
 |
 |
 |
 |
 |
Revisão
|
|
| Metodologias de síntese em fase líquida para a obtenção de nanomateriais inorgânicos à base de terras raras Liquid phase synthesis methodologies for the obtainment of rare earth-based inorganic nanomaterials |
|
Paulo C. de Sousa Filho*,#; Osvaldo A. Serra
Departamento de Química, Faculdade de Filosofia, Ciências e Letras de Ribeirão Preto, Universidade de São Paulo, Av. Bandeirantes, 3900, 14040-901 Ribeirão Preto - SP, Brasil Recebido em 04/11/2014 *e-mail: pcsfilho@usp.br Liquid phase synthesis techniques are commonly preferred for the preparation of nanosized inorganic solids due to several advantages over other methodologies, such as low energy consumption, safety, possibility of reactant recovery and, mainly, the homogeneity of the precursors and control of particle sizes and morphologies through bottom-up approaches. In this regard, Colloidal Chemistry plays a key role in the controlled production of different classes of nanoparticles, thus being a subject of growing interest in several fields of Materials, Inorganic, and Physical-Chemistry. Therefore, this article sought to present an introductory outline of Colloidal Chemistry, with a focus on the preparation of inorganic nanoparticles. Given the broadness of this subject, special attention was devoted to nanoparticles based on Rare Earth compounds, due to their current importance in several fields. Therefore, the work presents the principles involved in the most commonly applied methodologies for the liquid phase synthesis of nanoparticles, such as coprecipitations and hydro/solvothermal techniques, as well as precipitations into nanoreactors based on reverse microemulsions, with a brief survey of the main advances in these fields in recent years. INTRODUÇÃO O desenvolvimento da nanotecnologia permitiu, nas últimas décadas, a atribuição de novas e mais sofisticadas aplicações a materiais supostamente já conhecidos. A rigor, pode-se classificar como nanomateriais os sistemas que apresentam propriedades físicas e químicas diferentes dos seus equivalentes convencionais (bulk) como consequência de possuírem ao menos uma de suas dimensões espaciais entre 1 e 100 nm.1,2 Nessa faixa de tamanhos, os materiais tendem a apresentar propriedades intermediárias entre as moléculas ou agregados atômicos (< 1 nm) e materiais massivos (> 1 µm), sendo que características como reatividade,3-5 toxicidade,5,6 absorção/espalhamento de luz,7-9 band-gap,9 ponto de fusão e calor específico,10 por exemplo, podem ser bastante diferentes das exibidas em sistemas de maiores dimensões. Dessa forma, observam-se diversos efeitos quânticos e cooperativos na escala dos nanomateriais, cujos tamanhos permitem, ainda, uma convergência com o domínio biológico11,12 e com os princípios da química verde.13,14 Esse campo multidisciplinar do conhecimento exigiu, em particular, uma migração da tradicional química de volumes para uma química de superfícies, já que grande parte dos fenômenos observados na nanoescala envolve os átomos interfaciais em virtude da alta razão superfície/volume inerente a esses sistemas. Dessa forma, a necessidade de obtenção de novos materiais nanoestruturados impulsionou um grande desenvolvimento dos métodos de síntese de nanopartículas, bem como do aprimoramento das técnicas de caracterização. Nesse sentido, a química coloidal ocupa um papel chave, tendo produzido uma vasta literatura acerca da produção bottom-up de nanopartículas não aglomeradas, com o intuito de se permitir a acessibilidade à sua superfície bem como de possibilitar eventuais processos de passivação. Os métodos de síntese de partículas coloidais permitem a variação do tamanho e da composição dos materiais de modo a adaptá-las a um amplo leque de aplicações. A utilização de partículas coloidais engloba aplicações biológicas (sistemas de drug-delivery e marcação),11,12,15-19 analíticas (espectroscopias amplificadas em superfícies e detecção de uma molécula),20-23 em dispositivos fotovoltaicos,24 tecnologia da informação (transmissão e armazenamento de dados),25,26 catálise,13,24,27,28 entre outros, empregando-se, principalmente, diferentes composições de nanopartículas metálicas,29,30 de semicondutores (quantum-dots)9,31,32 e de óxidos/oxossais.33-35 A literatura é repleta de revisões consideravelmente detalhadas sobre a natureza, sobre as técnicas particulares para o preparo e sobre as aplicações tecnológicas de diferentes tipos de nanopartículas coloidais, possuindo, geralmente, um foco bastante específico nessa ampla área da química. Dessa forma, o presente trabalho tem o objetivo de apresentar uma visão introdutória à química dos coloides, com enfoque na síntese de nanopartículas inorgânicas. Tendo em vista a extensão do assunto, maior atenção é destinada a nanopartículas à base de compostos de terras raras (TR), dada a sua particular importância em diferentes áreas. Apresentam-se os fundamentos de algumas das metodologias mais frequentemente utilizadas, tais como coprecipitações e técnicas hidro/solvotermais, além de precipitações em nanorreatores (microemulsões reversas), com um breve levantamento sobre os principais avanços nessas áreas nos últimos cinco anos.
ASPECTOS GERAIS SOBRE A QUÍMICA DOS COLOIDES Define-se como um coloide um sistema composto por ao menos duas fases, sendo uma contínua (dispersante) e uma ou mais fases descontínuas (partículas dispersas) que possuem pelo menos uma de suas dimensões entre 1 nm e 1 µm, ou um sistema cujas descontinuidades encontram-se nessa faixa de tamanhos.36 Assim, grosso modo, o termo dispersão coloidal refere-se a sistemas intermediários entre as soluções (sistemas homogêneos cujas espécies dispersas têm dimensões moleculares) e as dispersões grosseiras (sistemas heterogêneos com tamanhos de partículas dispersas superiores a 10 µm).37 O termo "coloide" foi introduzido por Thomas Graham, considerado o pai da Química de Coloides, em 1861,38 significando "parecido com cola" (do grego "kolla" adicionado ao sufixo "oide", de "similar a"); o termo referia-se originalmente a materiais em solução/suspensão aquosa que possuíam baixa difusibilidade e eram incapazes de atravessar uma membrana de diálise. Originalmente, no entanto, atribui-se a Michael Faraday a invenção do termo em 1857, durante a apresentação de seu trabalho acerca dos efeitos da luz sobre suas famosas suspensões coloidais aquosas de ouro metálico.39 Os diferentes sistemas coloidais possíveis são atualmente classificados de acordo com a natureza das fases dispersa/dispersante envolvidas. Por exemplo, um coloide sólido/líquido é comumente chamado de "sol" (como as tintas); sistemas líquido/líquido são designados como "emulsões" (como o leite ou as microemulsões posteriormente descritas); coloides do tipo líquido/sólido são chamados de "géis" ou "emulsões sólidas"; sistemas líquido/gás são conhecidos como "aerossóis", entre outros.36,37 No que tange à síntese de nanopartículas inorgânicas, sistemas coloidais consistem em uma fase sólida, obtida por uma reação de precipitação, suspensa em uma fase contínua líquida, apresentando alto grau de dispersão das partículas e baixa taxa de aglomeração e sedimentação. Durante a síntese coloidal de nanopartículas, há dois processos, que ocorrem de maneira praticamente simultânea, a serem considerados: a nucleação (formação de numerosos cristalitos iniciais, em equilíbrio instável com o líquido parental) e o crescimento das partículas. A primeira teoria a tratar da influência de tais processos na obtenção de suspensões coloidais é o modelo de La Mer,40,41 originalmente desenvolvido para suspensões aquosas de enxofre coloidal. Atualmente, sabe-se que tal modelo é muito particular a esses sistemas e não se aplica diretamente à maioria dos coloides. Contudo, por sua ideia central de separação temporal entre nucleação e crescimento, o modelo serve de base para a elaboração de outros modelos bem como para o desenvolvimento de várias metodologias para a obtenção de coloides monodispersos. Para um sólido de fórmula AxBy, o produto de solubilidade (PS) das espécies iônicas em um determinado solvente pode ser escrito como:  em que γ e C representam os coeficientes de atividade e as concentrações molares, respectivamente, das espécies envolvidas na precipitação. Se o sólido AxBy encontra-se em equilíbrio termodinâmico com sua solução parental, tem-se que: 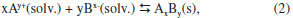 sendo que seu produto de solubilidade assume um valor constante (KPS) dado por: 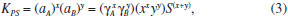 em que aA e aB são as atividades iônicas no equilíbrio termodinâmico, S é a solubilidade molar do sólido e x e y são os coeficientes estequiométricos. O grau de supersaturação (ΓS) pode ser definido como a razão entre o produto de solubilidade nas condições de saturação (nas quais as atividades são representadas por a') e o valor do KPS de equilíbrio termodinâmico: 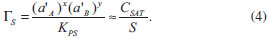 A equação anterior também pode ser expressa como a razão entre a concentração do soluto na saturação (CSAT) e sua concentração de equilíbrio (S). O grau de supersaturação pode ser considerado, portanto, como a força motriz do processo de precipitação, sendo que, quanto maior o valor da diferença CSAT-S, mais favorável será a formação do sólido. A Figura 1(a) ilustra o processo de precipitação segundo o modelo de La Mer com suas três etapas características: uma pré-nucleação, a nucleação e o crescimento. Inicialmente, partindo-se de uma solução insaturada em espécies precursoras do sólido, a concentração do soluto é inferior à concentração de supersaturação crítica, de modo que não se observa a formação de qualquer precipitado. A aplicação de uma ativação apropriada ou uma etapa de geração dos agentes precipitantes (por exemplo, por hidrólise ou (foto) oxidação/redução) leva ao aumento da concentração das espécies precursoras em valores acima da solubilidade de equilíbrio (S). Ao se atingir uma concentração de supersaturação mínima (Cmin), a fase de nucleação se inicia de maneira espontânea com a formação de germes iniciais (nucleação homogênea). A dimensão (R) dos núcleos formados nesse processo define o destino tomado por essas espécies. Os núcleos que possuírem um tamanho abaixo de um raio crítico de equilíbrio (R*) tendem a ser altamente instáveis e, dessa forma, são mais propensos a se redissolverem na solução. Já os núcleos com R>R* têm vida suficientemente longa para se submeterem à etapa de crescimento. O raio crítico de equilíbrio será tão menor quanto maior for o grau de supersaturação, ilustrando uma maior facilidade de nucleação para soluções de alto grau de supersaturação. Por outro lado, o valor de R* é diretamente proporcional à tensão interfacial sólido-líquido (entre o núcleo e a solução) e ao volume iônico do soluto; assim, o raio crítico de equilíbrio é um reflexo direto da energia livre de ativação para a formação do núcleo.42,43
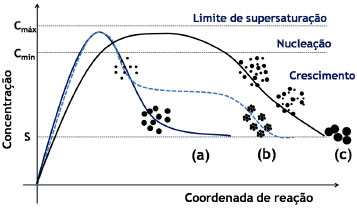 Figura 1. Ilustraçao de mecanismos de precipitaçoes coloidais que levam a partículas monodispersas: (a) modelo de La Mer, (b) modelo de Ocaña e (c) modelo de Sugimoto
Dessa forma, se a velocidade de nucleação for maior que a velocidade de geração das espécies precursoras, haverá uma queda rápida na concentração dos precursores abaixo de Cmin, cessando, assim, a nucleação. Nessas condições, a rápida etapa de nucleação é, então, seguida por uma etapa de crescimento uniforme dos germes formados. Tal etapa de crescimento é controlada pela difusão do soluto precursor em direção às partículas, podendo também ser controlada pela velocidade de geração dos precursores de modo a não se atingir novamente o valor de Cmin. Nesse sentido, parâmetros como taxas de adição e de agitação afetam diretamente as condições de supersaturação e, portanto, de nucleação ou crescimento de partículas. Por fim, a etapa de crescimento tende a prosseguir até que a concentração do soluto atinja o valor da solubilidade de equilíbrio termodinâmico. Por considerar uma etapa de nucleação rápida e de curta duração, o modelo de La Mer é normalmente inadequado à maioria dos sistemas, sobretudo os que apresentam outros equilíbrios em solução (complexação, por exemplo). Em tais casos a etapa de crescimento pode começar ao mesmo tempo em que a etapa de nucleação ainda ocorre, o que faz com que os diferentes germes formados experimentem diferentes tempos de crescimento. Tal diferença levará, em princípio, a um aumento da polidispersidade em relação ao tamanho das partículas, sendo que se desenvolveram vários modelos para se explicar o crescimento dos germes e a obtenção de coloides monodispersos.41 Alguns dos mais importantes são, por exemplo, os modelos de Ocaña44 e de Sugimoto.45 No modelo de Ocaña (Figura 1(b)), assume-se a ocorrência de um processo também rápido de nucleação, porém a geração das partículas finais ocorreria através da agregação dos germes iniciais e das partículas menores. No modelo de Sugimoto (Figura 1(c)), assume-se uma etapa muito lenta de nucleação, levando à formação de germes polidispersos. O crescimento é, então, explicado pela coalescência limitada por difusão dos germes menores (mais instáveis) em favor do aumento das partículas maiores, através do processo de Ostwald ripening ("maturação de Ostwald").37,46 O processo de Ostwald ripening consiste em um fenômeno em que partículas pequenas em suspensão tendem a se redissolver e depositar sua massa em partículas maiores, minimizando a área interfacial para que se atinja um sistema de maior estabilidade termodinâmica. Em outras palavras, a solubilidade das partículas aumenta com a diminuição do seu raio (da mesma maneira como a pressão de vapor de um líquido aumenta conforme diminui o tamanho da gota), o que pode ser expresso como uma variação da equação de Kelvin:37 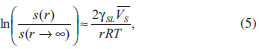 em que s(r) é a solubilidade da partícula em função de seu raio r, γSL é a tensão interfacial sólido-líquido e VS é o volume molar do soluto. As bases do tratamento matemático do fenômeno de Ostwald ripening são dadas pela teoria LSW (Lifshitz-Slyozov-Wagner), sendo que se recomenda uma literatura mais específica ao leitor interessado em tais detalhes.47-49 Contudo, fica clara a necessidade de que, para se obter uma baixa polidispersidade de partículas, os núcleos de todas as espécies sejam formados simultaneamente, sem haver a nucleação posterior de partículas menores. Além disso, para a obtenção de nanopartículas homogeneamente dispersas, o processo de nucleação deve ser consideravelmente rápido, enquanto o de crescimento deve ser lento. Assim, independentemente do modelo considerado para a formação do sólido, geralmente, uma reação de precipitação sem controle das etapas de nucleação e crescimento leva a um precipitado formado pela aglomeração não controlada das partículas. De maneira geral, o que diferencia uma precipitação comum de uma precipitação coloidal (de micro- ou nanopartículas bem dispersas) é uma etapa adicional de estabilização de superfície, de modo a se evitar seu crescimento posterior e sua agregação em suspensão. Dessa forma, a obtenção de uma suspensão coloidal estável exige que certas condições de estabilização sejam obedecidas, sendo elas sumarizadas pela chamada teoria DLVO (Deryagin, Landau, Verwey e Overbeek),37,46,50 que trata do balanço entre forças atrativas e repulsivas entre as partículas em suspensão (Figura 2(a)). Um coloide será tão mais estável quanto mais intensas forem as forças repulsivas atuantes entre suas partículas.
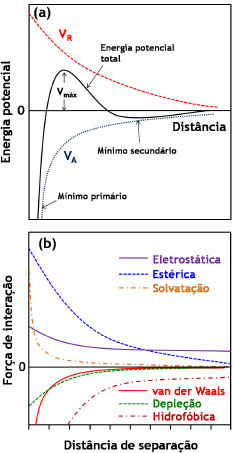 Figura 2. (a) Balanço dos potenciais de repulsao (VR) e atraçao (VA) entre partículas de acordo com a teoria DLVO. (b) Diagrama representando as possíveis forças atrativas (abaixo do eixo das abscissas) e repulsivas (acima das abscissas) atuantes entre partículas em suspensao
A teoria DLVO considera unicamente as forças de van der Waals (atrativas) e as forças eletrostáticas (repulsivas) para o tratamento da estabilidade coloidal. No entanto, deve-se ter em mente que várias outras interações podem estar presentes nos sistemas coloidais (Figura 2(b)), que são chamadas de forças não-DLVO, contribuindo com diferentes intensidades para a atração/repulsão de partículas. As forças atrativas, cuja tendência é a de agregar as partículas e de desestabilizar a suspensão coloidal, compreendem as forças de depleção, as interações hidrofóbicas e as forças de van der Waals. Forças de depleção são decorrentes da adição de um polímero não-adsorvente em suspensões concentradas, como resultado do aumento do volume livre na fase contínua (e consequente diminuição do volume disponível às partículas, que tendem a ser "excluídas" da suspensão). As interações hidrofóbicas também possuem contribuição apreciável apenas na presença de tensoativos adsorvidos à superfície das partículas ou no caso de coloides com superfície consideravelmente hidrofóbica. Tais interações são de natureza entrópica, decorrentes da introdução de uma organização adicional às moléculas do solvente de modo a excluir a cauda apolar do surfactante ou a superfície da partícula, aumentando, assim, a energia livre do sistema. Consequentemente, há a tendência da minimização da área interfacial entre as superfícies apolares e o solvente através de sua agregação mútua, o que pode levar à coagulação das partículas em suspensão. Por fim, as principais interações atrativas entre partículas coloidais, e únicas a serem intrínsecas ao sistema (isto é, independentes da presença de polímeros ou surfactantes adsorvidos), são as interações de van der Waals. Tais interações provêm de forças eletrostáticas decorrentes de flutuações na distribuição de cargas ao redor dos átomos. No caso de uma interação entre dois átomos separados por uma distância d (o que corresponde, portanto, a uma interação do tipo dipolo induzido/dipolo induzido), a força atrativa entre ambos é inversamente proporcional a d6. Ao se considerarem duas partículas esféricas idênticas separadas por uma distância d, o potencial atrativo de van der Waals é dado por:  em que A é a constante de Hamaker (dependente da natureza das partículas e da composição do meio, assumindo valores entre 0,25 e 25 kBT, sendo kB a constante de Boltzmann e T a temperatura absoluta) e r é o raio das partículas. As forças repulsivas, que têm a tendência de manter as partículas coloidais isoladas em suspensão, compreendem as interações de solvatação, as interações estéricas e as forças eletrostáticas. As interações de solvatação têm origem na repulsão causada pela presença de uma camada de moléculas de solvente nas proximidades da superfície da partícula. Contudo, por envolver as normalmente pouco volumosas moléculas de solvente, esse tipo de interação somente é efetiva quando se consideram superfícies altamente solvofílicas e pequenas distâncias de separação entre partículas, resultando em uma baixa contribuição para a estabilidade coloidal. A estabilização coloidal por repulsões estéricas, por outro lado, é consideravelmente importante para o balanço das forças do sistema quando da presença de (macro)moléculas adicionais. Tais interações envolvem a adsorção ou ligação química de moléculas orgânicas à superfície das partículas, de modo que o impedimento estérico entre essas espécies limita a distância entre as partículas a separações em que as interações de van der Waals são desprezíveis. Para que as repulsões estéricas sejam, de fato, efetivas para a estabilização coloidal, é fundamental que as moléculas orgânicas adsorventes tenham, ao mesmo tempo, uma intensa interação com a superfície das partículas e uma alta afinidade pelas moléculas do solvente (de modo a se minimizarem as interações hidrofóbicas). As mais importantes interações repulsivas são as interações eletrostáticas devidas à existência de uma carga líquida na superfície das partículas. A origem dessa carga líquida pode ser explicada através de diversos mecanismos, tais como reações com íons H3O+ e OH- (protonação/desprotonação), dissociação/ionização de grupos de superfície, adsorção preferencial de íons reticulares, quimissorção de íons, adsorção de surfactantes iônicos ou polieletrólitos etc. Como consequência, e a fim de se manter a neutralidade elétrica no sistema coloidal, os íons dissolvidos na fase contínua assumem uma organização preferencial nas proximidades das partículas, que pode ser descrita através de um modelo da dupla camada elétrica (Figura 3).
