
 |
 |
 |
 |
 |
Artigo
| Spectroscopic study of aluminum phthalocyanine chloride (AlPcCl) in homogeneous and micro-heterogeneous media consisting of P-123 and F-127 polymeric micelles |
|
Bruno H. VilsinskiI,*; Adriana P. GerolaII; Évelin O. LemosI; Patrícia M. BarbosaI; Katieli S. S. CampanholiI; Gabriel B. CésarI; André L. TessaroIII; Noboru HiokaI; Wilker CaetanoI
IDepartamento de Química, Universidade Estadual de Maringá, Av. Colombo 5.790, 87020-900 Maringá - PR, Brasil Recebido em 25/09/2014 *e-mail: vilsinski@yahoo.com.br Aluminum phthalocyanine chloride (AlPcCl) is a photoactive compound which has been used as a photosensitizer (PS) in photodynamic therapy (PDT). Its spectroscopic properties have been studied in solvents of different polarities (ethanol, acetone, dimethylsulfoxide and chloroform). Its solubility has been found to decrease with increasing solvent polarity, together with full self-aggregation in aqueous solution. The binding of AlPcCl to the copolymer PluronicTM micellar class P-123 and F-127 used as solubilizer/carriers was studied. Greater interaction between the more hydrophobic copolymer P-123 and AlPcCl was observed, besides a complex interaction profile involving different AlPcCl forms (self-aggregate/monomeric form) in the copolymers. Time- and temperature-dependent structural organization of AlPcCl in the copolymers was also observed. Thus, AlPcCl has a strong tendency to self-aggregate with increasing solvent polarity, an effect also observed in micellar media. INTRODUCTION Phthalocyanines are a versatile class of molecules used in many areas of knowledge, e.g., in the development of electronic devices, nonlinear optics, Langmuir-Blodgett films, gas sensors and Photodynamic Therapy (PDT).1-3 The basic principle of PDT is the association of a photosensitizing compound (PS) and light of a specific wavelength, which in the presence of molecular oxygen, generates cytotoxic species, such as singlet oxygen. Singlet oxygen has been reported to be responsible for cell damage.1,2,4,5 Although phthalocyanines (Figure 1a) have the main characteristics of a good PS, they are not very soluble in water and have a high tendency to self-aggregate in this medium.6 This phenomenon limits its use in PDT, since it diminishes the singlet oxygen quantum yield.7
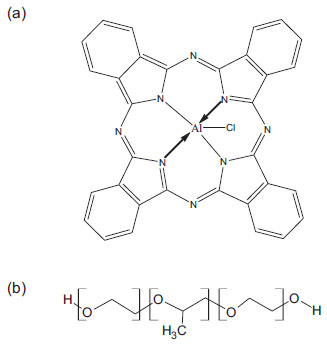 Figure 1. Molecular structure of (a) aluminum phthalocyanine chloride and (b) Pluronic® block copolymer. (x=z=20 and y =70 for P-123 and x=z=106 and y=70 for F-127)
Several factors affect self-aggregation process, such as the effects associated with temperature, solvent polarity, and ring substituents.8,9 They cause significant changes in the electronic absorption and fluorescence emission spectra.10,11 In order to minimize the self-aggregation, improve the bio transport and obtain controlled release, many PS have been incorporated into nanostructured systems, such as micelles, liposomes, polymer-photosensitizer conjugates, DNA-polymer complexes, nanogels, among others.12-15 Polymeric micelles have attracted great interest 16,17 due to their uniquely considerable biocompatibility and drug delivery site specificity.12,18 Additionally, due to their smaller particle size (10 to 100 nm) and greater uniformity, comparatively to carriers such as liposomes and microspheres, polymer micelles are not easily eliminated by the renal and reticuloendothelial systems, which renders greater circulation and accumulation of encapsulated drugs into targets of interest.18,19 Among the polymeric micelles, an important class is the constituted by PluronicTM triblock copolymers, such as F-127 and P-123 (Figure 1-b), which have been used in this study. These copolymers have been studied to solubilize several kinds of hydrophobic drugs as doxorubicin, paclitaxel and others, showing considerable efficiency at delivery and subsequently drug liberation in a specific cellular site.18,19 This kind of copolymer surfactant possesses others additional advantages: low toxicity and a relatively lower critical micelle concentration (around 10-6 mol L-1) when compared to usual surfactants as sodium dodecyl sulfate (SDS), cetyl trimethylammonium bromide (CTAB) and others.20 The mainly difference between both copolymers used in this study, is that P-123 is a more hydrophobic copolymer, once have a lower ratio hydrophobic PPO/hydrophilic PEO groups than F-127. It's known that these structural difference make each Pluronic® copolymer better to solubilize determined kind of molecule, depending of polarity degree of molecules, where more hydrophilic molecules are better solubilized by F-127 than P-123.20 So, in this study we examined the physicochemical properties of Aluminum phthalocyanine chloride (AlPcCl) (Figure 1-a) in homogeneous media and its association, solubility and molecular organization on copolymers with different degree of polarity (P-123 and F-127) at different temperatures in order to obtain a viable formulation AlPcCl/copolymers to application in PDT.
