NMR) The spectrum of the OH sample shows cellulose, traces of starch and other groups consistent with the expected proteins and lignins present in the raw material, including the typical cellulose signals of C1 at δ 106; C4 at δ 86 and δ 90 for amorphous and crystalline cellulose, respectively; and C6 at δ 64 and δ 66 (Figure 7). The integrated area of the C4 signals shows a concentration of 29.6% for the crystalline form of cellulose (Table 1). The direct integration of the C4 signal was hampered by the presence of the starch C4 at δ ~80; another starch signal could be assigned to C1 at δ 102.32 Residual protein was observed from the presence of CH2 signals at δ ~20 - 40, Cα signals at δ ~55, aromatic ring signals at δ ~120 - 140 and finally carbonyl group signals at δ ~175.33 These signals were not observed in the OHB sample. Two additional signals at δ 23.2 and δ 175 correspond to residual acetic acid from the bleaching step. The crystallinity indexes calculated from the NMR data are 33.7% for OHB, 40.0% for OHNF30 and 40.9% for OHNF60 (Table 1), which could be attributed to the selective hydrolysis of the amorphous regions of the cellulose fibrils. The CI values calculated from the NMR are consistent with the XRD values; both analytical techniques showed similar trends for the CI variations, although the CI values obtained using NMR were slightly higher than those of XRD (Table 1).
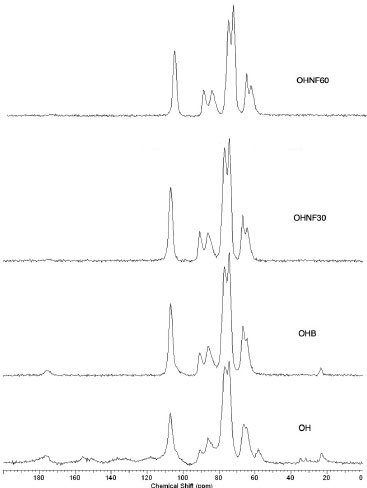 Figure 7. 13C solid state NMR spectra of OH, OHB, OHNF30 and OHNF60
CONCLUSIONS The peracetic acid bleaching treatment was effective for reducing the lignin content in the oat hulls. The compact structure around the cellulosic fibers was also removed, and the bundles become individualized. The extraction time did not affect the properties of the nanofibrillated cellulose, which presented a higher crystallinity index and thermal stability than the raw material, thus 30 min of hydrolysis could be used to produce these materials, requiring lower energy consumption. The nanocellulose crystallized as interconnected webs of tiny nanofibers with diameters of 70-100 nm and lengths of several micrometers, which results in a wide range of aspect ratios, indicating that these materials are suitable for polymer reinforcement. These results showed that this renewable source of agro-industrial residue has promising characteristics, especially as a reinforcing agent in polymer composites.
ACKNOWLEDGEMENTS The authors wish to thank the Laboratory of Microscopy and Microanalysis (LMEM) and the Laboratory of X-Ray Diffraction (LARX) - State University of Londrina for the analyses, and CNPq - Brazil (No. 479768-2012-9) for financial support.
REFERENCES 1. Alemdar, A.; Sain, M.; Bioresource Technol. 2008, 9, 1664. DOI: http://dx.doi.org/10.1016/j.biortech.2007.04.029 2. Purkait, B. S.; Ray, D.; Sengupta, S.; Kar, T.; Mohanty, A.; Misra, M.; Ind. Eng. Chem. Res. 2011, 50, 871. DOI: http://dx.doi.org/10.1021/ie101797d 3. Flauzino Neto, W. P.; Silvério, H. A.; Dantas, N. O.; Pasquini, D.; Ind. Crop. Prod. 2013, 42, 480. DOI: http://dx.doi.org/10.1016/j.indcrop.2012.06.041 4. Satyanarayana, K. G.; Arizaga, G. G. C; Wypych, F.; Progr. Polym. Sci. 2009, 4, 982. DOI: http://dx.doi.org/10.1016/j.progpolymsci.2008.12.002 5. Sun, X. F.; Xu, F.; Sun, R. C.; Fowler, P.; Baird, M. S.; Carbohydr. Res. 2009, 340, 97. DOI: http://dx.doi.org/10.1016/j.carres.2004.10.022 6. Galdeano, M. C.; Grossmann, M. V. E.; Cienc. Tecnol. Aliment. 2006, 26, 123. DOI: http://dx.doi.org/10.1590/S0101-20612006000100021 7. Teixeira, E. M.; Pasquini, D.; Curvelo, A. A. S.; Corradini, E.; Belgacem, M.; Dufresne, A.; Carbohydr. Polym. 