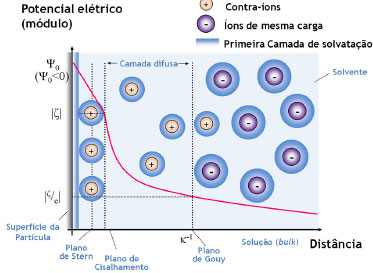 Figura 3. Ilustraçao do modelo de dupla camada elétrica e da variaçao do potencial elétrico (ψ0) em funçao distância a partir da superfície de uma partícula coloidal (no caso, de carga superficial negativa), com a representaçao do arranjo de contra-íons e co-íons em suas proximidades na presença das respectivas camadas de solvataçao37
A camada mais próxima da superfície da partícula (isto é, na vizinhança imediata da carga superficial) é conhecida como camada de Stern, sendo composta por moléculas de solvatação e por contra-íons fortemente adsorvidos. Nessa região, devido à presença desses íons de carga oposta, há uma queda brusca e linear no módulo do potencial elétrico em função da distância à superfície. Fundamentalmente, o diâmetro hidrodinâmico da partícula (medido, por exemplo, através de técnicas de espalhamento de luz)37,51,52 corresponde ao tamanho da partícula mais o tamanho da camada de Stern (isto é, soma das dimensões da partícula, da camada de solvatação por moléculas do solvente e de íons solvatados). O raio exterior da camada de Stern é conhecido como plano de cisalhamento, sendo que o potencial elétrico nesse plano corresponde ao potencial ζ (zeta) obtido através de medidas de mobilidade eletroforética.53,54 A camada iônica exterior ao plano de cisalhamento é conhecida como camada difusa, sendo composta por contra-íons submetidos ao potencial elétrico gerado pela superfície da partícula e à agitação térmica da própria solução. Nessa região, o potencial elétrico decai exponencialmente com a distância em relação à superfície carregada, com uma distância característica (κ-1, o comprimento de Debye) que é inversamente proporcional à força iônica do meio. O raio exterior da camada difusa (com distância de κ-1 da superfície) é chamado de plano de Gouy, a partir do qual a fase contínua apresenta propriedades isotrópicas (bulk). Ao se considerar a interação entre duas partículas carregadas em suspensão, haverá a interpenetração das camadas difusas de ambas as partículas, resultando em um aumento local da concentração de contra-íons. O potencial repulsivo entre as partículas é dado por: 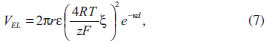 em que r é o raio das partículas, ε é a constante dielétrica do meio, z é a carga dos contra-íons, F é a constante de Faraday, ξ é o potencial reduzido de superfície (ζ = tanh Em suma, a combinação das interações atrativas e repulsivas resulta em uma curva de potencial com o perfil apresentado na Figura 2(a), com dois mínimos e uma barreira de potencial. Em curtas ou em longas distâncias, predominam as forças atrativas de van der Waals, que levam à ocorrência de um mínimo secundário e de um mínimo primário; esses pontos correspondem, respectivamente, aos pontos de floculação e coagulação do coloide. Nas proximidades do comprimento de Debye, no entanto, a repulsão eletrostática dá origem a um máximo de potencial, criando uma barreira de energia de valor Vmáx para a coagulação do coloide. Assim, pode-se demonstrar que a estabilidade coloidal é de natureza cinética (já que o estado de equilíbrio termodinâmico corresponde ao mínimo primário), sendo dependente da relação entre a altura da barreira (Vmáx) e da energia de ativação térmica kBT. Dessa forma, a taxa de agregação de partículas é proporcional a e
METODOLOGIAS DE PRECIPITAÇÃO COLOIDAL Coprecipitação As reações de coprecipitação envolvem a formação de espécies de baixa solubilidade em situações de supersaturação, sendo que tais condições são atingidas, geralmente, por meio de reação química. No âmbito da síntese de nanomateriais inorgânicos, a coprecipitação de nanopartículas a base de óxidos, oxossais, calcogenetos e haletos tende a envolver mecanismos consideravelmente mais complexos que as reações de formação de nanopartículas metálicas. Ao contrário dos metais, os compostos mencionados cristalizam-se com menor facilidade nas condições de síntese em solução que, normalmente, levam à formação de precipitados amorfos. Além disso, de maneira geral, agentes para o recobrimento de superfície ou estabilizantes são necessários para a minimização da aglomeração de partículas. Embora a cristalização direta dessas nanopartículas inorgânicas seja possível, normalmente a reação de coprecipitação leva à formação de um precursor que, após uma etapa de secagem ou calcinação, gera os produtos. A menos que metodologias especiais sejam aplicadas, tais como dispersão em matrizes inorgânicas (sílica ou alumina),55-57 a etapa de tratamento térmico invariavelmente leva a uma agregação, ainda que parcial, das partículas formadas. O preparo de óxidos não-amorfos através de reações diretas de coprecipitação em condições brandas normalmente se limita a estruturas de elevada estabilidade termodinâmica; a Tabela 1S sumariza os processos de obtenção direta de óxidos por reações de coprecipitação. Um dos exemplos mais comuns de óxidos com tais características são os espinélios, compostos com fórmula geral AB2O4 (em que A é um cátion divalente e B e um cátion trivalente), que normalmente são associados a estabilidades estruturais muito altas.58 Nesses casos, em que a cristalização efetiva do sólido é favorecida mesmo em temperaturas baixas, os hidróxidos formados são condensados durante etapas posteriores à precipitação, de modo que o tratamento térmico posterior dos materiais não é necessário. Recentemente, elucidaram-se alguns caminhos reacionais envolvidos na coprecipitação de nanopartículas de magnetita (Fe3O4), confirmando que a formação dos produtos depende, de fato, de transformações de fase de oxi-hidróxidos de ferro (akanganeíta, goetita, lepidocrocita e hidróxido de ferro(II)) ao invés da reação direta de íons Fe2+ e Fe3+ em solução aquosa.59 Por outro lado, Valenzuela et al.60 demonstraram que, nesses casos, mesmo fatores simples como a taxa de agitação mecânica são capazes de alterar tanto o tamanho de partícula quanto a composição final do sólido obtido na reação de coprecipitação. Pereira et al.61 descrevem, por exemplo, a obtenção de diferentes nanoestruturas de ferritas (MFe2O4, em que M=Fe, Co ou Mn) com tamanho controlável entre 4 e 12 nm, utilizando isopropanolamina e di-isopropanolamina como agentes precipitantes alcalinos. A possibilidade de inclusão de dopantes nas estruturas de ferritas é outra vantagem da aplicação de sínteses por coprecipitação, uma vez que os precursores encontram-se homogeneamente dispersos durante tais reações. Ferritas mistas de manganês e zinco [(Mn,Zn)Fe2O4] dopadas com íons Eu3+ (para aumento da resistividade elétrica) ou íons Ho3+ (aumento da coercitividade e diminuição da magnetização de saturação) são obtidas, por exemplo, através de coprecipitação dos cloretos metálicos com NaOH/Na2CO3.62,63 Meng et al.64 também obtêm nanopartículas de (Mn,Zn)Fe2O4 substituídas com La, Nd ou Gd na faixa de 10 a 20 nm, através da precipitação inicial da fase δ-FeOOH seguida de refluxo por 6 h. Alguns outros óxidos simples também podem ser precipitados diretamente de soluções aquosas, sem a necessidade de tratamentos posteriores, sendo que tais casos englobam, normalmente, óxidos binários de alta estabilidade como o TiO2 (fase rutilo) e o CeO2, que possui a estrutura cúbica da fluorita. Taniguchi et al.65 descrevem, por exemplo, o preparo de nanocristais de CeO2 com diâmetro de ~2,2 nm através da precipitação com amônia na presença de ácido oleico, que atua com agente estabilizante e permite a dispersão do sólido em meios apolares. Em um procedimento ainda mais simples, Almeida et al.66 demonstram a possibilidade de obtenção de nanopartículas de Ce1-xFexO2 através de precipitação direta com lenta adição de NaOH e secagem em dry-box por 72 h em temperatura ambiente. Xin et al.67 apresentam, ainda, uma rota sintética para nanobastões (10-50 nm de diâmetro, 30-250 nm de comprimento) de CeO2 dopado com Nd, Sm, Eu, Gd ou Y através da coprecipitação com amônia ou NaOH 2 mol L-1 seguida de envelhecimento da suspensão por 48 h em temperatura ambiente. Já a obtenção de outros óxidos ternários de composição mais complexa por coprecipitação direta também é possível, embora bem menos frequente. Esses casos (resumidos na Tabela 2S) também são associados a estruturas estáveis e de grande insolubilidade, em que a existência do ânion precipitante (ou de um precursor) em solução é possível. Esse é o caso, por exemplo, de fosfatos e vanadatos de terras raras, que possuem imensa importância em vários ramos da química do estado sólido, tais como biomarcadores,18,19 luminóforos,68 cerâmicas69 e estocagem de resíduos radioativos.70 Os fosfatos de terras raras precipitados de soluções aquosas podem se apresentar em quatro polimorfos, sendo duas estruturas anidras (monazita - monoclínica -, e xenotima/zircão - tetragonal), e duas hidratadas (rabdofano - hexagonal -, e churchita - monoclínica). A obtenção particular de cada uma dessas estruturas depende tanto da natureza do cátion envolvido (sobretudo seu raio iônico), quanto das condições de síntese. Os fosfatos das terras raras leves (La-Gd) assumem as estruturas da monazita ou do rabdofano (geometrias menos regulares e de maior número de coordenação), enquanto as terras raras pesadas (Tb-Lu, Y) cristalizam-se nas estruturas da xenotima e da churchita (embora também seja possível a ocorrência da fase rabdofano em alguns casos). Em condições comuns de coprecipitação, geralmente obtêm-se sólidos hidratados, sobretudo para as terras raras leves. É frequente, contudo, que as terras raras pesadas, mesmo em condições aquosas brandas, forneçam a estrutura anidra da xenotima. Por outro lado, a obtenção da fase monazita para os íons La-Gd é normalmente condicionada a condições mais enérgicas, como em baixos pHs ou altas temperaturas/pressões (sínteses hidrotermais, discutidas nas seções seguintes). Buissette et al.71 descrevem, por exemplo, a obtenção de nanopartículas de fosfatos de terras raras com a estrutura do rabdofano com tamanhos de partícula da ordem de ~5 nm através da reação de sais solúveis de TR com tripolifosfato de sódio (Na5P3O10). Este polifosfato atua tanto como precursor de íons ortofosfato quanto como agente complexante e estabilizante para as partículas, de modo que a reação a 90 ºC por 3 h gera coloides altamente estáveis de nanopartículas de LaPO4. Mais recentemente, Yang et al.72 descrevem a obtenção de nanofios de LaPO4 de ~15 nm de diâmetro e de comprimento controlável através de coprecipitação com (NH4)2HPO4 em meio de água/etanol. A variação da composição do solvente, do pH de síntese e do tempo de reação permite o controle do tamanho dos nanofios obtidos, sendo possível, ainda, a obtenção de estruturas core@shell por metodologia similar. Prasad et al.73 também descrevem a preparação de nanoestruturas core@shell contendo fosfatos de terras raras, envolvendo, nesse caso, o recobrimento de nanopartículas de Fe3O4 com fosfato de ítrio através de coprecipitação com NH4H2PO4 e agitação por ultrassom em presença de polietilenoglicol, o que gera nanopartículas bifuncionais do tipo Fe3O4@YPO4:Eu3+. A literatura traz inúmeros outros procedimentos para a síntese de fosfatos de terras raras e estruturas correlatas (tais como apatitas e hidroxiapatitas,74 por exemplo), sendo que se recomendam leituras mais amplas sobre esse tema particular aos leitores interessados.75,76 Os vanadatos de terras raras são estruturalmente similares aos fosfatos, ocorrendo nas estruturas monoclínica e tetragonal. No entanto, apenas lantânio e cério formam fases estáveis de estrutura da monazita, sendo que os demais elementos desse grupo produzem, nas sínteses em solução aquosa, sólidos com a estrutura da xenotima. Ao contrário dos fosfatos, o pH da solução precursora é crucial não apenas para a morfologia, mas para a composição dos vanadatos precipitados. Em condições ácidas ocorre, normalmente, a condensação reversível dos precursores em polivanadatos (HxV3O93-x, HxV10O286-x etc.), que dão coloração amarelada à solução e aos produtos. Embora as reações sejam comumente realizadas em condições básicas (em que há o favorecimento da formação das espécies VO43-), elevados pHs podem levar à competição entre precipitação de ortovanadatos e hidróxidos, que podem contaminar o sólido final. Huignard et al.77 descrevem a coprecipitação de YVO4 através da adição gota a gota de soluções de terras raras sobre soluções de Na3VO4, sendo fundamental a manutenção do pH entre 12 e 12,5 para o controle adequado da morfologia e da composição das nanopartículas. Os mesmos autores mostram que o uso de íons citrato como estabilizantes (repulsão estérica e eletrostática) permite a obtenção de coloides altamente concentrados (~400 g L-1) de nanopartículas ainda menores (~10 nm) de YVO4:Eu3+, com o estudo da evolução do sistema precursor em função do tempo de reação.78 Já Kumar et al.79 obtêm nanocristais de YVO4 dopados com Sm3+, Eu3+ ou Bi3+ na faixa de 10 a 50 nm, através da dissolução dos óxidos correspondentes (TR2O3, Bi2O3 e V2O5) em HNO3/H2O2 e posterior adição gota a gota de solução de NH3/H2O2 até regulagem do pH a 10; nesse caso, contudo, os precipitados são possivelmente contaminados com polivanadatos remanescentes, uma vez que os autores mencionam uma coloração amarelada nos sólidos finais. Outras abordagens sintéticas para nanopartículas de YVO4 serão abordadas nas seções posteriores. Outra importante classe de compostos de terras raras obtidos diretamente a partir de reações de precipitação são os fluoretos, que formam sólidos insolúveis de fórmula TRF3 ou MTRF4 (em que M é um metal alcalino). A composição dos sólidos formados é normalmente controlada através da escolha do agente precipitante e das razões empregadas. Para a obtenção dos trifluoretos emprega-se, geralmente, HF, NH4F ou NH4HF2, enquanto que, para o preparo dos sistemas MF.TRF3, utilizam-se NaF, KF ou os respectivos tetrafluoroboratos (MBF4). A alteração do pH permite o controle da morfologia dos sólidos finais, embora, em determinadas situações, as condições aplicadas possam levar a contaminações por oxifluoretos (TROF). O ácido trifluoroacético também é utilizado como precursor para fluoretos de terras raras, uma vez que sua hidrólise ou termodecomposição são capazes de gerar as espécies TRF3 ou MTRF4. Contudo, como a decomposição do ácido trifluoroacético é lenta em condições normais, seu emprego como precursor em solução é frequentemente limitado a condições hidro- ou solvotérmicas, posteriormente apresentadas. Os processos de obtenção de nanopartículas a base de fluoretos de TR foram recentemente discutidos por Rahman e Green,80 sendo que tanto os métodos de síntese como as propriedades fundamentais de diversos fluoretos inorgânicos foram extensivamente detalhados por Fedorov et al.81 Com relação aos métodos de coprecipitação direta recentemente empregados para a obtenção de tais compostos, Grzyb e Lis82 obtêm, por exemplo, nanopartículas de LaF3:Eu3+ e GdF3:Eu3+ de 12 e 17 nm, respectivamente, utilizando-se NH4F como agente precipitante e ácido cítrico como estabilizante em meio aquoso, com regulagem de pH através de adição de NH3. Já Fedorov et al.83 descrevem a obtenção de nanopartículas de Ba1-xYxF2+x com a estrutura da fluorita através de coprecipitação direta dos nitratos com ácido fluorídrico diluído. Estruturas do tipo core@shell também são amplamente estudadas no caso de fluoretos, tanto para a minimização da influência de defeitos de superfície quanto para que se confiram múltiplas funcionalidades às nanoestruturas.84 Nesse sentido, Cui et al.85 descrevem, por exemplo, a obtenção de estruturas LaF3:Nd3+@LaF3 com tamanhos de partícula de ~10 nm através de coprecipitações sucessivas com NH4F em presença de ácido oleico como estabilizante em meio de água/etanol. Nesses casos, o recobrimento com a camada de LaF3 chega a resultar em aumentos de 140% na intensidade de luminescência em comparação às partículas de LaF3:Nd3+ não recobertas. Grzyb et al.86 apresentam, ainda, uma metodologia para o preparo de nanopartículas do tipo TbF3@CeF3, que são obtidas através de um processo de auto-organização nas soluções precursoras. O processo consiste na coprecipitação em excesso de NH4F na presença de Triton® X-100 a 50 ºC, o que dá origem a nanopartículas de TbF3@CeF3 de ~10 nm. As partículas podem ser recobertas com uma camada adicional de SiO2, dando origem a estruturas do tipo TbF3@CeF3@SiO2 que, tal como as nanopartículas não recobertas, mostram-se neutras dos pontos de vista de toxicidade e de proliferação celular. Nesse caso, contudo, o recobrimento com a camada de sílica resulta em uma diminuição da intensidade de luminescência em virtude da absorção e espalhamento de parte da radiação excitante pelo SiO2. Com relação à grande maioria dos óxidos e oxossais, a coprecipitação de precursores que, por tratamento térmico, são convertidos nos produtos finais é bem mais frequente, sendo que vários outros compostos são também obtidos através da mesma abordagem, como mostra a Tabela 3S. Por exemplo, nanopartículas de CdS puras e dopadas com Mn2+ ou TR3+ (Ho3+, Er3+) podem ser obtidas por coprecipitação de precursores solúveis com Na2S em presença de polivinilpirrolidona (PVP) em temperatura ambiente.87 As partículas precursoras obtidas nesse processo são recobertas pelo estabilizante orgânico, de modo que a eliminação dessas espécies e a cristalização efetiva do sólido, necessária para o aprimoramento das propriedades de upconversion, é realizada por calcinação a 650 ºC. Em virtude dessa etapa, constata-se, contudo, uma oxidação parcial das partículas com a formação da fase contaminante de CdSO4. Além de sulfetos, alguns óxidos ternários de difícil preparo direto também podem ser obtidos a partir de precursores coprecipitados de soluções. Por exemplo, o niobato de ítrio (YNbO4) não é precipitado diretamente de soluções já que os precursores