EXPERIMENTAL Materials Aluminum phthalocyanine chloride: Chloro (29H,31H-phthalocyaninato) aluminum (AlPcCl 95%, MM= 574.96 g mol-1) and polymeric surfactants F-127 (MM = 12600 g mol-1 and P-123 (MM = 5800 g mol-1) (Figure 1-b) were purchased from Sigma-Aldrich and used without additional purification. All the used organic solvents (ethanol, dimethylsulfoxide, acetone and chloroform) were of high degree and utilized as purchased. Bi-distilled water was used. Spectroscopic characterization of AlPcCl in homogeneous media The AlPcCl stock solution (1.0x10-5 mol L-1) was prepared by addition of 1.0x10-4 g in 25.0 ml of ethanol. The spectroscopy properties of phthalocyanine were studied in a variety of solvents (ethanol, acetone, water, dimethyl sulfoxide and chloroform) by electronic absorption, fluorescence emission and resonance light scattering (RLS) spectrum analysis. The absorbance and fluorescence experiments were carried out in spectrophotometers DU-800 Beckman and Cary Eclipse, respectively. The resonance light scattering (RLS) was assessed at Δλ= 0 nm in regular 1.00-cm cuvettes and triangular cuvettes. Studies of self-aggregation process of AlPcCl in ethanol/water mixture Mixtures of solvents were prepared in volumetric flasks (10.0 mL) by adding of appropriate volumes of ethanol and filling them up with distilled water. The AlPcCl concentration for the experiments in homogeneous systems was kept at 3.0x10-6 mol L-1 (for absorption) and 3.0x10-7 mol L-1 (for fluorescence), except for the Lambert-Beer plots. All the experiments were made at 25.0 ºC. Evaluation of interaction process between AlPcCl and copolymers F-127 and P-123 Interaction studies of AlPcCl with P-123 and F-127 surfactant followed the procedure reported by Caetano and Tabak, 1999.35 Based on this methodology, aliquots of these surfactants were added in an aqueous medium containing AlPcCl (8.0x10-7 mol L-1). The titrations were followed by acquisition of fluorescence emission spectra. The experiments were carried out at 15.0, 25.0 and 40.0 ºC. In order to evaluate the incorporation of phthalocyanine into polymeric micelles, the kinetic profile of AlPcCl at 677 nm (monomer band) was monitored at various [surfactant]/[AlPcCl] ratios.
RESULTS AND DISCUSSION Spectroscopic properties and solubility of AlPcCl in homogeneous media: electronic absorption and fluorescence studies Firstly, AlPcCl is insoluble in water, being precipitated without showing any absorption band in this media (Figure 2-a). Poor solubility was also observed in other solvents, where there was a verified decrease and enlargement in different degree of principal bands of AlPcCl in monomeric state in the following order: CHCl3 > acetone > DMSO (Dimethyl Sulfoxide). On the other hand, their solubility is significantly increased in ethanol, as can be seen in Figure 2-a. The absorption spectrum shows two major bands, attributed to π-π* HOMO-LUMO transitions, centered at 353 (Soret band, S0 → S2) and 672 nm (QI band, S0 → S1), which are characteristic of phthalocyanines in monomeric state.22 Besides the QI band a1u → eg symmetry, there were two other Q bands, labeled QII (606 nm) and QIII (640 nm), which were assigned to vibrational transitions, Q (1,0) and Q (2,0), respectively.22 The emission spectrum of AlPcCl in ethanol shows an emission maximum at a wavelength of 680 nm (Figure 2-b), relative to the S1 → S0 transition and two additional bands of lesser intensity at 713 and 748 nm, related to vibrational state transitions.23 The emission spectra of AlPcCl in acetone and DMSO (not shown) were similar to that obtained in ethanol, showing only a lower intensity, probably due to dye self-aggregation, which was more evident in these solvents.