2009, 78, 422. DOI: http://dx.doi.org/10.1016/j.carbpol.2009.04.034 8. Zhou, Y. M.; Fu, S. Y.; Zheng, L. M.; Zhan, H. Y.; eXPRESS Polym. Lett. 2012, 6, 794. DOI: http://dx.doi.org/10.3144/expresspolymlett.2012.85 9. Zimmermann, T.; Bordeanu, N.; Strub, E.; Carbohydr. Polym. 2010, 79, 1086. DOI: http://dx.doi.org/10.1016/j.carbpol.2009.10.045 10. Abdul Khalil, H. P. S.; Bhat, A. H.; Ireana Yusra, A. F.; Carbohydr. Polym. 2012, 87, 963. DOI: http://dx.doi.org/10.1016/j.carbpol.2011.08.078 11. Herrick, F. W.; Casebier, R. L.; Hamilton, J. K.; Sandberg, K. R.; J. Appl. Polym. Sci. 1983, 37, 797. 12. Turbak, A. F.; Snyder, F. W.; Sandberg, K. R.; J. Appl. Polym. Sci. 1983, 37, 815. 13. Jonoobi, M.; Harun, J.; Shakeri, A.; Misra, M.; Oksman, K.; BioResources 2009, 4, 626. 14. Hassan, M. L.; Mathew, A. P.; Hassan, E. A.; El-Wakil, N. A.; Oksman, K.; Wood Sci. Technol. 2012, 46, 193. DOI: http://dx.doi.org/10.1007/s00226-010-0373-z 15. Ferrer, A.; Filpponen, I.; Rodríguez, A.; Laine, J.; Rojas, O. J.; Bioresour. Technol. 2012, 125, 249. DOI: http://dx.doi.org/10.1016/j.biortech.2012.08.108 PMID: 23026341 16. Wang, B.; Sain, M.; Polym. Int. 2007, 56, 538. DOI: http://dx.doi.org/10.1002/pi.2167 17. Wang, B.; Sain, M.; Compos. Sci. Technol. 2007, 67, 2521. DOI: http://dx.doi.org/10.1016/j.compscitech.2006.12.015 18. Qua, E. H.; Hornsby, P. R.; Sharma, H. S. S.; Lyons, G.; Mater. Sci. 2011, 46, 6029. DOI: http://dx.doi.org/10.1007/s10853-011-5565-x 19. Abdul-Khalil, H. P. S.; Davoudpour, Y.; Nazuruyl Islam, M.; Asniza, M.; Sudesh, K.; Dungani, R.; Carbohydr. Polym. 2014, 99, 649. DOI: http://dx.doi.org/10.1016/j.carbpol.2013.08.069 PMID: 24274556 20. Abraham, E.; Deepa, B.; Pothan, L. A.; Jacob, M.; Thomas, S.; Cvelbar, U.; Anandjiwala, R.; Carbohydr. Polym. 2011, 86, 1468. DOI: http://dx.doi.org/10.1016/j.carbpol.2011.06.034 21. Szczesna-Antczak, M.; Kazimierczak, J. Antczak, T.; Fibres Text. East. Eur. 2012, 20, 8. 22. Zimmermann, T.; Pohler, E.; Geiger, T.; Adv. Eng. Mat. 2004, 6, 754. DOI: http://dx.doi.org/10.1002/adem.200400097 23. Tappi Test Method T222 om-88, Acid-insoluble lignin in wood and pulp. In: Tappi Test Methods. Atlanta: Tappi Press, 1999. 24. Ruland, W.; Acta Crystallogr. 1961, 14, 180. 25. Besbes, I.; Alila, S.; Boufi, S.; Carbohydr. Polym. 2011, 84, 975. DOI: http://dx.doi.org/10.1016/j.carbpol.2010.12.052 26. Križman, P.; Kovac, F.; Tavcer, P. F.; Color. Technol. 2005, 121, 304. DOI: http://dx.doi.org/10.1111/j.1478-4408.2005.tb00373.x 27. Lojewska, J.; Miskowiec, P.; Lojewski, T.; Pronienwicz, L. M.; Polym. Degrad. Stab. 2005, 88, 512. DOI: http://dx.doi.org/10.1016/j.polymdegradstab.2004.12.012 28. Sun, J. X.; Sun, X. F.; Zhao, H.; Sun, R. C.; Polym. Degrad. Stab. 2004, 84, 331. DOI: http://dx.doi.org/10.1016/j.polymdegradstab.2004.02.008 29. Kakurácová, M.; Capek, P.; Sasinková, V.; Wellner, N.; Ebringerová, A.; Carbohydr. Polym. 2000, 43, 195. DOI: http://dx.doi.org/10.1016/S0144-8617(00)00151-X 30. Yang, H.; Yan, R.; Chen, H.; Lee, D. H.; Zheng, C.; Fuel 2009, 86, 1781. DOI: http://dx.doi.org/10.1016/j.fuel.2006.12.013 31. Deepa, B.; Abraham, E.; Cherian, B. M.; Bismarck, A.; Blaker, J.; Pothan, L. A.; Leao, A. L.; Souza, S. F.; Kottaisamy, M.; Bioresour. Technol. 2011, 102, 1988. DOI: http://dx.doi.org/10.1016/j.biortech.2010.09.030 PMID: 20926289 32. Paris, M.; Bizot, H.; Emery, J.; Buzaré, J. Y.; Buléon, A.; Carbohydr. Polym. 1999, 39, 327. DOI: http://dx.doi.org/10.1016/S0144-8617(99)00022-3 33. Wishart, D. S.; Sykes, B. D.; J. Biomol. NMR 1994, 4, 171. PMID: 8019132 |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access