solúveis contendo nióbio(V) não geram o ânion NbO43- em meio aquoso. Assim, Xiao e Yan88 descrevem o preparo de sólidos do tipo YNb0,5M0,5O4:TR3+ (M=P,V e TR3+=Tb3+,Er3+,Sm3+) através da coprecipitação de precursores híbridos, que consistem em polímeros de coordenação com ácido salicílico em presença de polietilenoglicol (PEG); os precursores são convertidos nos sólidos finais através de calcinação a 1000 ºC. Já Chawla et al.89 relatam a obtenção de nanopartículas de ortofosfatos do tipo K2GdZr(PO4)3:Eu3+ através da coprecipitação dos sais solúveis com (NH4)2HPO4 em diferentes condições de pH, o que gera partículas esféricas com menos de 50 nm de diâmetro. Posteriormente, os sólidos são tratados a 800 ºC, dando origem a estruturas agregadas de ~0,5 µm com propriedades luminomagnéticas. Outra classe de compostos que requer a precipitação de precursores são os boratos de terras raras (TRBO3), pois o ânion BO33- também não é diretamente gerado em meio aquoso a partir dos precursores comuns (H3BO3, B(OR)3 etc.). Nesse sentido, Szczeszak et al.90 descrevem, por exemplo, a obtenção de boratos de ítrio (YBO3:Eu3+) através da coprecipitação dos nitratos de TR3+ com ácido bórico em pH=9, com posterior calcinação do sólido obtido a 900 ºC. Nesse caso, a ação estabilizante de diferentes correagentes foi avaliada, concluindo-se que o uso de glicerina leva aos melhores resultados quanto ao controle morfológico. Tungstatos também são frequentemente obtidos a partir do tratamento térmico de precursores coprecipitados, normalmente utilizando-se sais solúveis do tipo M6W7O24 (M=Na+, K+, NH4+ etc.) como fontes de ânions WO42-. Zhang et al.91 obtêm, por exemplo, partículas de CaWO4:TR3+ (TR3+=Y3+,Eu3+,Gd3+) pela coprecipitação de um precursor utilizando TR(NO3)3 e (NH4)6W7O24 em presença de NH4HCO3/NH3 em água, com posterior calcinação a 800 ºC. A obtenção de óxidos binários e ternários a partir de precursores coprecipitados normalmente envolve a formação de hidróxidos ou (hidroxi)carbonatos intermediários, de modo que o tratamento térmico posterior promova a eliminação de CO2 e condensação dos grupos OH. Essa metodologia geral consiste no procedimento mais tradicionalmente empregado para o preparo de óxidos. Por exemplo, Mahmood et al.92 descrevem o preparo de nanoestruturas do tipo LaMnO3 dopadas com Gd3+ e Cr3+ pela coprecipitação dos precursores solúveis com a adição de NH3 2 mol L-1 até pH=11-12 a 70 ºC, seguida de calcinação a 700 ºC. Um procedimento similar para a obtenção de óxidos mistos de cério(IV) e zircônio(IV) consiste na coprecipitação dos nitratos de cério(III) e zirconilo com amônia concentrada em presença de peróxido de hidrogênio, em proporção 4:1:4 (v/v) por 7 h em condições ambientes; os hidróxidos precipitados são secos a 80 ºC por 24 h e calcinados em ar a 550 ºC por 3 h para a obtenção dos óxidos mistos finais, que possuem propriedades catalíticas na redução da emissão de fuligem oriunda da combustão do diesel.93 Uma das mais importantes classes de intermediários para o preparo de óxidos de compostos contendo terras raras são os hidroxicarbonatos (TR(OH)CO3.nH2O), que são classicamente obtidos por precipitação homogênea com ureia.94 Mais recentemente, Sorbello et al.95 descreveram a obtenção de partículas esféricas e monodispersas de (Ce,Gd,Eu)(OH)CO3.H2O com ~200 nm de diâmetro. As partículas de hidroxicarbonatos são posteriormente submetidas a um tratamento térmico a 1000 ºC gerando os óxidos de interesse (Ce0,95-xGdxEu0,05O1,975-x/2, com 0<x<0,95), com a manutenção de sua homogeneidade química e sua dispersibilidade. Contudo, o aquecimento leva a uma sinterização parcial das partículas, que perdem sua textura superficial e são convertidas em esferoides facetados, com ~150 nm. Uma metodologia similar é descrita por Raju et al.96 para a obtenção de hidroxicarbonatos de gadolínio dopados com íons lantanídeos, envolvendo a precipitação homogênea dos nitratos aquosos de TR com ureia em recipiente fechado a 90 ºC. Nesse caso, obtêm-se nanoestruturas em forma de flor (segundo os autores, partículas com aparência de crisântemos), que são convertidas em esferas de ~200 nm de Gd2O3 após tratamento térmico a 750 ºC. Como a perda da morfologia dos sólidos iniciais é comum nas etapas de tratamento térmico, as partículas de hidroxicarbonatos de TR também são frequentemente empregadas como templates para o preparo de outras nanoestruturas derivadas através de reações em suspensão. Nesses casos, partículas de TR(OH)CO3.nH2O são tratadas com precursores que tendem a formar produtos de maior estabilidade termodinâmica em relação aos hidroxicarbonatos (ex. PO43- VO43-, F- etc.), permitindo a manutenção parcial da morfologia dos sólidos iniciais e minimizando a agregação de partículas. Embora essas situações sugiram que a precipitação do produto mais insolúvel ocorra apenas na superfície das partículas, o que formaria uma espécie de camada protetora e impediria a exposição do material contido no interior das estruturas, frequentemente observam-se reações completas com a obtenção de estruturas ocas.97,98 Tal comportamento é explicado com base nas diferentes mobilidades dos íons envolvidos nas reações na interface sólido-líquido. Inicialmente, o contato da partícula de hidroxicarbonato com o agente precipitante leva à formação de uma camada sobre as partículas precursoras, que inibe a reação química direta e faz com que a precipitação do produto final passe a ser limitada pela difusão das espécies envolvidas. Como a taxa de difusão dos cátions (TR3+) do interior da partícula em direção à superfície tende a ser maior que a dos ânions desde a interface até o interior, geralmente observa-se uma produção mais rápida do precipitado na superfície externa (interface sólido-líquido) em relação à superfície interna (interface sólido-sólido), com o consumo contínuo da fase de TR(OH)CO3.nH2O. Trata-se, portanto, de uma observação e aplicação do efeito Kirkendall (originalmente relacionado à diferença de mobilidades atômicas/iônicas em ligas metálicas) em interfaces sólido-líquido nos sistemas nanoestruturados.99,100 Assim, por exemplo, Lv et al.97 relatam a obtenção de esferas ocas de GdF3 com propriedades luminescentes e capacidade de aplicação em liberação controlada de drogas. Para isso, partículas de TR(OH)CO3 de ~100 nm são expostas a uma solução contendo NaBF4 a 50 ºC em diferentes valores de pH. Por outro lado, Jia et al.98 propõem um método universal para a síntese de nanopartículas ocas a base de TR com a utilização de um template de sacrifício baseado em hidroxicarbonatos. Nesse caso, obtêm-se partículas ocas de YVO4, YPO4 e NaYF4 com diferentes morfologias após o tratamento das partículas iniciais com NH4VO3, NH4H2PO4 e NaBF4 em condições hidrotermais, que são processos discutidos na próxima seção. Sínteses hidro- e solvotermais Chamam-se, genericamente, de processos hidrotermais ou hidrotérmicos as técnicas que envolvam reações em solventes aquosos ou mineralizadores em altas temperaturas e pressões de vapor, utilizando-se, normalmente, recipientes selados especiais (autoclaves, tubos de pressão etc., Figura 4).101 Tais condições podem conduzir a processos de dissolução e recristalização de materiais que são relativamente insolúveis em condições normais, permitindo o controle das etapas de nucleação, crescimento e aumento de cristalinidade para que as características dos sólidos de interesse sejam modeladas. Embora não haja definição precisa quanto às condições de temperatura e pressão, a maioria dos autores fixa um limite mínimo de 100 ºC e 1 atm para que se considere uma síntese como hidrotermal. Essa terminologia tem origem na descrição geoquímica de processos de formação de minerais, caracterizados por combinarem a ação da água e do calor sob pressões elevadas; isso passou a restringir, portanto, o uso do termo para os casos em que o meio da reação seja aquoso. Assumindo-se a possibilidade de se utilizarem outros solventes em processos de síntese, pode-se definir um termo mais amplo, considerando-se um processo solvotermal como uma reação heterogênea na presença de um solvente (aquoso ou não aquoso) em temperaturas maiores que a temperatura ambiente e em pressões maiores que 1 atm em um sistema fechado.101,102 Na grande maioria dos experimentos de reações hidro e solvotermais, a pressão do sistema é autógena (isto é, gerada a partir da volatilização do solvente e da eventual formação de gases). Dessa forma, a pressão final é determinada tanto pela temperatura do processo, quanto pelo grau de preenchimento do recipiente em tais situações. Alguns casos envolvem, ainda, o emprego de solventes em condições supercríticas, em que, por apresentarem características associadas tanto a líquidos como a gases, obtêm-se condições de alta viscosidade e de baixas tensões interfaciais sólido-líquido.103 Embora alguns processos solvotermais sejam realizados com solventes supercríticos visando à síntese de nanoestruturas, a imensa maioria da literatura relata o emprego de condições abaixo do ponto crítico do solvente, que tendem a resultar no aumento da solubilidade e da reatividad, e , dos precu, rsores metálicos. Isso permite que se observe a cristalização de sólidos inorgânicos em temperaturas consideravelmente mais baixas do que as normalmente empregadas em reações de estado sólido. Além disso, contrariamente aos processos de coprecipitação, as sínteses solvotermais normalmente levam a materiais de alta cristalinidade em virtude das condições mais enérgicas, de modo que os produtos não necessitam obrigatoriamente de um tratamento térmico posterior.
 Figura 4. Alguns sistemas utilizados em reaçoes hidro- e solvotermais: (a) tubos selados de paredes de vidro espesso (com tampas de Teflon vedadas por O-rings), (b) autoclaves de aço inox com recipientes internos de Teflon, e (c) tubos de Teflon jaquetados com invólucros cerâmicos para reaçoes com irradiaçao por micro-ondas
De maneira geral, utilizam-se, nessas técnicas de fase líquida, soluções precursoras de cátions metálicos, que posteriormente formarão precipitados quando em contato com agentes precipitantes. Para garantir precipitações homogêneas e controladas, são frequentemente utilizados agentes complexantes e/ou estabilizantes. Em alguns casos utilizam-se, ainda, solventes coordenantes de alto ponto de ebulição.104 Na maioria dos procedimentos que visam à produção de micro e nanopartículas, para haver condições adequadas de precipitação, é necessário que a concentração dos reagentes seja mantida relativamente alta, de modo a garantir uma rápida precipitação, o que impede o crescimento das partículas sólidas. Os métodos solvotermais têm sido amplamente utilizados para a obtenção de catalisadores, sensores, carreadores de drogas, pigmentos, luminóforos, entre outros; a funcionalidade desses materiais depende intimamente das características morfológicas dos sólidos, conferindo ainda mais importância a essa metodologia de síntese de nanoestruturas. Alguns procedimentos hidrotermais simples, que não envolvem o emprego de estabilizantes ou outros aditivos, podem ser aplicados para a obtenção de uma variedade de sólidos nanoestruturados, desde óxidos binários e ternários até fluoretos e oxossais complexos. Tais exemplos estão sumarizados na Tabela 4S. Por exemplo, Barrera et al.105 relatam a obtenção de partículas alongadas de Lu2O3:Yb3+,Er3+ e Lu2O3:Yb3+,Tm3+ com emissões por upconversion e catodoluminescência, com a investigação de seu comportamento como guias de onda. Nesse caso, a síntese consiste na dissolução do óxido comercial em HCl e regulagem do pH com amônia (pH=7 ou 10), com posterior tratamento hidrotermal a 185 ºC por 24 h, obtendo-se partículas de 90 nm de diâmetro e 45 µm de comprimento. Já Zhong et al.106 descrevem o preparo de soluções sólidas do tipo Ce1-xZnxO2-δ, que possuem atividade catalítica na oxidação de CO, através da precipitação inicial de uma mistura de hidroxicarbonatos de cério (Ce(OH)CO3) e de zinco (Zn5(CO3)2(OH)6) em condições hidrotermais. Nesse caso, utilizam-se os nitratos metálicos em presença de ácido clorídrico e ureia, com tratamento a 160 ºC por 24 h. A metodologia leva à obtenção de nanodiscos de 400-500 nm de diâmetro e ~65 nm de espessura que, após tratamento a 600 ºC, são convertidos na solução sólida. Mei e Wu107 também obtêm óxidos ternários através de síntese hidrotermal; no caso, preparam-se estruturas alongadas de YMn2O5 com propriedades magnéticas. A síntese utiliza nitrato de ítrio, permanganato de potássio e cloreto de manganês(II) como precursores, que são misturados em solução aquosa e têm seu pH ajustado entre 3 e 8, sendo posteriormente submetidos a tratamento hidrotermal por 64 h em diferentes temperaturas (190-250 ºC). A alteração do valor de pH permite o controle morfológico do sólido, com a formação de nanobastões cada vez maiores em baixos pHs. Observou-se, ainda, o efeito da concentração dos precursores, o que permitiu a obtenção de materiais unidimensionais (nanofios) ao se utilizarem baixas concentrações dos metais. Com relação à síntese hidrotermal de estruturas alongadas em processos livres de surfactante, uma abordagem interessante é a precipitação inicial de hidróxidos de terras raras e seu posterior tratamento com agentes precipitantes. Por exemplo, nanotubos e nanobastões de NaTRF4 podem ser obtidos através do tratamento apropriados de partículas alongadas de TR(OH)3, dada a similaridade estrutural entre os hidróxidos hexagonais de terras raras e os fluoretos mistos em seu polimorfo do tipo β. Por exemplo, Zhang e Zhao108 descrevem o preparo de nanotubos de β-NaTRF4 (TR=Y,Pr,Sm,Gd,Tb,Dy,Er) em um processo de troca iônica in situ, que consiste na precipitação hidrotermal (120 ºC, 12 h) de TR(OH)3 em solução aquosa a pH=14, seguida de tratamento com NaF e HF em água por a 120 ºC por 12 h adicionais. Assim, obtêm-se nanotubos com dimensões controláveis, sendo o diâmetro entre 80 e 500 nm e as espessuras entre 25 e 80 nm. Em um procedimento bastante similar, Xu et al.109 produzem nanobastões e nanofios de β-NaTRF4 (TR=Y,Sm,Eu,Gd,Tb,Dy,Ho). As estruturas precursoras de TR(OH)3 são obtidas através da mesma metodologia utilizada por Zhang e Zhao,108 contudo a utilização de concentrações ligeiramente menores durante o tratamento das partículas de TR(OH)3, bem como o emprego de maiores temperaturas (nesse caso, 180 ºC), resultam no fechamento completo dos nanotubos e conduzem à obtenção de nanobastões com dimensões controláveis. Oxifluoretos de terras raras (TROF) também podem ser controladamente sintetizados em condições hidrotermais sem a necessidade de adição de estabilizantes. Zhang et al.110 descrevem, por exemplo, o preparo de YOF:TR3+ (TR=Eu,Tb,Tm,Yb) a partir de nitratos de TR e em presença de amônia (pH=9, 11 ou 14), utilizando-se diferentes precursores de fluoreto (NH4F, LiF, NaF, KF), a 180 ºC por 12 h; os sólidos obtidos nesse processo são posteriormente calcinados a 600 ºC. As diferentes condições de síntese permitem um controle da morfologia dos materiais finais, que normalmente se apresentam como microbastões de 5 µm de comprimento. Outras classes de partículas, tais como vanadatos de TR, também podem ser obtidas com morfologia controlada em condições hidrotermais na ausência de aditivos. Nesse sentido, Calderón-Villajos et al.111 demonstram a possibilidade de preparo de partículas de GdVO4:TR3+ (TR=Ho,Er,Tm,Yb) através da mistura de nitratos de TR com NH4VO3 em pH=7 (ajustado com NH3) em reação hidrotermal a 185 ºC por 24 h. As partículas possuem entre 30 e 45 nm e formas retangulares e quadradas, podendo ser posteriormente recobertas com uma camada de 5 nm de sílica para o aumento de usa eficiência de upconversion sob excitação no infravermelho próximo. Por outro lado, a utilização de diferentes agentes estabilizantes em solução durante as sínteses hidrotermais tende a permitir um controle mais amplo da morfologia dos sólidos finais. Preferencialmente, tais aditivos devem tanto possuir alta afinidade pelos cátions em solução quanto capacidade de se adsorver à superfície das partículas e impedir seu crescimento e agregação posteriores, sendo, ainda, estáveis ou inócuos em relação à hidrólise (isto é, não sofrer hidrólise ou, se sofrer, gerar produtos que não contaminem as partículas finais). Nesse contexto, diversos estabilizantes podem preencher tais características e se adaptar às condições de síntese, como, por exemplo, ácido cítrico (H3cit), ácido láctico (Hlat), ácido etilenodiaminotetraacético (H4edta), acetilacetona (Hacac), além de polímeros como poli(ácido acrílico) (PAA) e poli(vinilpirrolidona) (PVP), entre outros,112 como esquematizado na Tabela 5S. Uma abordagem sintética interessante é descrita por Lima e Serra,113 que utilizam ácido tripolifosfórico (H5P3O10) como agente estabilizante e como precursor de grupos ortofosfato na síntese de nanopartículas de CePO4, caso que apresenta a vantagem de não haver necessidade de eliminação do estabilizante ou possibilidade de contaminação. Com relação a estabilizantes orgânicos, acido cítrico, por exemplo, é extensivamente utilizado em sínteses em meio aquoso, possibilitando um controle preciso da morfologia dos produtos de interesse. J. Lin e seus colaboradores descrevem, por exemplo, o preparo de micro e nanoestruturas de Y(P,V)O4:TR3+114 e NaYF4:TR3+115 com o uso de citrato de sódio como estabilizante, sendo que a modificação dos parâmetros de síntese (pH, precursor, razão TR3+/cit3-, temperatura) permite a obtenção de diversas morfologias. Como ambas as fases (fosfato e fluoreto de ítrio) podem ocorrer com estrutura cristalina hexagonal, a presença de citrato leva, nos dois casos, a microcristais de hábito hexagonal. No caso das partículas de Y(P,V)O4:TR3+,114 na ausência de citrato durante a síntese hidrotermal (180 ºC, 24 h), realizada a partir de solução de