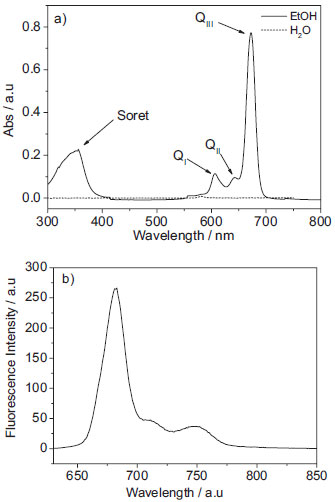 Figure 2. a) absorption spectra of AlPcCl (4.0x10-6 mol L-1) in ethanol and water and b) fluorescence emission spectra of AlPcCl (4.0x10-6 mol L-1) in ethanol, λexc: 604 nm at 25.0 ºC
The electronic absorption spectra obtained for AlPcCl at different concentrations solubilized in ethanol (Figure 1S) showed an increase in the absorbance with the increase in the AlPcCl concentration, without any change in the spectral profile. This linear relationship between the absorbance and the concentration suggests the presence of monomeric species. This pattern was not observed for the increase in the concentration of AlPcCl in acetone or DMSO. In these cases, a decrease of absorptivity was detected with changes in the maximum absorption wavelength and band broadening (mainly in QIII band, not shown), which suggest AlPcCl self-aggregation in these solvents.24,25 The self-aggregation was even more drastic in water and chloroform, which showed a considerable band broadening. Thus, ethanol was chosen for the preparation of the AlPcCl stock solution used in all the experiments performed in this work. In order to determine the molar absorptivity coefficient of the phthalocyanine in ethanol, the absorbance for AlPcCl at various concentrations was plotted against its concentration (Figure 1S-b). The plot shows a perfect linear relationship between the AlPcCl absorbance and concentration, which may be an indication of the presence of only monomeric species. Although the data obey the Lambert-Beer law, this does not guarantee the absence of aggregates in the medium. Thus, the same adjustment was made for emission data (Figure 1S-b), which showed a negative deviation from the concentration equal to 1.0x10-6 mol L-1. These breaks in the linearity were attributed to the presence of self-aggregates, since they have their energy self-suppressed. Therefore, only concentrations below 1.0x10-6 mol L-1 were used in the determination of the molar absorptivity coefficient. The same procedure was adopted for AlPcCl in acetone and DMSO, but in these cases, the concentrations in which the phthalocyanine remained in a monomeric state were lower than that observed with ethanol. The molar absorptivity coefficient obtained of principal bands of AlPcCl in ethanol was 205, 257, 305 and 634 x103 L mol-1cm-1 (to QIII, QII, QI and Soret respectively). Characterization of AlPcCl aggregation process in homogeneous media: spectroscopic studies of AlPcCl in ethanol-water mixtures To gain a greater insight about the AlPcCl self-aggregation process, the stability of a fixed concentration of AlPcCl was evaluated in several water/ethanol mixtures (V/V) by absorbance and fluorescence spectroscopy (Figure 3). Initially, the increase in the water content caused a reduction in the absorbance intensity, with a slightly bathochromic displacement of their maxima (Figure 3-a), suggestive of the existence of self-aggregates. The spectral overlap showed drastic changes in the spectral profile around 50% of water, with the extinction of the three Q bands (672, 641 and 606 nm), which is more evident in Figure 3-c. Additionally, displacement of the Soret band and the appearance of a new band at 610 nm also took place. These alterations are associated with the self-aggregation process and suggest the formation of J aggregates.26 The analysis of the fluorescence data shows that by increasing the water content, even in low percentages, the fluorescence intensity decreased. As aforementioned, this phenomenon may be attributed to the self-aggregation process, leading to the formation of small species, such as dimers. Additionally, water may be acting as an energy suppressor, as suggested in the literature.27 Once again, striking differences were observed for a water content of around 50%, such as a drastic reduction in emission intensity (Figure 3-b). This content signalizes a critical water percentage in which strong aggregation takes place, yielding aggregates of high order, such as trimers, tetramers. In order to prove this supposition, resonance light scattering experiments were applied, since this technique is utilized to detect large aggregates. Figure 3-d depicts intense RLS signs in the 430 nm region and above 700 nm for solutions with high water contents, which confirms the formation of high order J aggregates.