cloretos de TR em presença do precursor de fosfato (NaH2PO4) e com pH ajustado com HCl, há a formação de fosfovanadatos de estrutura tetragonal, com partículas alongadas (nanospindles, 45 nm de diâmetro e 180 nm de comprimento) em que se observa crescimento preferencial na direção [001]. Já na presença de citrato, em combinação com a escolha apropriada do precursor, obtêm-se fosfovanadatos de estrutura hexagonal; empregando-se Na3PO4 em pH=6, observa-se a formação de partículas com morfologia de prisma hexagonal, com diâmetro de 120 nm e comprimento de 130 nm, com crescimento preferencial nos planos [100] e [010]. O mesmo ocorre ao se utilizar NH4H2PO4 em pH=4 como precursor, sendo que, nesse caso, obtêm-se prismas hexagonais de maiores dimensões (diâmetro de 930 nm e comprimento de 710 nm). Por fim, ao se utilizar Na5P3O10 como precursor em pH=6, observa-se a formação de nanobastões com diâmetro de 80 nm e comprimento de 180 nm, com crescimento preferencial na direção do plano [001]. Já no caso das partículas de NaYF4:TR3+,115 a síntese hidrotermal é realizada a 80-220 ºC por 24 h após a mistura dos precursores (YCl3, NaF e Na3cit) nas proporções desejadas. A variação das razões molares e da temperatura permite o controle da estrutura cristalina (cúbica (α) ou hexagonal (β)), da morfologia (nanopartículas, microprismas hexagonais, microbastões) e da razão comprimento/diâmetro observada para as partículas (desde 0,7 até 5), normalmente, com crescimento preferencial na direção [0001] das estruturas hexagonais. Han et al.116 também descrevem o preparo de fluoretos de TR micro e nanoestruturados, no caso ScF3:TR3+ (TR=Tb,Eu,Yb,Er,Tm,Ho), avaliando o efeito de outros estabilizantes além do ácido cítrico. A reação (180 ºC, 24 h) é realizada com a adição de NH4F à mistura entre TR(NO3)3 e estabilizantes (H3cit, H4edta ou CTAB), com regulagem do pH a 1. Nesse caso, enquanto o emprego cit3- resulta em grandes aglomerados de estruturas em forma de agulha (4 µm), o uso de edta resulta em estruturas cúbicas bem definidas com tamanhos entre 500 nm e 1 µm; o uso de CTAB também leva à formação de cubos, que passam a assumir menores tamanhos (200 nm) com maior homogeneidade de dimensões. A obtenção de outra classe de fluoretos por via hidrotermal, em particular de sistemas ternários xMF2-yTRF3, em que M é um metal alcalino-terroso, também é descrita por Xia et al..117 Nesse caso, emprega-se apenas edta como agente estabilizante na obtenção de Sr2YF7:TR3+ (TR=Yb,Er,Gd), que é preparado a partir dos nitratos de TR e Sr e com o uso de (NH4)HF2 como agente precipitante a 180 ºC por 12 h. O procedimento leva à obtenção de nanocristais retangulares com tamanho entre 30 e 50 nm, com alto grau de cristalinidade. O uso do estabilizante apropriado permite, inclusive, que o processo hidrotermal resulte em estruturas meso e macroporosas. Madhusudan et al.118 relatam, por exemplo, o preparo de estruturas meso/macroporosas de BiVO4 para fotocatálise, a partir do tratamento hidrotermal de misturas (previamente envelhecidas por 24 h) de Bi(NO3)3 e NH4VO3 em pH=7 a 180 ºC por 2 h. Constata-se que a adição de melamina (1,3,5-triazina-2,4,6-triamina) como estabilizante permite que se gerem estruturas com diâmetro de poro entre 5 e 200 nm, que consistem em canais interconectados às partículas. O efeito do estabilizante adicionado também pode ser modelado através da variação da composição do solvente durante uma síntese solvotermal; tais casos encontram-se resumidos na Tabela 6S. Por exemplo, o uso de etanol (em combinação ou não com a água) tende a permitir uma maior dispersibilidade dos estabilizantes, bem como a geração de uma maior pressão autógena. Cui et al.119 destacam a obtenção de submicroesferas (140-430 nm) de Y2O3 através de precipitação solvotermal (150 ºC, 12 h) de Y(NO3)3 com ureia em presença de PVP e etanol em pH=1,72, com posterior tratamento térmico das partículas a 600 ºC. O emprego de tais condições permite que se obtenham coloides monodispersos de tamanho controlável, cujo mecanismo de formação segue o modelo de La Mer. Já Hirano e Dozono120 descrevem uma metodologia solvotermal para o preparo de soluções sólidas do tipo YNbO4-TbNbO4 em toda a faixa de concentrações de Y e Tb com estrutura da fergusonita. A síntese é conduzida através da mistura dos nitratos de TR com NbCl5 em etanol e ajuste de pH com NH3, com tratamento a 240 ºC por 4 h, gerando partículas de morfologia elipsoidal. Ainda que na ausência de estabilizantes, a morfologia das partículas pode ser controlada unicamente pela razão entre solventes, como é o caso da síntese elaborada por Ma et al..121 No relato, descreve-se preparo de partículas de LaPO4 monoclínico a partir de La(NO3)3 e NaH2PO4 em meio de etanol e água, com tratamento a 200 ºC por 24 h, sendo que a variação da razão EtOH/H2O possibilita o controle da forma dos aglomerados obtidos. Outro solvente comumente utilizado em condições solvotermais, combinado ou não com água ou aditivos, é o etilenoglicol. Suas principais vantagens consistem, inicialmente, em seu poder coordenante; por se tratar de um 1,2-diol, sua ação quelante é capaz de estabilizar eficientemente cátions em solução, além de sua habilidade em se adsorver à superfície das partículas e diminuir sua taxa de agregação. Além disso, seu alto ponto de ebulição (197 ºC) permite que as sínteses sejam realizadas em temperaturas bastante elevadas com relativa segurança (devido às menores pressões geradas). Entretanto, tal como outros dióis (propilenoglicol, PG, dietilenoglicol, DEG, trietilenoglicol, TEG, etc.), seu uso normalmente deve considerar a atividade oxidorredutora das espécies em solução, pois, trata-se de um solvente de fácil oxidação. Pelo mesmo motivo, comumente é conveniente que o sistema de reação solvotermal seja desaerado (com fluxo de argônio ou nitrogênio), a fim de que se elimine o O2 dissolvido no solvente e presente no volume morto do frasco reacional. Contudo, o emprego do etileno glicol normalmente resulta em um alto controle da morfologia e cristalinidade dos materiais, sendo, portanto, um dos solventes mais empregados em sínteses solvotermais. Nesse sentido, por exemplo, Song et al.122 utilizam EG como solvente em presença de PVP como estabilizante para a obtenção de partículas altamente monodispersas e uniformes (170 nm-1,25 µm) de Gd2O2S:TR3+. O agente precipitante empregado nesse caso é a tioureia, previamente dissolvida em EtOH, realizando-se a síntese a 200 ºC por 24 h; o controle do tamanho das partículas é realizado pela variação da razão EtOH/EG. Liao et al.123 também utilizam a combinação de EG e PVP para o controle do tamanho de partículas na síntese solvotermal, nesse caso para a obtenção de luminóforos do tipo YVO4:Eu3+,Ba2+. Obtêm-se partículas submicrométricas (140 - 150 nm) bem dispersas e com morfologia quasi-octaédrica, com reação a 150 ºC (10 h) a partir de TR(NO3)3 e Na3VO4 (em pH=10) como precursores. Em condições similares, embora com o uso de NaVO3 como precursor de vanadato, Liang et al.124 sintetizam partículas monodispersas de TRVO4 (TR=Y,La,Pr-Lu) com diferentes morfologias (esferas, elipses ou plaquetas) e tamanhos (150-200 nm, 500 nm), que são aparentemente dependentes do raio iônico do íon lantanídeo utilizado. Já Yang et al.125 relatam o preparo de partículas ovais monodispersas de LaPO4:TR3+ (TR=Eu,Ce,Tb), que adquirem altas cristalinidades sem necessidade de tratamento térmico posterior, em meio de EG/H2O=37/3 (v/v) a 180 ºC. Em um procedimento muito similar, autores do mesmo grupo descrevem126 a obtenção de molibdatos de estrôncio ou bário dopados com lantanídeos (AMoO4:TR3+, A=Sr, Ba, TR=Eu,Tb) com estrutura tetragonal da scheelita, empregando a mesma razão EG/H2O. Nesse caso, obtêm-se partículas ovais ou com formato de casca de amendoim (peanut-like), com baixo grau de aglomeração e estreita distribuição de tamanhos. Outros importantes solventes coordenantes de alto ponto de ebulição compreendem aminas e ácidos carboxílicos de cadeia longa, como é o caso de oleilamina (P.E. 364 ºC) e ácido oleico (P.E. 360 ºC), que normalmente são utilizados em conjunto ou combinados com um hidrocarboneto de cadeia longa (como octadeceno, P.E. 315 ºC, por exemplo). O uso de oleilamina e ácido oleico na obtenção de diferentes classes de nanopartículas e as características intrínsecas associadas e esses solventes/ligantes são extensivamente discutidos por Mourdikoudis e Liz-Marzán,127 demonstrando-se que comumente se obtêm sólidos altamente cristalinos e com excelente dispersibilidade em meios apolares. Li et al.,128 por exemplo, relatam a obtenção de KTR3F10 (TR=Sm-Lu,Y) em condições solvotermais utilizando ácido oleico em presença de KOH como solvente/estabilizante e TR(NO3)3 e KF aquosos como precursores, com reação a 180 ºC por 36 h. Nesse caso, obtêm-se partículas coloidais de diversas morfologias (esferas, cubos e folhas hexagonais) e de alta cristalinidade, capazes de formar coloides límpidos em ciclo-hexano e clorofórmio. De maneira similar, Bu et al.129 descrevem a síntese de molibdatos mistos de TR e sódio (NaLa(MoO4)2) através do controle cooperativo da cristalização com ácido oleico e oleilamina, obtendo-se nanocristais monodispersos com morfologia octaédrica ou alongada após reação a 140 ºC por 6 h. O uso de ácido oleico também permite a obtenção de nanoesferas monodispersas de SrYbF5 com ~10 nm de diâmetro, com a reação dos nitratos metálicos aquosos com NH4F a 220 ºC por 24 h, conforme descrito por Gong et al.130 Reações com aquecimento por micro-ondas Uma abordagem bastante interessante e que consiste em uma área de pesquisa de rápido crescimento é o uso de irradiação por micro-ondas como método de aquecimento em diferentes tipos de reações químicas, seja em recipientes abertos, seja em condições hidro ou solvotermais. O campo apresenta uma grande abrangência com relação à síntese de compostos orgânicos ou inorgânicos e tende a continuar crescendo nos próximos anos, sendo possível, inclusive, o uso de tais métodos para escalas não-laboratoriais. O aquecimento por micro-ondas consiste em uma alternativa às metodologias tradicionais, as quais se baseiam na convecção e condução de energia na forma de calor, permitindo rápidos aquecimentos volumétricos e menores tempos de processo. Isso leva a maiores velocidades e seletividades de reação, ao mesmo tempo em que se diminuem os custos e se poupa energia. A frequência normalmente utilizada em laboratório (e também em aparelhos domésticos) é de 2,45 GHz, que corresponde a um comprimento de onda de ~12,2 cm. Na presença das micro-ondas nessa frequência, moléculas polares ou íons em um solvente ou em um sólido tendem a se orientar ou se movimentar de acordo com o campo eletromagnético da radiação, o que leva ao aquecimento de qualquer material contendo esse tipo de espécies (Figura 5). No caso de solventes polares, as moléculas tendem a se orientar com o campo alternante, de modo que se gera calor devido à rotação, fricção e colisão entre as moléculas, o que corresponde ao chamado mecanismo aquecimento por polarização dipolar. No caso de íons, a flutuação do campo elétrico faz com que as espécies carregadas se movam em solução, mudando constantemente de direção, o que resulta em um aumento local de temperatura devido a fricção e colisões. Esse efeito é chamado de aquecimento por mecanismo de condução iônica, e também é observado em amostras semicondutoras e condutoras, em que os elétrons constituintes formam uma corrente elétrica que dissipa energia na forma de calor devido à resistência elétrica intrínseca do material.131 Com tais características de aquecimento, as micro-ondas são capazes de aumentar a taxa de colisões efetivas (levando a uma nucleação mais rápida e homogênea), além de aumentar a cristalinidade dos materiais graças às altas temperaturas e pressões atingidas. Em outras palavras, a irradiação por micro-ondas leva a um aquecimento rápido, uniforme e energeticamente eficiente do sistema, o que torna o método absolutamente vantajoso para o preparo de diferentes materiais, tendo sido adaptada para a síntese de vários tipos de nanopartículas.
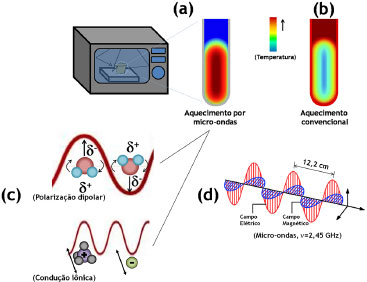 Figura 5. Ilustraçao (a) do perfil de aquecimento de amostras sob irradiaçao de micro-ondas em comparaçao ao (b) perfil de aquecimento convencional (conduçao/convecçao), e (c) representaçao dos mecanismos de aquecimento (polarizaçao dipolar e conduçao iônica) envolvidos nos processos químicos induzidos pelas micro-ondas, descritas figurativamente em (d)
Nesse sentido, recentemente, Zhu e Chen131 e Baghbanzadeh et al.132 discutiram extensivamente a obtenção de nanoestruturas em fase líquida com irradiação por micro-ondas, recomendando-se tal literatura ao leitor interessado em maiores detalhes. Já Kitchen et al.133 discutem com profundidade o emprego de irradiação por micro-ondas em sínteses de materiais inorgânicos, com maior ênfase nos mecanismos envolvidos e no desenvolvimento de técnicas in-situ para o estudo de reações desse tipo com resolução temporal. Tais trabalhos lidam com aparelhagens de micro-ondas especificamente desenvolvidas para reações químicas, embora, há um razoável tempo, já houvesse grande interesse no uso dessa ferramenta, ainda que através da adaptação de aparelhos domésticos.134 Em todo caso, o uso da irradiação por micro-ondas permite a utilização das mesmas abordagens aplicadas nas sínteses hidro- e solvotermais convencionais (uso de estabilizantes, variação de solventes etc.), porém se observam, geralmente, tempos de reação expressivamente menores. Algumas metodologias de síntese com aquecimento por micro-ondas estão descritas na Tabela 7S. É caso, por exemplo, da obtenção de diferentes estruturas esféricas de TROOH (TR=Gd,Eu) descrita por Majeed e Shivashankar,135 que utilizam acetilacetonatos ([TR(acac)3(H2O)2]) em EtOH como precursores, realizando a reação a 150 ºC com tempo total de cerca de 7 min. A evolução da morfologia das partículas é avaliada com respeito ao tempo de reação, gerando estruturas finais que são praticamente monodispersas e possuem cerca de 1,3 µm de diâmetro, sendo constituídas pela agregação de nanopartículas menores que 20 nm; as partículas obtidas podem ser convertidas nos óxidos correspondentes sem que se perca a morfologia esférica, apresentando, contudo, um forte incremento de suas propriedades luminescentes. Por outro lado, Jin et al.136 utilizam a irradiação por microondas (30 W) para a síntese de fosfovanadatos de TR (Y(P,V)O4:TR3+, TR=Eu,Sm,Dy) a 150 ºC em diferentes tempos (5-60 min), através da mistura dos cloretos precursores com Na3VO4 e Na3PO4 em presença de PVP. Nesse caso, obtêm-se partículas com alto grau de cristalinidade, com tamanhos de grão entre 20 e 40 nm. Runowski et al.137 também obtêm nanobastões de LaPO4:Tb3+ que são posteriormente recobertos com camada de sílica e modificados com grupos NH2 com o intuito de aumentar a biocompatibilidade das partículas. Nesse caso, o tratamento hidrotermal com micro-ondas (600 W, 180 ºC, 40 bar, 2 h) tem a função de aumentar a cristalinidade das partículas previamente sintetizadas por coprecipitação (adição gota a gota de solução de NH4H2PO4 à solução de TR(NO3)3, ambas em presença de PEG 6000). Há também os casos em que as reações de precipitação são realizadas em refluxo, mesmo em aparelhos domésticos adaptados. Esse é o caso descrito por Ma et al.,138 que preparam nanobastões similares aos de Runowski et al.137 utilizando-se os mesmos precursores, porém com regulagem a pH=2 com HNO3 e reação por 20 min sob irradiação a ~400 W de potência em um micro-ondas doméstico. Fluoretos de TR também são muito frequentemente sintetizados com aquecimento por micro-ondas, sendo que sua facilidade de cristalização acaba por exigir temperaturas e tempos razoavelmente baixos. Wang et al.,139 por exemplo, sintetizam nanocristais de GdF3 dopado com diferentes íons TR a partir da mistura de nitratos metálicos e NaBF4 em meio aquoso, com aquecimento a 85 ºC e tempo total de 10 min (aplicando-se potência de 200 W). Já Mi et al.140 obtêm nanocristais de LaF3:Ce3+,Tb3+ em meio de etilenoglicol em presença de polietilenoimina (PEI10000), através da mistura dos cloretos de lantanídeos com NaF a 120 ºC por 30 min, gerando partículas uniformes de estrutura hexagonal e diâmetros de ~12 nm. Outro procedimento bastante interessante é apresentado por Li et al.,141 que propõem o uso de líquidos iônicos, particularmente [BMIM][BF4], em combinação à irradiação por microondas (300 W, 150 ºC, 20 min) para a obtenção de TRF3. Nesse caso, o líquido iônico atua como absorvedor de microondas, como solvente e como precursor de íons fluoreto, permitindo o controle da morfologia e da estrutura dos sólidos pela variação de parâmetros como tempo de reação e composição. A obtenção de fluoretos mistos (NaTRF4) com aquecimento por micro-ondas também é frequentemente descrita na literatura, utilizando-se por exemplo, etilenoglicol como solvente.142,143 Enquanto Niu et al.142 realizam a reação em sistema aberto a 160 ºC (300 W, 50 min) utilizando nitratos de TR e NH4F como precursores, Mi et al.143 conduzem a reação à mesma temperatura em recipiente selado, utilizando acetatos de TR e diferentes precursores de fluoreto (NH4F, NH4HF2, NaF e [BMIM][BF4]) em presença de NaCl; ambas as metodologias levam a partículas de ~50 nm de alta cristalinidade com seletividade estrutural. Ding et al.144 também descrevem o preparo de microbastões de β-NaYF4:TR3+ (TR=Yb,Er,Tm,Ho) em ~12 min, utilizando uma metodologia baseada na reação entre os nitratos de TR sólidos e NaF em presença de NaNO3 e KNO3. O aquecimento da amostra é realizado utilizando-se grafita como absorvedor de micro-ondas, de modo que o recipiente contendo os reagentes é imerso em outro recipiente contendo o absorvedor, possibilitando um elevado aquecimento in situ.
MICROEMULSÕES REVERSAS COMO NANORREATORES O conceito de microemulsão deriva dos experimentos de Hoar e Schulman em 1943, quando da obtenção de uma fase límpida após a titulação, com 1-hexanol, de uma emulsão turva de água, hidrocarbonetos e CTAB.43,46,145 Já o termo "microemulsão" foi criado em 1959, fazendo alusão ao tamanho reduzido das gotas que compunham o sistema. Esse termo foi constantemente retificado até 1981, quando Danielsson e Lindman definiram uma microemulsão como sendo "um sistema contendo água, óleo e tensoativo(s), que consiste em uma dispersão líquida opticamente isotrópica e termodinamicamente estável".146 Contudo, tal definição leva, em princípio, ao entendimento de que as microemulsões podem ser descritas como as soluções convencionais, o que não é correto uma vez que a orientação das moléculas de tensoativo não é aleatória. Assim, modernamente, uma microemulsão deve ser tratada como uma mistura termodinamicamente estável e macroscopicamente homogênea de água, óleo e tensoativo(s) que, em nível microscópico, consiste em domínios individuais de água e óleo separados por uma monocamada de tensoativo(s). Note-se que os termos água e óleo referem-se, na verdade, a quaisquer líquidos de polaridades substancialmente diferentes que sejam, portanto, imiscíveis em condições normais. As propriedades das microemulsões tornam-se mais claras ao se traçarem paralelos com as emulsões. Ao contrário do que inicialmente se pode pensar, por exemplo, a diferença mais marcante entre os dois tipos de sistema não é o tamanho das gotas, mas sim a estabilidade termodinâmica das microemulsões. Tal como as soluções comuns, as microemulsões são sistemas que tendem a se formar espontaneamente (ΔG<0), mantendo suas propriedades inalteradas após a sua formação, uma vez que se atinge o equilíbrio químico entre seus componentes. Já as (macro)emulsões são sistemas intrinsecamente instáveis (ou cuja estabilidade é mantida cineticamente), sendo que suas gotas eventualmente sofrerão coalescência e suas fases se segregarão. Enquanto as emulsões apresentam gotas relativamente grandes (de 1 a 10 µm, capazes de espalhar eficientemente a luz visível - daí seu aspecto turvo), os agregados nas microemulsões são cerca de mil vezes menores (~10 nm - que dão aspecto homogêneo ao sistema). Deve-se mencionar, ainda, que o termo "nanoemulsão" não se refere a esse tipo de sistema, sendo tradicionalmente empregado para descrever emulsões (termodinamicamente instáveis) com tamanhos de gota menores que 100 nm.147 Além disso, emulsões convencionais são sistemas relativamente estáticos, enquanto as microemulsões são sistemas altamente dinâmicos (com frequentes trocas de moléculas entre as diferentes fases e agregados). Por possuírem superfícies internas muito elevadas e por apresentarem filmes interfaciais óleo/água com curvaturas muito altas em comparação com as emulsões convencionais, as microemulsões requerem quantidades de surfactantes consideravelmente maiores. A termodinâmica de formação das microemulsões pode ser explicada levando-se em conta sua estabilidade, de modo que a energia livre de dispersão de uma fase em outra deve ser negativa (ΔGdisp<0).148,149 A energia livre de dispersão pode ser expressa como sendo um balanço entre a energia livre necessária para a formação das microestruturas componentes e a contribuição entrópica envolvida no processo:37 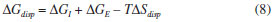 O termo ΔGI corresponde à energia livre associada ao aumento da área superficial com a formação da microemulsão, portanto:  em que γOA é a tensão interfacial entre a fase aquosa e a fase óleo e ΔA é a variação de área com a formação das gotas dispersas. Como ΔA >> 0, o termo ΔGI não favorecerá a formação da microemulsão a menos que γOA assuma valores ultrabaixos (tipicamente na faixa de 10-5 a 10-2 mN m-1) ou mesmo negativos. O tratamento do ponto de vista termodinâmico para a ocorrência de valores negativos de tensão interfacial é tratado em várias revisões.150-152 A diminuição da tensão interfacial é proporcionada pela presença de um surfactante e, muitas vezes, de um cossurfactante (normalmente, um álcool de cadeia média). O papel do cossurfactante é diminuir as repulsões eletrostáticas e/ou estéricas entre as cabeças dos surfactantes, aumentando, assim, a estabilidade dos agregados. O tipo da cadeia do surfactante também é determinante para as propriedades da microemulsão, uma vez que a curvatura da gota é da mesma ordem do tamanho das cadeias carbônicas. Dessa forma, não apenas o balanço hidrófilo-lipófilo (BHL) é importante, mas também a geometria da molécula anfifílica e seu parâmetro de empacotamento são fundamentais para o design de tais sistemas (Figura 6).37,46 Cadeias lineares são mais adequadas à formação de microemulsões óleo-em-água; cadeias altamente ramificadas favorecem a formação de sistemas água-em-óleo; cadeias de ramificação intermediária são propícias à formação de microemulsões bicontínuas.
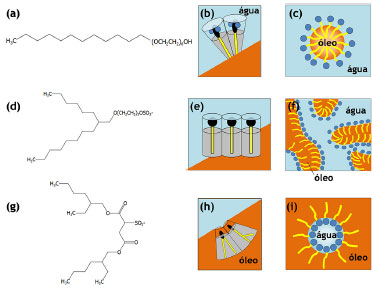 Figura 6. Ilustraçao da influência da cadeia carbônica do surfactante [(a),(d),(g)] na organizaçao interfacial [(b),(e),(h)] e na estrutura dos agregados micelares [(c),(f),(i)] em microemulsoes: (a)-(c) álcool etoxilado formando microemulsoes óleo/água; (d)-(f) éter-sulfato de cadeia ramificada para a composiçao de microemulsoes bicontínuas; e (g)-(i) sulfonato de cadeia dupla gerando microemulsoes água/óleo46
O termo ΔGE da equação (8) é associado às interações atrativas e repulsivas entre os agregados micelares, de modo que sua parcela atrativa é devida às forças de van der Waals entre as micelas, enquanto a fração repulsiva é dada pelas interações eletrostáticas entre as duplas camadas elétricas e pelas repulsões estéricas envolvidas na formação da microemulsão. Tais fatores são relacionados à natureza dos componentes e eletrólitos presentes na microemulsão, sendo, também, relacionados à curvatura do filme interfacial e à razão surfactante/cossurfactante. De acordo com as equações (6) e (7), ao se considerarem altos valores de separação entre as estruturas micelares, tanto o potencial atrativo quanto o repulsivo tornam-se desprezíveis em comparação às outras contribuições à energia livre de dispersão. Já em baixas separações, o potencial atrativo de van der Waals é muito pequeno frente ao potencial repulsivo das duplas camadas elétricas, de modo que a contribuição das interações de van der Waals ao termo ΔGE pode ser desprezada.148 Dessa forma, a contribuição dos efeitos de atrativos e repulsivos à energia livre de dispersão pode ser associada apenas à repulsão entre as gotas dispersas no sistema, de modo que:  em que a integral representa o trabalho necessário para a aproximação de duas gotas de mesma carga nos volumes disponíveis para as fases macroscópicas contra o potencial elétrico de repulsão. Esse termo também é positivo, uma vez que é necessário trabalho externo para confinar um grande número de partículas de carga similar em um volume finito. Dessa forma, a formação da microemulsão somente será favorável se o termo entrópico compensar a soma dos efeitos de aumento de área e de repulsão representados pelas equações (9) e (10). O termo entrópico é dado por 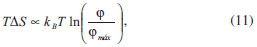 em que φ e φmáx correspondem à fração de volume ocupada pelas gotas no sistema e a fração volume que a fase dispersa ocuparia considerando-se o máximo empacotamento possível para esferas rígidas (arranjo cúbico centrado nas faces ou hexagonal densamente empacotado). Como o volume excluído pelo movimento térmico e pelas repulsões não é desprezível, tem-se que φ >φmáx em qualquer situação, de modo que o termo entrópico é sempre positivo. Assim, a microemulsão se manterá termodinamicamente estável quando ΔGI + ΔGΕ < TΔS. Dentre a vasta gama de aplicações das microemulsões, que vão desde sistemas de drug-delivery à recuperação terciária de petróleo,37,46,153-155 uma de suas mais elegantes utilizações consiste em seu emprego como meios micro-heterogêneos para a realização de diversas reações químicas.156-162 Nesses casos, são frequentemente utilizadas microemulsões reversas (ou "inversas"), em cujo núcleo aquoso acontecem as reações de precipitação, sendo que o sólido tem seu tamanho e forma modulados pelas propriedades estruturais dos agregados micelares, no que se pode chamar de "química de zeptolitros". Tais microemulsões reversas podem ser caracterizadas pela razão molar entre água e surfactante, ω0:43 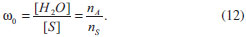 Esta razão pode ser relacionada ao raio da micela reversa, considerando-se que o volume micelar VM é dado pela soma dos volumes ocupados pela água e pelo surfactante: 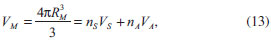 em que nS e nA são os números de mols de surfactante e água por micela e VS e VA seus volumes molares. Assumindo que a área superficial da micela AM é definida apenas pelo surfactante, temos que:  Combinando-se as equações (12), (13) e (14), obtém-se uma expressão para o raio micelar: 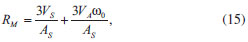 que relaciona diretamente o tamanho da micela reversa à razão água/surfactante utilizada para a composição da microemulsão (sendo os demais parâmetros constantes características do surfactante). Embora tal relação seja bastante objetiva, sua aplicação em sistemas reais é consideravelmente complexa, uma vez que as microemulsões se formam somente em faixas relativamente estreitas de composição, além de frequentemente ocorrer a presença de um quarto ou quinto componente na mistura (cossurfactantes). Além disso, fatores como força iônica e temperatura também influenciam o equilíbrio de fases. Dessa forma, a relação entre o raio micelar e a razão água/surfactante é geralmente determinada de forma empírica, de onde se derivam os parâmetros do surfactante para cada sistema. A realização de reações em microemulsões deve considerar a natureza dinâmica de tais sistemas, uma vez que diversos tipos de processos são envolvidos em uma ampla faixa de escalas de tempo. Por exemplo, os processos dinâmicos envolvidos na formação de microemulsões compreendem desde a escala de picossegundos (movimento das cadeias alquílicas nos agregados) à escala de microssegundos a minutos (dissolução de água ou óleo entre as fases).163 Os processos dinâmicos de interesse particular à realização de reações químicas em microemulsões são a troca de moléculas de surfactante entre o agregado e a fase contínua (~100 ns) e a colisão/coalescência das gotas (µs a ms). As reações em microemulsões reversas envolvem o movimento browniano a que estão sujeitos os agregados, que resulta em frequentes colisões entre as micelas. Em parte dessas colisões, há a formação de dímeros micelares de vida curta através da remoção de algumas moléculas de surfactante para o interior da fase contínua. Nessas ocasiões, ocorre a troca dos conteúdos (aquosos, no caso de microemulsões reversas) das duas micelas durante o período de vida do dímero (~100 ns a poucos microssegundos), levando a uma distribuição homogênea de todos os componentes após sua descoalescência (Figura 7). Assumindo-se que a colisão, coalescência e troca de conteúdo (também chamado de "solubilizato") entre agregados envolvam um tempo característico τex e que a reação química de interesse envolva um tempo característico τR, três situações podem ser traçadas com respeito à influência da microemulsão no andamento da reação. Se a reação de interesse ocorre em tempos muito baixos, da ordem de poucos nanossegundos (τR << τex), as gotas podem ser consideradas como sistemas isolados, de modo que a troca de conteúdos dos agregados pode ser desconsiderada. No caso de reações com tempo característico da mesma ordem do tempo de colisão/coalescência (τR /τexc ≈1), a fusão entre agregados pode ser tratada como uma etapa limitante para a ocorrência da reação, sendo que a dinâmica da microemulsão afetará a cinética e o equilíbrio da reação química. Por fim, se a reação ocorrer em tempos maiores que os envolvidos na comunicação entre as gotas (τR >> τex), a fase dispersa pode ser tradada como sendo um contínuo (pseudo-fase), de modo que a microemulsão pode ser considerada como um sistema "estático" com respeito à reação química.163 Para solventes comuns de baixa viscosidade, a taxa de encontro entre as partículas (ken) pode ser estimada como sendo da ordem de 106 s-1, o que resulta em um tempo médio para colisão (τcol = ken-1) estimado em ~1 µs. Como nem todas as colisões levam à troca do solubilizato, a taxa de coalescência com troca de conteúdo (kex = τex-1) pode ser tratada como sendo uma fração da taxa total de encontro, sendo que kex = γken. O termo γ é, portanto, um fator de encontro que depende tanto da natureza dos reagentes contidos nas gotas quanto da rigidez do filme de surfactantes. O valor de γ varia desde 10-3 em filmes rígidos (o que significa que, a cada mil colisões entre agregados, uma resulta na troca de conteúdo) a até 10-1 em filmes flexíveis, levando, assim, a diferentes tempos característicos τex anteriormente discutidos.162-164
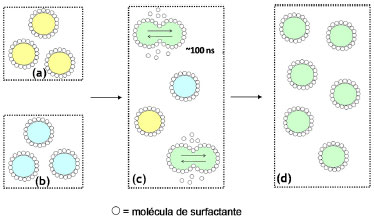 Figura 7. Representaçao esquemática do processo de dispersao dos solubilizatos das microemulsoes precursoras (a) e (b) através da colisao entre micelas reversas, com a formaçao dos dímeros de vida curta nos quais ocorre a mistura das fases aquosas (c), o que é seguido pela dissociaçao das micelas e formaçao de agregados com conteúdo homogeneizado (d)