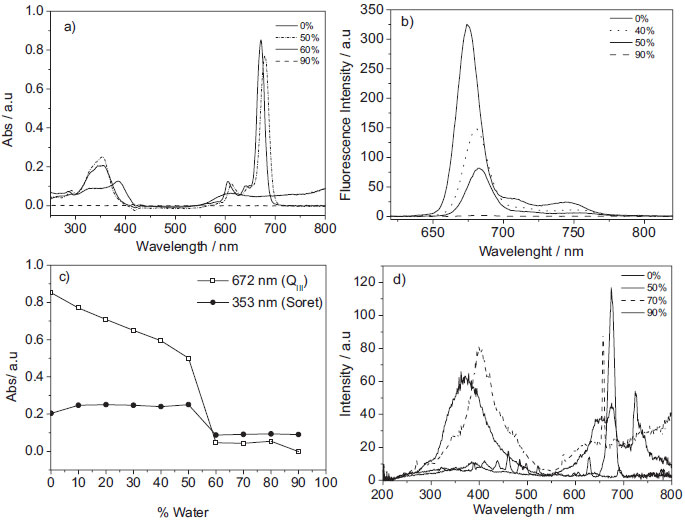 Figure 3. Spectras of AlPcCl in different percentages of water/ethanol (V/V). a) Electronic Absorption spectra of AlPcCl (3.0x10-6 mol L-1) at 25.0 ºC, b) emission spectra of AlPcCl (3.0x10-7 mol L-1) λexc= 604 nm, c) absorption at QIII and Soret bands in different percentages of water (V/V), d) RLS of AlPcCl (3.0x10-7 mol L-1) at 25.0 ºC
The tendency of AlPcCl to self-aggregate in the water-rich medium is attributed to an unbalanced hydrophilic/hydrophobic system due to the presence of water, which promotes hydrophobic interactions between AlPcCl molecules. This phenomenon has been reported in the literature for similar compounds.25,28,29 These that involve the spectroscopic properties of AlPcCl in homogeneous media constituted of binary mixtures water/ethanol are important to understand better the aggregation process of AlPcCl in media of high content of water. These studies also are important to understand the self-aggregates forms of AlPcCl in micro-heterogeneous media constituted of polymeric micelles of P-123 and F-127, describe after. Studies of physicochemical properties of AlPcCl in micro-heterogeneous nanostructured systems: P-123 and F-127 Polymeric Micelles Binding Isotherms of AlPcCl in polymeric micellar systems of P-123 and F-127 The UV-Vis absorption spectra obtained for the titrations of AlPcCl (8.0x10-7 mol L-1) with both copolymers at the initial time at 25.0 ºC are shown in Figure 4. Without copolymer addition, the spectral profile of self-aggregated AlPcCl becomes evident mainly through the presence of the characteristic aggregate band at 610 nm (Figure 3-a). No significant spectral changes were observed for the AlPcCl/F-127 system (Figure 4-a), but the system AlPcCl/P-123 showed a slight recovery in the band related to the absorption by AlPcCl monomers (λ = 675 nm) (Figure 4-b) and a decrease in the band related to the self-aggregates at λ = 610 nm with the addition of the copolymer. Additionally, in the inset shown in Figure 4-b, the increased concentration of P-123 induced a hypsochromic shift of the wavelength of maximum absorption at 675 nm (Δλ = 7 nm). These results suggest that the interaction of AlPcCl molecules with P-123 induced a greater breakdown effect and a structural reorganization of the different types of AlPcCl self-aggregates by the more hydrophobic copolymer.
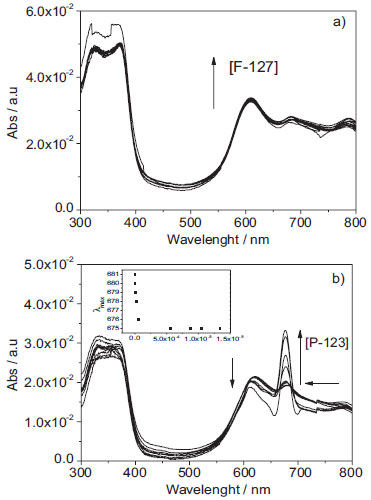 Figure 4. Electronic absorption spectra of AlPcCl (8.0x10-7 mol L-1) in the presence of a) [F-127] (from 0 to 6.4x10-4 mol L-1) b) [P-123] (from 0 to 1.5x10-3 mol L-1) at pH = 6.9, 25.0 ºC. Inset: variation of the maximum absorbance wavelength monitored through variations in the intensity of the QIII band of AlPcCl (λmax = 675 nm)
The same titrations were performed by monitoring fluorescence emission. The emission spectra of AlPcCl (8.0x10-7 mol L-1) at different concentrations of F-127 (a) and P-123 (b) at the initial time are shown in Figure 5.
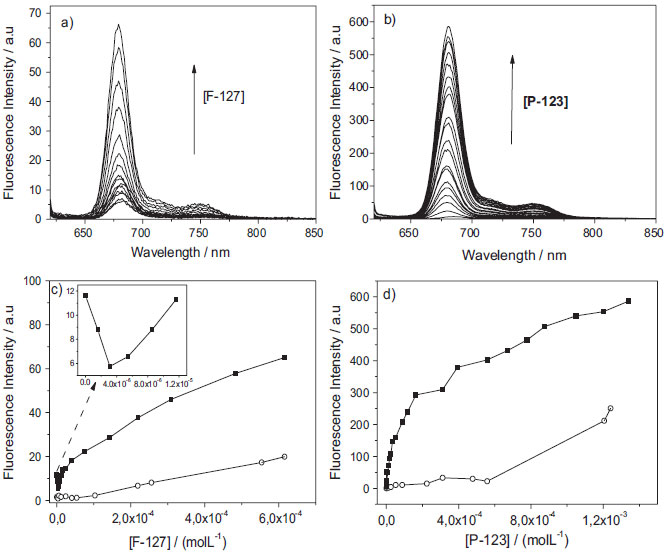 Figure 5. Fluorescence emission spectra of AlPcCl (8.0x10-7 mol L-1) and binding isotherm as a function of the addition of copolymers being: a) and c) F-127 (0 to 6.5x10-4 mol L-1), b) and d) P-123 (0 to 1.5x10-3 mol L-1) at pH = 6.9, λexc = 604 nm, 25.0 ºC. The arrow shows the increase in fluorescence emission with concentration of copolymers. Fluorescence emission obtained immediately after the addition of the copolymer (■; t= 0) and after equilibrium (○; t > 48 h) at pH 6.9, 25.0 ºC. The inset in Figure 5-c shows the isotherm region for low concentrations of F-127 (0 to 1.2x10-5 mol L-1)
The initial emission intensity of AlPcCl of approximately zero was attributed mainly to the AlPcCl self-aggregation effect in aqueous medium and/or a AlPcCl fluorescence suppression effect by water molecules.27,30 Subsequently, with the addition of the copolymers, the fluorescence emission intensity increased, a fact indicative of the association of AlPcCl with the polymeric micelles. The increase in the fluorescence emission intensity can be explained by the association of AlPcCl with the micelle, which leads to a disaggregation effect and the redistribution of the phthalocyanines molecules in the microenvironment during this primary binding process.31 For P-123, a higher emission intensity (on average 10 times greater) was observed when compared to the AlPcCl/F-127 system, which can be attributed to an AlPcCl disaggregation effect in parallel with the micelle association process, favored by the existence of a more hydrophobic microenvironment for the copolymer P-123 in micellar form (Figure 1-b).21,32 This enables the copolymer to stabilize the PS in the monomeric in hydrophobic core of the micelles more efficiently than F-127.20,32 Figures 5-c and 5-d show emission intensity graphs for AlPcCl (λmax = 681 nm) versus the P-123 and F-127 concentrations, respectively, added to the system at the initial time and in infinite time (spectra acquired 48 h after the additions). The inset in Figure 5-c shows the low concentration region of F-127 (between 0 and 1.5x10-5 mol L-1) at the initial time. The AlPcCl/copolymer isotherms showed a complex behavior, different from that usually observed, which prevented the use of simple binding models 33,34 that consider the interaction between the photosensitizer molecule in solution initially in the monomeric state with the micellar system.35,36 However, it was noted that the AlPcCl emission intensities for both systems were relatively smaller in the equilibrium time than at the initial time. This fact highlights the structural reorganization upon the addition of AlPcCl in the micellar microenvironment with the time. At the initial time and the equilibrium time, there seems to be more than one step in the association process between AlPcCl and the polymeric micelles for