Ao se realizarem reações nos nanorreatores aquosos, garante-se não só o controle do tamanho dos agregados através da variação de ω0, como também a distribuição eficiente dos conteúdos devido ao movimento browniano, além de uma alta homogeneidade das nanopartículas formadas. Entretanto, tais situações envolvem complexidade adicional, uma vez que as constantes de equilíbrio e as velocidades de reação podem ser alteradas em até uma ordem de grandeza em relação aos sistemas não nanoestruturados. Além disso, os íons presentes na solução afetam não só a estabilidade da micela reversa (efeitos de solvatação e interação com a cabeça polar do surfactante), como também o equilíbrio de fases. A escolha do surfactante também deve ser cuidadosa, levando-se em conta tanto seu efeito na formação da microemulsão, quanto sua possível interferência nos produtos formados. Com respeito à síntese de materiais inorgânicos, microemulsões são extensivamente aplicadas para o preparo de nanopartículas metálicas através de reações de oxidorredução,165 a síntese de óxidos metálicos por coprecipitação ou processos sol-gel e para a obtenção de partículas core@shell.160-162,166 Com relação aos surfactantes empregados para a composição de microemulsões aplicadas à síntese de nanomateriais, o bis(2-etil-hexil)sulfossuccinato de sódio (também conhecido como Aerosol-OT ou NaAOT, representado na Figura 6(g)) é o surfactante mais extensivamente utilizado. De natureza aniônica e com cadeia bastante ramificada, o AOT permite a solubilização de grandes volumes de água na fase não-polar mesmo sem a utilização de cossurfactantes, sendo, inclusive, bastante estável à hidrólise e a reações paralelas. Contudo, como o AOT forma filmes bastante rígidos, a adição de moléculas de álcoois de cadeia intermediária (como 1-butanol ou 1-hexanol) normalmente é realizada, o que tende a aumentar a fluidez da superfície e diminuir a rigidez do filme. Além disso, moléculas estabilizantes com relação às nanopartículas formadas também são utilizadas em conjunto com o AOT e seus eventuais cossurfactantes; tais estabilizantes compreendem moléculas neutras com grupos polares de alta capacidade de interação com nanopartículas, como o trioctilfosfinóxido (TOPO), tributilfosfato (TBP) dodecanotiol (DDT) ou bis(2-etil-hexil)amina (BEA).162,163,166 Outros surfactantes aniônicos, como laurilsulfato de sódio também são relatados na literatura, embora em frequência muito menor que o AOT.160-162 Os surfactantes catiônicos mais utilizados são os sais de amônio quaternário como o brometo de hexadeciltrimetilamônio (CTAB) ou o brometo de didodecildimetilamônio (DDAB). Embora o DDAB seja, em virtude de sua geometria molecular, muito mais efetivo para a geração de microemulsões, seu custo relativamente elevado em comparação ao CTAB diminui a extensão de seu uso, de modo que o CTAB, de cadeia alifática simples, é o surfactante catiônico mais utilizado na composição de nanorreatores. O CTAB normalmente forma filmes muito flexíveis, com uma elevada dinâmica de troca de solubilizato entre as micelas, sendo também capaz de solubilizar elevadas quantidades de soluções aquosas de altas forças iônicas.162 Quanto aos surfactantes não-iônicos, os mais frequentemente utilizados são o Triton®-X100, o NP-5 (polioxietileno(5)nonilfeniléter), e o NP-9 (polioxietileno(9)nonilfeniléter), sendo que os tensoativos Brij® 52,56 ou 58 e Tween® 20, 60 e 80 também têm seu uso descrito; o leitor é referido a uma literatura mais específica para a discussão das diferentes características dos surfactantes iônicos e não-iônicos empregados em microemulsões.46,160,162,163,166 No caso da síntese de compostos de TR, cujos exemplos são listados na Tabela 8S, um dos problemas da utilização do AOT (bem como de outros surfactantes aniônicos) é a interação do tensoativo com os íons metálicos em solução, levando, por exemplo, à formação de compostos insolúveis. Isso, de forma alguma, inviabiliza o emprego desses surfactantes aniônicos, porém é muito mais frequente a utilização de surfactantes não-iônicos ou catiônicos como o CTAB. Nesse caso, os íons brometo interagem fracamente com os cátions TR3+ aquosos, de modo que as únicas restrições são quanto a interações com outras espécies ou reações redox no caso particular de íons Ce3+/Ce4+. Nesse sentido, o CTAB é bastante aplicado para a composição de microemulsões aplicadas na síntese de fluoretos de TR de diferentes morfologias. Por exemplo, Wang et al.167 obtêm estruturas similares a feixes de fibras de YF3 dopadas com Yb3+ e Tm3+, que são compostas por fios de ~700 nm de comprimento e ~2 nm de diâmetro. Utilizam-se, nesse caso, fluoreto de sódio e cloretos de TR aquosos como precursores, dispersos em uma microemulsão que tem ciclo-hexano como fase apolar contínua e CTAB/1-pentanol como surfactante/cossurfactante, com reação à temperatura ambiente. Nesse caso, controla-se a morfologia final dos sólidos através da variação da concentração de TRCl3 e NaF e da quantidade de água (razão ω0), além de que a aplicação de maiores temperaturas de síntese (até 160 ºC) conduz à obtenção de estruturas de menor comprimento e menor diâmetro. Janssens et al.168 descrevem a obtenção de nanobastões que se agregam em estruturas com morfologia similar, porém com composição BaMgF4:M (M=Ce3+,Eu3+,Nd3+ e Mn2+), utilizando-se nitratos metálicos e fluoreto de amônio como precursores. Nesse caso, a microemulsão consiste na fase aquosa confinada por CTAB e 2-octanol, que parece atuar como cossurfactante e fase apolar, de modo que os autores afirmam obter fases transparentes. Já Huang et al.169 empregam uma microemulsão similar (CTAB/1-butanol/ciclo-hexano/solução aquosa) para o preparo de nanoestruturas de NaTRF4 (TR=Y,Yb,Er), utilizando nitratos de TR e NH4HF2 como precursores em presença de acetato de sódio como controlador da força iônica. Nesse caso, combinam-se as vantagens das microemulsões com as das sínteses solvotermais, realizando-se a reação a 210 ºC por períodos entre 5 h e 40 h. Com a variação do tempo de reação, é possível controlar a morfologia das partículas, que varia desde esferas de ~100 nm de diâmetro (5 h de reação) a microtubos semiabertos de ~10 µm de comprimento e 1 µm de diâmetro (em tempos superiores a 30 h), com transformação dos sólidos isotrópicos de estrutura cúbica em estruturas anisotrópicas hexagonais com os maiores tempos de reação. A utilização de surfactantes diferentes do CTAB para a obtenção de fluoretos é também possível, tal como descrito por Lemyre e Ricey na obtenção de partícuals de YF3.170 Os autores empregam o surfactante NP-5 e ciclo-hexano como fase apolar com YCl3 e NH4HF2 aquosos como precursores, conduzindo a reação de duas formas: (i) reação convencional entre duas microemulsões ou (ii) adição direta do precursor de fluoreto aquoso à microemulsão contendo TR. Tal variação permite um controle muito interessante da morfologia das nanopartículas, que são esferas monodispersas (com tamanho controlável entre 6 e 50 nm) e amorfas no caso da reação convencional entre duas microemulsões. Já no caso da adição direta de NH4HF2 à microemulsão de TR, observa-se a formação de produtos monocristalinos com morfologia quadrilateral ou hexagonal, com tamanhos entre 50 e 350 nm, sendo que se obtêm partículas maiores ao se utilizar menores concentrações de surfactante. Essa diferença é atribuída ao fato de que, com a adição direta, a incorporação do agente precipitante às micelas torna-se mais lenta, diminuindo a velocidade de formação do sólido. Com relação à obtenção de fosfatos de TR, a literatura relata tanto o emprego de microemulsões à base de CTAB quanto de AOT, que normalmente levam à obtenção de nanoestruturas alongadas.171-173 O emprego de CTAB é possível, por exemplo, utilizando-se 1-butanol como cossurfactante e heptano como fase apolar, com NaH2PO4 e TR(NO3)3 como precursores aquosos.171 A realização da reação em refluxo por 24 h leva à obtenção de nanoestruturas alongadas (~30 nm de diâmetro) com comprimento ajustável de acordo com a razão água/CTAB utilizada. Constata-se que, nesse caso, a taxa de precipitação das partículas inorgânicas é maior que a taxa de coalescência/descoalescência das micelas reversas, o que leva à obtenção das estruturas alongadas. A adição de ácido cítrico como agente estabilizante inibe esse crescimento, permitindo a produção de nanobastões com menor diferença entre comprimentos e diâmetros e com menor grau de agregação. Nanobastões de fosfatos de TR também podem ser obtidos com o uso de AOT como surfactante, tal como descrito por S. Mann e seus colaboradores.172,173 Por exemplo, obtêm-se nanofios de CePO4 com diâmetro médio de ~3,7 nm e comprimento de ~250 nm, que são amorfos quando de sua obtenção mas adquirem alta cristalinidade após os procedimentos de remoção do solvente e surfactante.172 Nesse caso, emprega-se NaAOT em meio de iso-octano, sendo Na2HPO4 e Ce(NO3)3 os precursores em pH=5,5; as microemulsões são misturadas e deixadas em temperatura ambiente por períodos de até um mês. Outra abordagem bastante interessante descreve o preparo de nanobastões uniformes de CePO4 com 4-5 nm de diâmetro e comprimento controlável até 1200 nm.173 Nesses casos, os precursores são os próprios contra-íons dos surfactantes utilizados, formando agregados que os autores chamam de micelas reversas "cataniônicas". Para isso, prepara-se o bis(2-etil-hexil)sulfossuccinato de cério(III) (Ce(AOT)3) e o ortofosfato de hexadeciltrimetilamônio ((CTA)3PO4), que são dissolvidos em iso-octano em presença de água, com razão Ce(AOT)3/(CTA)3PO4=1:1. As partículas de CePO4 assim obtidas são altamente cristalinas e de superfície hidrofóbica, sendo morfologicamente invariantes com respeito aos parâmetros de reação.173 Zhang et al.174 também descrevem a obtenção de nanoestruturas alongadas de fosfatos de TR, utilizando, contudo, um método que combina as características das microemulsões com as de reações hidrotermais utilizando líquidos iônicos. Além disso, emprega-se tributilfosfato (TBP) como agente estabilizante, num caso que consiste em microemulsões "óleo-em-água" à base de líquidos iônicos. Para isso, nitratos de TR (TR=La-Tb) são dissolvidos em TBP e dispersos em uma solução contendo o líquido iônico [Omim]Cl (cloreto de 1-octil-3-metilimidazólio), o que forma uma microemulsão transparente; a esse sistema, adiciona-se solução aquosa de NaH2PO4, de modo que a mistura é mantida a 100 ºC por 48 h em autoclave. A síntese conduz à obtenção de fosfatos de TR de estrutura hexagonal, que têm diâmetros de 5-15 nm e comprimentos de até 10 µm. Surfactantes não iônicos também podem ser utilizados na elaboração de estruturas de fosfatos, como é descrito, por exemplo, por Sun et al.,175 que prepara nanopartículas de hidroxiapatita dopadas com TR em condições hidrotermais. A síntese consiste em uma microemulsão ciclo-hexano/Triton® X-100/1-butanol/solução aquosa, utilizando nitratos de cálcio, lantânio e európio e (NH4)2HPO4 em pH=8-9 como precursores, com tratamento hidrotermal a 160 ºC por 12 h após a mistura das microemulsões iniciais. Além disso, o emprego de condições hidro- e solvotermais também fornece bons resultados de controle morfológico no caso de microemulsões baseadas em ácido oleico/oleilamina, que atuam tanto como estabilizantes como surfactantes. Um exemplo dessa aplicação é descrito por Deng et al.,176 que sintetizam nanopartículas de vanadatos de TR com morfologias de prato quadrado ou em forma de H. A síntese é conduzida dissolvendo-se os nitratos de TR precursores (TR=Ce,Nd) em água e posterior dispersão em 1-octadeceno em presença de ácido oleico e oleilamina; essa mistura é inicialmente tratada a 100 ºC por 30 min e, após esse período, adiciona-se o precursor de vanadato (Na3VO4) em presença de NaOH, sendo que a mistura é mantida a 180 ºC por 16-24 h. Tanto o tempo de reação quanto a quantidade de NaOH permitem o controle morfológico, sendo que em períodos mais curtos obtêm-se as estruturas quadradas, enquanto longos tempos de reação e maiores valores de pH conduzem às nanopartículas em forma de H. Como descrito, as microemulsões são bastante apropriadas para a síntese de compostos de TR de alta insolubilidade, capazes de cristalizar em condições bastante brandas. Contudo, mesmo óxidos de mais difícil cristalização também podem ser preparados através do emprego de microemulsões. Por exemplo, Rill et al.177 preparam coloides de óxidos/hidróxidos de TR a partir de microemulsões contendo CTAB, Triton® X-100 ou dodecilsulfato de sódio (SDS), com a avaliação do efeito dos parâmetros da microemulsão no tamanho de partícula e na cristalinidade. Utiliza-se, nesse caso, amônia como agente precipitante e cloretos de TR como precursores, sendo que a fase orgânica da microemulsão consiste em ciclo-hexano e o cossurfactante é, em todos os casos, 1-hexanol. A mesma metodologia, particularmente com o uso de Triton® X-100, pode ser estendida e adaptada para a obtenção de partículas de outros óxidos notadamente estáveis, como é o caso das ferritas como a magnetita (Fe3O4).178 Além disso, mesmo estruturas de cristalização consideravelmente complexa, como é o caso do Y3Al5O12 (YAG) podem ser preparadas pela adaptação dessas metodologias, tal como é descrito por Hsu et al.,179 que empregam um protocolo diferente da reação tradicional de duas microemulsões. Nesse caso, prepara-se uma microemulsão baseada em núcleos aquosos, contendo os íons metálicos precursores (no caso, Y3+, Al3+ e Eu3+), dispersos em ciclo-hexano, com polioxietileno(10)octilfeniléter (OP-10) e 1-hexanol como surfactante e cossurfactante. Essa microemulsão é adicionada gota a gota em querosene aquecida (~180 ºC), sendo que o sistema é ligado a uma bomba para a evaporação da água presente; essa solução final é seca a 500 ºC por 2 h, gerando as nanopartículas precursoras, que são posteriormente tratadas termicamente. Nesse caso, observa-se que a diminuição da razão água/surfactante na microemulsão inicial de fato resulta na diminuição do tamanho das partículas finais (de 80 a 30 nm).
CONSIDERAÇÕES FINAIS O presente texto revisa os conceitos fundamentais relacionados a sistemas coloidais envolvendo nanopartículas inorgânicas, abrangendo técnicas tradicionalmente consolidadas ou mais recentemente desenvolvidas sobre os aspectos preparativos de materiais à base de terras raras. De maneira geral, todos os protocolos discutidos consistem, em sua essência, na aplicação dos princípios físico-químicos básicos que regem os fenômenos de nucleação e crescimento, agregação e adsorção a estruturas sólidas num meio líquido reacional. Com base nesses conceitos, é possível a elaboração de metodologias de síntese em fase líquida de nanopartículas inorgânicas com as mais variadas características, às quais se associa um imenso leque de aplicações. O emprego de tais técnicas consiste, sem dúvida, nas metodologias mais importantes para a obtenção de nanomateriais, visto que as mesmas possibilitam uma distribuição homogênea de todos os precursores envolvidos, permitindo, ainda, a introdução de diferentes substâncias (estabilizantes, complexantes) no sistema reacional. Dessa forma, diferentes estratégias bottom-up de síntese podem ser aplicadas para a manipulação das propriedades morfológicas dos materiais e adaptação de suas características às aplicações vislumbradas. Além disso, tratam-se de processos simples, seguros, relativamente rápidos e de baixo consumo energético, o que torna essas abordagens consideravelmente vantajosas com respeito a outras técnicas frequentemente aplicadas. Isso se constata, por exemplo, pela enorme fração a que essas metodologias correspondem em comparação ao total de publicações envolvendo síntese de nanopartículas, como se pode constatar no portal ISI Web of Knowledge. O Brasil, tal como em outras áreas, ocupa um papel cada vez mais sólido com relação ao desenvolvimento e aplicação de sínteses de nanomateriais inorgânicos em fase líquida, ganhando mais e mais espaço nesse campo frente à comunidade internacional. Em virtude da posição que o país ocupa também com relação ao seu patrimônio de minérios de terras raras, há que se considerar que se devem destinar esforços especiais não apenas à prospecção e separação desses elementos, mas também às suas aplicações em diferentes campos tecnológicos, com a busca por uma produção científica e tecnológica cada vez maior e de maior impacto na área da obtenção de nanomateriais de terras raras em fase líquida.
MATERIAL SUPLEMENTAR Tabelas adicionais acerca das metodologias descritas ao longo do texto estão disponíveis em http://quimicanova.sbq.org.br, na forma de arquivo PDF, com acesso livre.
AGRADECIMENTOS Os autores agradecem à CAPES, ao CNPq (Proc. 4791290/2011, OAS), ao inct-INAMI e à FAPESP (Proc. 2013/01669-1, PCdSF) pelos auxílios financeiros. Os autores agradecem, ainda, à Dr.ª J. F. Lima pelo auxílio durante a elaboração de algumas imagens.
REFERÊNCIAS 1. Goesmann, H.; Feldmann, C.; Angew. Chem. Int. Ed. 2010, 49, 1362. DOI: http://dx.doi.org/10.1002/anie.200903053 2. Martins, M. A.; Trindade, T.; Quim. Nova 2012, 35, 1434. DOI: http://dx.doi.org/10.1590/S0100-40422012000700026 3. Bastús, N. G.; Casals, E.; Ojea, I.; Varon, M.; Puntes, V. In The Delivery of Nanopaticles; Hashim, A. A., ed. InTech (Online), 2012. Capítulo 18. 4. Cuyena, B. R.; Thin Solid Films 2010, 518, 3127. DOI: http://dx.doi.org/10.1016/j.tsf.2010.01.018 5. Casals, E.; Gonzalez, E.; Puntes, V. F.; J. Phys. D: Appl. Phys. 2012, 45, 443001. DOI: http://dx.doi.org/10.1088/0022-3727/45/44/443001 6. Stern, S. T.; McNeil, S. E.; Toxicol. Sci. 2008, 101, 4. DOI: http://dx.doi.org/10.1093/toxsci/kfm169 PMID: 17602205 7. Irmer, G.; Moneke, J.; Verma, P. In Encyclopedia of Nanoscience and Nanotechnology; Nalwa, H. S., ed., American Scientific Publishers (Online), 2003, Volume 4. 8. Ringe, E.; Langille, M. R.; Sohn, K.; Zhang, J.; Huang, J.; Mirkin, C. A.; J. Phys. Chem. Lett. 2012, 3, 1479. DOI: http://dx.doi.org/10.1021/jz300426p 9. Steigerwald, M. L.; Brus, L. E.; Acc. Chem. Res. 1990, 23, 183. DOI: http://dx.doi.org/10.1021/ar00174a003 10. Lai, S. L.; Guo, J. Y.; Petrova, V.; Ramanath, G.; Allen, L. H.; Phys. Rev. Lett. 1996, 77, 99. DOI: http://dx.doi.org/10.1103/PhysRevLett.77.99 PMID: 10061781 11. Nel, A. E.; Mädler, L.; Velegol, D.; Xia, T.; Hoek, E. M. V.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M.; Nat. Mater. 2009, 8, 543. DOI: http://dx.doi.org/10.1038/nmat2442 PMID: 19525947 12. Obonyo, O.; Fisher, E.; Edwards, M.; Douroumis, D.; Crit. Rev. Biotechnol. 2010, 30, 283. DOI: http://dx.doi.org/10.3109/07388551.2010.487184 PMID: 20528252 13. Polshettiwar, V.; Varma, R. S.; Green Chem. 2010, 12, 743. DOI: http://dx.doi.org/10.1039/b921171c 14. Mulvihill, M. J.; Beach, E. S.; Zimmerman, J. B.; Anastas, P. T.; Annu. Rev. Environ. Resour. 2011, 36, 271. DOI: http://dx.doi.org/10.1146/annurev-environ-032009-095500 15. Colombo, M.; Carregal-Romero, S.; Casula, M. F.; Gutiérrez, L.; Morales, M. O, Böhm, I. B.; Heverhagen, J. T.; Prosperi, D.; Parak, W. J.; Chem. Soc. Rev. 2012, 41, 4306. DOI: http://dx.doi.org/10.1039/c2cs15337h PMID: 22481569 16. Alivisatos, P.; Nat. Biotech. 2004, 22, 47. DOI: http://dx.doi.org/10.1038/nbt927 17. Medintz, I. L.; Uyeda, H. T.; Goldman, E. R.; Mattoussi, H.; Nat. Mater. 2005, 4, 435. DOI: http://dx.doi.org/10.1038/nmat1390 PMID: 15928695 18. Mazumder, S.; Dey, R.; Mitra, M. K.; Mukherjee, S.; Das, G. C. ; J. Nanomater. 2009, 815734. 19. Shen, J.; Sun, L.-D.; Yan, C.-H.; Dalton Trans. 2008, 42, 5687. DOI: http://dx.doi.org/10.1039/b805306e PMID: 18941653 20. Aroca, R. F.; Phys. Chem. Chem. Phys. 2013, 15, 5355. DOI: http://dx.doi.org/10.1039/c3cp44103b PMID: 23493861 21. Pieczonka, N. P. W.; Aroca, R. F.; Chem. Soc. Rev. 2008, 37, 946. DOI: http://dx.doi.org/10.1039/b709739p PMID: 18443680 22. Brus, L.; Acc. Chem. Res. 2008, 41, 1742. DOI: http://dx.doi.org/10.1021/ar800121r PMID: 18783255 23. Wang, Y.; Schlücker, S.; Analyst 2013, 138, 2224. DOI: http://dx.doi.org/10.1039/c3an36866a PMID: 23420174 24. Sevonkaev, I.; Privman, V.; Goia, D.; J. Solid State Electrochem. 2013, 17, 279. DOI: http://dx.doi.org/10.1007/s10008-012-1954-6 25. Ebbesen, T. W.; Genet, C.; Bozhevolnyi, S. I.; Phys. Today 2008, 61, 44. DOI: http://dx.doi.org/10.1063/1.2911177 26. Dionne, J. A.; Sweatlock, L. A.; Atwater, H. A.; Polman, A.; Phys. Rev. B 2006, 73, 035407. DOI: http://dx.doi.org/10.1103/PhysRevB.73.035407 27. Zahmakiran, M.; Özkar, S.; Nanoscale 2011, 3, 3462. DOI: http://dx.doi.org/10.1039/c1nr10201j PMID: 21833406 28. Li, Y.; Somorjai, G.A.; Nano Lett. 2010, 10, 2289. DOI: http://dx.doi.org/10.1021/nl101807g PMID: 20524636 29. Zhou, J.; Ralston, J.; Sedev, R.; Beattie, D. A.; J. Colloid Interface Sci. 2009, 331, 251. DOI: http://dx.doi.org/10.1016/j.jcis.2008.12.002 PMID: 19135209 30. Xia, Y.; Xiong, Y.; Lim, B.; Skrabalak, S. E.; Angew. Chem. Int. Ed. 2009, 48, 60. DOI: http://dx.doi.org/10.1002/anie.200802248 31. Silva, F. O.; Viol, L. C. S.; Ferreira, D. L.; Alves, J. L. A.; Schiavon, M. A.; Quim. Nova 2010, 33, 1933. DOI: http://dx.doi.org/10.1590/S0100-40422010000100025 32. Reiss, P.; Protière, M.; Li, L.; Small 2009, 5, 154. DOI: http://dx.doi.org/10.1002/smll.200800841 PMID: 19153991 33. Buissette, V.; Giaume, D.; Gacoin, T.; Boilot, J.-P.; J. Mater. Chem. 2006, 16, 529. DOI: http://dx.doi.org/10.1039/B508656F 34. Yan, Z.-G.; Yan, C.-H.; J. Mater. Chem. 2008, 18, 5046. DOI: http://dx.doi.org/10.1039/b810586c 35. Laurent, S.; Forge, D.; Port, M.; Roch, A.; Robic, C.; Vander Elst, L.; Muller, R. N.; Chem. Rev. 2008, 108, 2064. DOI: http://dx.doi.org/10.1021/cr068445e PMID: 18543879 36. McNaught, A. D.; Wilkinson, A.; IUPAC Compendium of Chemical Terminology ("Gold Book") 2nd Ed. (online), Blackwell Scientific Publications: Oxford, 2006. (http://goldbook.iupac.org, acessada em Julho 2013). 37. Goodwin, J.; Colloids and Interfaces with Surfactants and Polymers - An Introduction, John Wiley & Sons: Chichester, 2004. 38. Graham, T.; Philos. Trans. R. Soc. London 1861, 151, 183. DOI: http://dx.doi.org/10.1098/rstl.1861.0011 39. Faraday, M.; Philos. Trans. R. Soc. London 1857, 147, 145. DOI: http://dx.doi.org/10.1098/rstl.1857.0011 40. La Mer, V. K.; Dinegar, R. H.; J. Am. Chem. Soc. 1950, 72, 4847. DOI: http://dx.doi.org/10.1021/ja01167a001 41. Viswanatha, R.; Sarma, D. D. In Nanomaterials Chemistry: Recent Developments and New Directions; Rao, C. N. R.; Müller, A.; Cheetham, A. K., eds., Wiley: Weinhein, 2007. Capítulo 4. 42. Füredi-Milhofer, H.; Pure Appl. Chem. 1981, 53, 2041. 43. Cushing, B. L.; Kolesnichenko, V. L.; O'Connor, C. J.; Chem. Rev. 2004, 104, 3893. DOI: http://dx.doi.org/10.1021/cr030027b PMID: 15352782 44. Ocaña, M.; Rodriguez, C.; Serna, C. J.; Adv. Mater. 1995, 7, 212. DOI: http://dx.doi.org/10.1002/adma.19950070225 45. Sugimoto, T.; Adv. Colloid Interface Sci. 1987, 28, 65. DOI: http://dx.doi.org/10.1016/0001-8686(87)80009-X 46. Holmberg, K.; Jönsson, B.; Kronberg, B.; Lindman, B.; Surfactants and Polymers in Aqueous Solutions, 2nd ed., John Wiley & Sons: Chichester, 2002. 47. Lifshitz, I. M.; Slyozov, V. V.; J. Phys. Chem. Solids. 1961, 19, 35. DOI: http://dx.doi.org/10.1016/0022-3697(61)90054-3 48. Yao, J. H.; Elder, K. R.; Guo, H.; Grant, M.; Phys. Rev. B 1993, 47, 14110. DOI: http://dx.doi.org/10.1103/PhysRevB.47.14110 49. Niethammer, B.; Arch. Rational Mech. Anal. 1999, 147, 119. DOI: http://dx.doi.org/10.1007/s002050050147 50. Pashley, R. M.; Karaman, M. E.; Applied Colloid and Surface Chemistry, John Wiley & Sons: Chichester, 2004. 51. Alexander, M.; Dalgleish, D. G.; Food Biophys. 2006, 1, 2. DOI: http://dx.doi.org/10.1007/s11483-005-9000-1 52. Hoo, C. M.; Starostin, N.; West, P.; Mecartney, M. L.; J. Nanopart. Res. 2008, 10, 89. DOI: http://dx.doi.org/10.1007/s11051-008-9435-7 53. Sennett, P.; Olivier, J. P.; Ind. Eng. Chem. 1965, 57, 32. DOI: http://dx.doi.org/10.1021/ie50670a007 54. Doane, T.; Chuang, C.-H.; Hill, R. J.; Burda, C.; Acc. Chem. Res. 2012, 45, 317. DOI: http://dx.doi.org/10.1021/ar200113c PMID: 22074988 55. Mialon, G.; Türkan, S.; Dantelle, G.; Collins, D. P.; Hadjipanayi, M.; Taylor, R. A.; Gacoin, T.; Alexandrou, A.; Boilot, J.-P.; J. Phys. Chem. C 2010, 114, 22449. DOI: http://dx.doi.org/10.1021/jp107900z 56. Reveaux, A.; Dantelle, G.; George, N.; Seshadri, R.; Gacoin, T.; Boilot, J.-P.; Nanoscale 2011, 3, 2015. DOI: http://dx.doi.org/10.1039/c0nr01000f 57. Bertail, C.; Maron, S.; Buissette, V.; Le Mercier, T.; Gacoin, T.; Boilot, J.-P.; Chem. Mater. 2011, 23, 2961. DOI: http://dx.doi.org/10.1021/cm2005902 58. Sickafus, K. E.; Wills, J. M.; Grimes, N. W.; J. Am. Ceram. Soc. 1999, 82, 3279. DOI: http://dx.doi.org/10.1111/j.1151-2916.1999.tb02241.x 59. Ahn, T.; Kim, J. H.; Yang, H.-M.; Lee, J. W.; Kim, J.-D.; J. Phys. Chem. C. 2012, 116, 6069. DOI: http://dx.doi.org/10.1021/jp211843g 60. Valenzuela, R.; Fuentes, M. C.; Parra, C.; Baeza, J.; Duran, N.; Sharma, S. K.; Knobel, M.; Freer, J.; J. Alloys Compd. 2008, 488, 227. DOI: http://dx.doi.org/10.1016/j.jallcom.2009.08.087 61. Pereira, C.; Pereira, A. M.; Fernandes, C.; Rocha, M.; Mendes, R.; Fernández-García, M. P.; Guedes, A.; Tavares, P. B.; Grenèche, J.-M.; Araújo, J. P.; Freire, C.; Chem. Mater. 2012, 24, 1496. DOI: http://dx.doi.org/10.1021/cm300301c 62. Asif Iqbal, M.; ul-Islam, M.; Ali, I.; Khan, H. M.; Mustafa, G.; Ali, I.; Ceram. Int. 2013, 39, 1539. DOI: http://dx.doi.org/10.1016/j.ceramint.2012.07.104 63. Ali, I.; ul-Islam, M.; Ishaque, M.; Khan, H. M.; Ashiq, M. N.; Rana, M. U.; J. Magn. Magn. Mater. 2012, 324, 3773. DOI: http://dx.doi.org/10.1016/j.jmmm.2012.06.008 64. Meng, Y. Y.; Liu, Z. W.; Dai, H. C.; Yu, H. Y.; Zeng, D. C.; Shukla, S.; Ramanujan, R. V.; Powder Technol. 2012, 229, 270. DOI: http://dx.doi.org/10.1016/j.powtec.2012.06.050 65. Taniguchi, T.; Watanabe, T.; Sakamoto, N.; Matsushita, N.; Yoshimura, M.; Cryst. Growth Des. 2008, 8, 3725. DOI: http://dx.doi.org/10.1021/cg800363w 66. Almeida, J. M. A.; Santos, P. E. C.; Cardoso, L. P.; Meneses, C. T.; J. Magn. Magn. Mater. 2013, 327, 185. DOI: http://dx.doi.org/10.1016/j.jmmm.2012.09.007 67. Xin, Y.; Qi, Y.; Ma, X.; Wang, Z. Zhang, Z.; Zhang, S.; Mater. Lett. 2010, 64, 2659. DOI: http://dx.doi.org/10.1016/j.matlet.2010.08.061 68. Feldmann, C.; Jüstel, T.; Ronda, C.; Schmidt, P.; Adv. Funct. Mater. 2003, 13, 511. DOI: http://dx.doi.org/10.1002/adfm.200301005 69. Davis, J. B.; Marshall, D. B.; Housley, R. M.; Morgan, P. E. D.; J. Am. Ceram. Soc. 1998, 81, 2169. DOI: http://dx.doi.org/10.1111/j.1151-2916.1998.tb02602.x 70. Terra, O.; Clavier, N.; Dacheux, N.; Podor, R.; New J. Chem. 2003, 27, 957. DOI: http://dx.doi.org/10.1039/b212805p 71. Buissette, V.; Moreau, M.; Gacoin, T.; Boilot, J.-P.; Chane-Ching, J.-Y.; Le Mercier, T.; Chem. Mater. 2004, 16, 3767. DOI: http://dx.doi.org/10.1021/cm049323a 72. Yang, M.; You, H.; Liu, K.; Zheng, Y.; Guo, N.; Zhang, H.; Inorg. Chem. 2010, 49, 4996. DOI: http://dx.doi.org/10.1021/ic100099w PMID: 20462235 73. Prasad, A. I.; Parchur, A. K.; Juluri, R. R.; Jadhav, N.; Pandey, B. N.; Ningthoujam, R. S.; Vatsa, R. K.; Dalton Trans. 2013, 42, 4885. DOI: http://dx.doi.org/10.1039/c2dt32508j PMID: 23370409 74. Liu, Z.; Wang, Q.; Yao, S.; Yang, L.; Yu, S.; Feng, X.; Li, F.; Ceram. Inter. 2014, 40, 2613. DOI: http://dx.doi.org/10.1016/j.ceramint.2013.06.101 75. Yan, Z.-G.; Yan, C.-H.; J. Mater. Chem. 2008, 18, 5046. DOI: http://dx.doi.org/10.1039/b810586c 76. Wang, G.; Peng, Q.; Li, Y.; Acc. Chem. Res. 2011, 44, 322. DOI: http://dx.doi.org/10.1021/ar100129p PMID: 21395256 77. Huignard, A.; Gacoin, T.; Boilot, J.-P.; Chem. Mater. 2000, 12, 1090. DOI: http://dx.doi.org/10.1021/cm990722t 78. Huignard, A.; Buissette, V.; Laurent, G.; Gacoin, T.; Boilot, J.-P.; Chem. Mater. 2002, 14, 2264. DOI: http://dx.doi.org/10.1021/cm011263a 79. Kumar, V.; Khan, A. F.; Chawla, S.; J. Phys. D: Appl. Phys. 2013, 46, 365101. DOI: http://dx.doi.org/10.1088/0022-3727/46/36/365101 80. Rahman, P.; Green, M.; Nanoscale 2009, 1, 214. DOI: http://dx.doi.org/10.1039/b9nr00089e PMID: 20644840 81. Fedorov, P. P.; Luginina, A. A.; Kuznetsov, S. V.; Osiko, V. V.; J. Fluorine Chem. 2011, 132, 1012. DOI: http://dx.doi.org/10.1016/j.jfluchem.2011.06.025 82. Grzyb, T.; Lis, S.; J. Rare Earths 2009, 27, 588. DOI: http://dx.doi.org/10.1016/S1002-0721(08)60294-X 83. Fedorov, P. P.; Mayakova, M. N.; Kuznetsov, S. V.; Voronov, V. V.; Ermakov, R. P.; Samarina, K. S.; Popov, A. I.; Osiko, V. V.; Mater. Res. Bull. 2012, 47, 1794. DOI: http://dx.doi.org/10.1016/j.materresbull.2012.03.027 84. Lezhnina, M. M.; Jüstel, T.; Kätker, H.; Wiechert, D. U.; Kynast, U. H.; Adv. Funct. Mater. 2006, 16, 935. DOI: http://dx.doi.org/10.1002/adfm.200500197 85. Cui, X.; She, J.; Gao, C.; Cui, K.; Hou, C.; Wei, W.; Peng, B.; Chem. Phys. Lett. 2010, 494, 60. DOI: http://dx.doi.org/10.1016/j.cplett.2010.05.070 86. Grzyb, T.; Runowski, M.; Dąbrowska, K.; Giersig, M.; Lis, S.; J. Nanopart. Res. 2013, 15, 1958. DOI: http://dx.doi.org/10.1007/s11051-013-1958-x PMID: 24273438 87. Sekhar, H.; Rao, T.; Reddy, P. H.; Rao, D. N.; J. Alloys Compd. 2013, 562, 38. DOI: http://dx.doi.org/10.1016/j.jallcom.2013.01.194 88. Xiao, X.-Z.; Yan, B.; J. Alloys Compd. 2008, 448, 298. DOI: http://dx.doi.org/10.1016/j.jallcom.2006.10.088 89. Chawla, S.; Rajkumar, R.; Khan, A. F.; Kotnala, R. K.; J. Lumin. 2013, 136, 328. DOI: http://dx.doi.org/10.1016/j.jlumin.2012.11.039 90. Szczeszak, A.; Lis, S.; Naginyi, V.; J. Rare Earths 2011, 29, 1142. DOI: http://dx.doi.org/10.1016/S1002-0721(10)60613-8 91. Zhang, W.; Long, J.; Fan, A.; Li, J.; Mater. Res. Bull. 2012, 47, 3479. DOI: http://dx.doi.org/10.1016/j.materresbull.2012.06.075 92. Mahmood, A.; Warsi, M. F.; Ashiq, M. N.; Sher, M.; Mater. Res. Bull. 2012, 47, 4197. DOI: http://dx.doi.org/10.1016/j.materresbull.2012.09.003 93. Silva, R. F.; DeOliveira, E.; de Sousa Filho, P. C.; Neri, C. R.; Serra, O. A.; Quim Nova 2011, 34, 759. DOI: http://dx.doi.org/10.1590/S0100-40422011000100027 94. D'assunçao, L. M.; Giolito, I.; Ionashiro, M.; Thermochim. Acta 1989, 137, 319. DOI: http://dx.doi.org/10.1016/0040-6031(89)87224-7 95. Sorbell, C.; Barja, B. C.; Jobbágy, M.; J. Mater. Chem. C 2014, 2, 1010. DOI: http://dx.doi.org/10.1039/C3TC31952K 96. Raju, G. S. R.; Pavitra, E.; Yu, J. S.; Dalton Trans. 2013, 42, 11400. DOI: http://dx.doi.org/10.1039/c3dt51154e PMID: 23817306 97. Lv, R.; Gai, S.; Dai, Y.; Niu, N.; He, F.; Yang, P.; ACS Appl. Mater. Interfaces 2013, 5, 10806. DOI: http://dx.doi.org/10.1021/am4041652 PMID: 24106741 98. Jia, Y.; Sun, T.-Y.; Wang, J.-H.; Huang, H.; Li, P.; Yu, X.-F.; Chu, P. K.; CrystEngComm 2014, 16, 6141. DOI: http://dx.doi.org/10.1039/C4CE00440J 99. Yin, Y.; Rioux, R. M.; Erdonmez, C. K.; Hughes, S.; Somorjai, G. A.; Alivisatos, P.; Science 2004, 304, 711. DOI: http://dx.doi.org/10.1126/science.1096566 PMID: 15118156 100. Fan, H. J.; Gösele, U.; Zacharias, M.; Small 2007, 3, 1660. DOI: http://dx.doi.org/10.1002/smll.200700382 PMID: 17890644 101. Byrappa, K.; Adschiri, T.; Prog. Cryst. Growth Charact. 2007, 53, 117. DOI: http://dx.doi.org/10.1016/j.pcrysgrow.2007.04.001 102. Rabenau, A.; Angew. Chem. Int. Ed. 1985, 24, 1026. DOI: http://dx.doi.org/10.1002/anie.198510261 103. Reverchon, E.; Adami, R.; J. Supercrit. Fluids 2006, 37, 1. DOI: http://dx.doi.org/10.1016/j.supflu.2005.08.003 104. Feldmann, C.; Adv. Funct. Mater. 2003, 13, 101. DOI: http://dx.doi.org/10.1002/adfm.200390014 105. Barrera, E. W.; Pujol, M. C.; Díaz, F.; Choi, S. B.; Rotermund, F.; Park, K. H. Jeong, M. S.; Cascales, C.; Nanotechnology 2011, 22, 075205. DOI: http://dx.doi.org/10.1088/0957-4484/22/7/075205 PMID: 21233550 106. Zhong, S.-L.; Zhang, L.-F.; Wang, L.; Huang, W.-X.; Fan, C.-M.; Xu, A.-W.; J. Phys. Chem. C 2012, 116, 13127. DOI: http://dx.doi.org/10.1021/jp3017826 107. Mei, Y.; Wu, S.; RSC Adv. 2013,3, 11888. DOI: http://dx.doi.org/10.1039/c3ra41671b 108. Zhang, F.; Zhao, D.; ACS Nano 2009, 3, 159. DOI: http://dx.doi.org/10.1021/nn800533v PMID: 19206262 109. Xu, Z.; Li, C.; Yang, P.; Zhang, C.; Huang, S.; Lin, J.; Cryst. Growth Des. 2009, 9, 4752. DOI: http://dx.doi.org/10.1021/cg800287e 110. Zhang, Y.; Li, X.; Geng, D.; Shang, M.; Lian, H.; Cheng, Z.; Lin, J.; CrystEngComm. 2014, 16, 2196. DOI: http://dx.doi.org/10.1039/c3ce42323a 111. Calderón-Villajos, R.; Zaldo, C.; Cascales, C.; Nanotechnology 2012, 23, 505205. DOI: http://dx.doi.org/10.1088/0957-4484/23/50/505205 PMID: 23196274 112. Casavola, M.; Buonsanti, R.; Caputo, G.; Cozzoli, P.D.; Eur. J. Inorg. Chem. 2008, 837. DOI: http://dx.doi.org/10.1002/ejic.200701047 113. Lima, J. F.; Serra, O. A.; Dyes Pigm. 2013, 57, 291. DOI: http://dx.doi.org/10.1016/j.dyepig.2012.12.020 114. Li, C.; Hou, Z.; Zhang, C.; Yang, P.; Li, G.; Xu, Z.; Fan, Y.; Lin, J.; Chem. Mater. 2009, 21, 4598. DOI: http://dx.doi.org/10.1021/cm901658k 115. Li, C.; Zhang, C.; Hou, Z.; Wang, L.; Quan, Z.; Lian, H.; Lin, J.; J. Phys. Chem. C 2009, 113, 2332. DOI: http://dx.doi.org/10.1021/jp808280z 116. Han, L.; Wang, Y.; Guo, L.; Zhao, L.; Tao, Y.; Nanoscale 2014, 6, 5907. DOI: http://dx.doi.org/10.1039/c4nr00512k PMID: 24759954 117. Xia, Z.; Du, P.; Liao, L.; Phys. Status Solidi A 2013, 210, 1734. 118. Madhusudan, P.; Kumar, M. V.; Ishigaki, T.; Toda, K.; Uematsu, K.; Sato, M.; Environ. Sci. Pollut. Res. 2013, 20, 6638. DOI: http://dx.doi.org/10.1007/s11356-013-1694-x 119. Cui, Y.; Lai, X.; Li, L.; Hu, Z.; Wang, S.; Halpert, J. E.; Yu, R.; Wang, D.; ChemPhysChem 2012, 13, 2610. DOI: http://dx.doi.org/10.1002/cphc.201100806 PMID: 22213203 120. Hirano, M.; Dozono, H.; Mater. Chem. Phys. 2014, 143, 860. DOI: http://dx.doi.org/10.1016/j.matchemphys.2013.10.025 121. Ma, M.-G.; Deng, F.; Yao, K.; Mater. Lett. 2014, 124, 173. DOI: http://dx.doi.org/10.1016/j.matlet.2014.03.050 122. Song, Y.; You, H.; Huang, Y.; Yang, M.; Zheng, Y.; Zhang, L.; Guo, N.; Inorg. Chem. 2010, 49, 11499. DOI: http://dx.doi.org/10.1021/ic101608b PMID: 21105693 123. Liao, Y.; Chen, N.; Du, G.; J. Alloys Comp. 2013, 561, 214. DOI: http://dx.doi.org/10.1016/j.jallcom.2013.01.185 124. Liang, X.; Kuang, S.; Li, Y.; J. Mater. Res. 2011, 26, 1168. DOI: http://dx.doi.org/10.1557/jmr.2011.36 125. Yang, P.; Quan, Z.; Li, C.; Hou, Z.; Wang, W.; Lin, J.; J. Solid State Chem. 2009, 182, 1045. DOI: http://dx.doi.org/10.1016/j.jssc.2008.10.014 126. Yang, P.; Li, C.; Wang, W.; Quan, Z.; Gai, S.; Lin, J.; J. Solid State Chem. 2009, 182, 2510. DOI: http://dx.doi.org/10.1016/j.jssc.2008.10.014 127. Mourdikoudis, S.; Liz-Marzán, L.; Chem. Mater. 2013, 25, 1465. DOI: http://dx.doi.org/10.1021/cm4000476 128. Li, C.; Xu Z.; Yang, D.; Cheng, Z.; Hou, Z.; Ma, P.; Lian, H.; Lin, J.; CrystEngComm 2012, 14, 670. DOI: http://dx.doi.org/10.1039/C1CE05810J 129. Bu, W.; Chen, Z.; Chen, F.; Shi, J.; J. Phys. Chem. C 2009, 113, 12176. DOI: http://dx.doi.org/10.1021/jp901437a 130. Gong, L.; Yang, J.; Li, Y.; Ma, M.; Xu, C.; Ren, G.; Lin, J.; Yang, Q.; J. Mater. Sci. 2013, 48, 3672. DOI: http://dx.doi.org/10.1007/s10853-013-7163-6 131. Zhu, Y.-J.; Chen, F.; Chem. Rev. 2014, 114, 6462. DOI: http://dx.doi.org/10.1021/cr400366s PMID: 24897552 132. Baghbanzadeh, M.; Carbone, L.; Cozzoli, P. D.; Kappe, C. O.; Angew. Chem. Int. Ed. 2011, 50 11312. DOI: http://dx.doi.org/10.1002/anie.201101274 133. Kitchen, H. J.; Vallance, S. R.; Kennedy, J. L.; Tapia-Ruiz, N.; Carassiti, L.; Harrison, A.; Whittaker, A. G. Drysdale, T. D.; Kingman, S. W.; Gregory, D. H.; Chem. Rev. 2014, 114, 1170. DOI: http://dx.doi.org/10.1021/cr4002353 PMID: 24261861 134. Pecoraro, E.; Davolos, M. R,; Jafelicci, Jr., M.; Quim Nova 1997, 20, 89. DOI: http://dx.doi.org/10.1590/S0100-40421997000100011 135. Majeed, S.; Shivashankar, S. A.; J. Mater. Chem. C 2014, 2, 2965. DOI: http://dx.doi.org/10.1039/c3tc32492c 136. Jin, Y.; Li, C.; Xu, Z.; Cheng, Z.; Wang, W.; Li, G.; Lin, J.; Mater. Chem. Phys. 2011, 129, 418. DOI: http://dx.doi.org/10.1016/j.matchemphys.2011.04.035 137. Runowski, M.; Dąbrowska, K.; Grzyb, T.; Miernikiewicz, P.; Lis, S.; J. Nanopart. Res. 2013, 15, 2068. DOI: http://dx.doi.org/10.1007/s11051-013-2068-5 PMID: 24307860 138. Ma, L.; Xu, L.-M.; Chen, W.-X.; Xu, Z.-D.; Mater. Lett. 2009, 63, 1635. DOI: http://dx.doi.org/10.1016/j.matlet.2009.04.038 139. Wang, S.; Su, S.; Song, S.; Deng, R.; Zhang, H.; CrystEngComm 2012, 14, 4266. DOI: http://dx.doi.org/10.1039/C1CE05810J 140. Mi, C.-C.; Tian, Z.-H.; Han, B.-F.; Mao, C.-B.; Xu, S.-K.; J. Alloys Compd. 2012, 525, 154. DOI: http://dx.doi.org/10.1016/j.jallcom.2012.02.095 PMID: 22879690 141. Li, C.; Ma, P.; Yang, P.; Xu, Z.; Li., G.; Yang, D.; Peng, C.; Lin, J.; CrystEngComm 2011, 13, 1003 DOI: http://dx.doi.org/10.1039/C0CE00093K 142. Niu, N.; He, F.; Gai, S.; Li, C.; Zhang, X.; Huang, S.; Yang, P; J. Mater. Chem. 2012, 22, 21613. DOI: http://dx.doi.org/10.1039/c2jm34653b 143. Mi, C.; Tian, Z.; Cao, C.; Wang, Z.; Mao, C.; Xu, S.; Langmuir 2011, 27, 14632. DOI: http://dx.doi.org/10.1021/la204015m PMID: 22029665 144. Ding, M.; Lu, C.; Ni, Y.; Xu, Z.; Chem. Eng. J. 2014, 241, 477. DOI: http://dx.doi.org/10.1016/j.cej.2013.10.045 145. de Oliveira, A. G.; Scarpa, M. V.; Correa, M. A.; Cera, L. F. R.; Formariz, T. P.; Quim. Nova 2004, 27, 131. DOI: http://dx.doi.org/10.1590/S0100-40422004000100023 146. Danielsson, I.; Lindman, B.; Colloids Surf. 1981, 3, 391. DOI: http://dx.doi.org/10.1016/0166-6622(81)80064-9 147. McClements, D. J.; Soft Matter 2012, 8, 1719. DOI: http://dx.doi.org/10.1039/C2SM06903B 148. Ruckenstein, E.; Chi, J. C.; J. Chem. Soc., Faraday Trans. 2 1975, 71, 1690. DOI: http://dx.doi.org/10.1039/f29757101690 149. Nagarajan, R.; Ruckenstein, E.; Langmuir 2000, 16, 6400. DOI: http://dx.doi.org/10.1021/la991578t 150. Patashinski, A. Z.; Orlik, R.; Ratner, M. A.; Grzybowski, B. A.; Soft Matter 2012, 8, 1601. DOI: http://dx.doi.org/10.1039/C1SM06590D 151. Corti, D. S.; Kerr, K. J.; Torabi, K.; J. Chem. Phys. 2011, 135, 024701. DOI: http://dx.doi.org/10.1063/1.3609274 PMID: 21766963 152. Prince, L. M.; J. Colloid Interface Sci. 1967, 23, 165. DOI: http://dx.doi.org/10.1016/0021-9797(67)90099-9 153. Schwuger, M.-J.; Stickdorn, K.; Schomäcker, R.; Chem. Rev. 1995, 95, 849. DOI: http://dx.doi.org/10.1021/cr00036a003 154. Israelachvili, J.; Colloids Surf., A 1994, 91, 1. DOI: http://dx.doi.org/10.1016/0927-7757(94)02743-9 155. Paul, B. K.; Moulik, S. P.; J. Dispersion Sci. Technol. 1997, 18, 301. DOI: http://dx.doi.org/10.1080/01932699708943740 156. Pileni, M.-P.; J. Phys. Chem. 1993, 97, 6961. DOI: http://dx.doi.org/10.1021/j100129a008 157. Pileni, M.-P.; Langmuir 1997, 13, 3266. DOI: http://dx.doi.org/10.1021/la960319q 158. Pileni, M.-P.; Nat. Mater. 2003, 2, 145. DOI: http://dx.doi.org/10.1038/nmat817 PMID: 12612669 159. Ganguli, A. K.; Ganguly, A.; Vaidya, S.; Chem. Soc. Rev. 2010, 39, 474. DOI: http://dx.doi.org/10.1039/B814613F PMID: 20111772 160. Uskoković, V.; Drofenik, M.; Adv. Colloid Interface Sci. 2007, 133, 23. DOI: http://dx.doi.org/10.1016/j.cis.2007.02.002 PMID: 17400166 161. López-Quintela, M. A.; Curr. Opin. Colloid Interface Sci. 2003, 8, 137. DOI: http://dx.doi.org/10.1016/S1359-0294(03)00019-0 162. López-Quintela, M. A.; Tojo, C.; Blanco, M. C.; García Rio, L.; Leis, J. R.; Curr. Opin. Colloid Interface Sci. 2004, 9, 264. DOI: http://dx.doi.org/10.1016/j.cocis.2004.05.029 163. Tondre, C. Em Dynamics of Surfactants Self-Assemblies: Micelles, Microemulsions, Vesicles and Lyotropic Phases; Zana, R., ed., CRC Press (Online), 2005. Capítulo 5. 164. Fletcher, P. D. I.; Howe, A. M.; Robinson, B. H.; J. Chem. Soc. Faraday Trans. 1 1987, 83, 985. DOI: http://dx.doi.org/10.1039/f29878300985 165. Capek, I.; Adv. Colloid Interface Sci. 2004, 110, 49. DOI: http://dx.doi.org/10.1016/j.cis.2004.02.003 PMID: 15142823 166. Eastoe, J.; Hollamby, M. J.; Hudson, L.; Adv. Colloid Interface Sci. 2006, 128, 5. DOI: http://dx.doi.org/10.1016/j.cis.2006.11.009 PMID: 17254535 167. Wang, G.; Quin, W.; Zhang, J.; Zhang, J.; Yan, W.; Cao, C.; Wang, L.; Wei, G.; Zhu, P.; Kim, R.; J. Phys. Chem. C 2008, 112, 12161. DOI: http://dx.doi.org/10.1021/jp8004713 168. Janssens, S.; Williams, G. V. M. Clarke, D. ; J. Lumin. 2013, 134, 277. DOI: http://dx.doi.org/10.1016/j.jlumin.2012.08.032 169. Huang, Y.; You, H.; Song, Y.; Jia, G.; Yang, M.; Zheng, Y.; Zhang, L.; Liu, K.; J. Cryst. Growth 2010, 312, 3214. DOI: http://dx.doi.org/10.1016/j.jcrysgro.2010.07.045 170. Lemyre, J.-L.; Ricey, A. M.; Chem. Mater. 2005, 17, 3040. DOI: http://dx.doi.org/10.1021/cm0502065 171. de Sousa Filho, P. C.; Serra, O. A.; J. Phys. Chem. C 2011, 115, 636. DOI: http://dx.doi.org/10.1021/jp109988a 172. Xing, Y.; Li, M.; Davis, S. A.; Mann, S.; J. Phys. Chem. B 2006, 110, 1111. DOI: http://dx.doi.org/10.1021/jp0564896 PMID: 16471651 173. Xing, Y.; Li, M.; Davis, S. A.; Patil, A. J.; Mann, S.; Soft. Matter 2006, 2, 603. DOI: http://dx.doi.org/10.1039/b604831e 174. Zhang, C.; Chen, J.; Zhu, X.; Zhou, Y.; Li, D.; Chem. Mater. 2009, 21, 3570. DOI: http://dx.doi.org/10.1021/cm8027516 175. Sun, Y.; Yang, H.; Tao, D.; Ceram. Int. 2011, 37, 2917. DOI: http://dx.doi.org/10.1016/j.ceramint.2011.03.030 176. Deng, H.; Yang, S.; Xiao, S.; Gong, H.-M.; Wang, Q.-Q.; J. Am. Chem. Soc. 2008, 130, 2032. DOI: http://dx.doi.org/10.1021/ja0778141 PMID: 18198875 177. Rill, C.; Bauer, M.; Bertagnolli, H.; Kickelbick G.; J. Colloid Interface Sci. 2008, 325, 179. DOI: http://dx.doi.org/10.1016/j.jcis.2008.05.008 178. Fernandes, M. T. C.; Kawachi, E. Y.; Quim. Nova 2010, 33, 1242. DOI: http://dx.doi.org/10.1590/S0100-40422010000600004 179. Hsu, C.-H.; Huang, C.-H.; Cheng, B.-M.; Lu, C.-H.; Mater. Chem. Phys. 2010, 124, 632. DOI: http://dx.doi.org/10.1016/j.matchemphys.2010.07.026 |
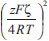 ), κ é o inverso do comprimento de Debye e d é a distância entre as partículas.
), κ é o inverso do comprimento de Debye e d é a distância entre as partículas.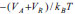 , sendo que a estabilidade coloidal é atingida quando (VA+VR)>>kBT. A altura da barreira Vmáx é, portanto, relacionada à carga superficial (potencial ζ) das partículas e à força iônica do meio, de modo que, desconsiderando-se a adição de outras espécies ao sistema coloidal, tais situações de estabilidade são atingidas com elevadas cargas superficiais (|ζ|>30 mV) ou de baixo valor de força iônica.37,41,46,50
, sendo que a estabilidade coloidal é atingida quando (VA+VR)>>kBT. A altura da barreira Vmáx é, portanto, relacionada à carga superficial (potencial ζ) das partículas e à força iônica do meio, de modo que, desconsiderando-se a adição de outras espécies ao sistema coloidal, tais situações de estabilidade são atingidas com elevadas cargas superficiais (|ζ|>30 mV) ou de baixo valor de força iônica.37,41,46,50On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access