both copolymers. For the isothermal spectrophotometric titration with F-127 (Figure 5-c), there clearly is a first step that displays a fall in the intensity of the initial AlPcCl emission to approximately 5.0x10-6 mol L-1 of this copolymer (inset in Figure 5-c). It is worth mentioning that this region of concentration is below the CMC of this surfactant at this temperature.37 This behavior was attributed to a combination of self-aggregated AlPcCl with the copolymer micro-domains, resulting from a relatively low amount of binding sites available for the monomers suitable for this phthalocyanine in these conditions. One also has to consider the differences between a monomer (unimer) of copolymer relative to a usual surfactant monomer (SDS, CTAB, etc.). Even though the surfactant concentration is below its CMC, it is known that only one unimer of a copolymer itself can self-organize in a system with certain defined structure.38 This decrease in emission intensity probably results from increased AlPcCl self-aggregation due to the localization of the few and relatively small micro-environments in an unimer. During the equilibrium time, the behavior in this range of concentration of copolymer F-127 was the same, with only the isotherm having lower emission intensity when compared to that of the initial time in these conditions. In turn, when above 5.0x10-6 mol L-1 of F-127 (therefore, above the CMC), a second phase (up to about 5.0x10-5 mol L-1) began, which tended to redistribute the AlPcCl molecules in this system. However, rival association by disaggregated species in the copolymer is not ruled out.38 At higher concentrations, the isotherm had continuous behavior and was the slowest; however, without achieving complete saturation. By analyzing the isotherm of the system AlPcCl/P-123 (Figure 5-d), one observes that there was the initial step with an emission intensity drop at the lowest concentrations of P-123, as observed for the AlPcCl/F-127 system. Interestingly, the range of concentrations studied in the P-123 system was above its CMC at this temperature (25.0 ºC).31 Thus, the isotherm behavior was similar to that obtained in the AlPcCl/F-127 system (Figure 5-c). Up to the P-123 concentration of approximately 2.0x10-4 mol L-1, an AlPcCl binding tendency (in the self-aggregated and monomeric forms) with the polymeric micelles was observed. Above this concentration, there was a step that presented a more continuous behavior and slower AlPcCl emission range without saturation. The binding profiles of these complexes observed in the isotherms for both copolymers suggest that different forms of self-aggregated AlPcCl may self-reorganize during the association with the micellar microenvironment. The time dependence of the AlPcCl interaction with the micellar copolymers during the PS/polymeric micelles association led to further investigation of its kinetic profile of incorporation. This study is also important to know better the time-dependence of ratio copolymer/AlPcCl in structural organization of AlPcCl in micro domains of polymeric micelles. Kinetic studies of association of AlPcCl in P-123 and F-127 polymeric micelles The kinetics of incorporation of AlPcCl (2.5x10-6 mol L-1) was obtained with the addition of various concentrations of F-127 and P-123 to the PS aqueous solution. The quantities of copolymers added, corresponded to the experimental points in the different regions of the binding isotherm (beginning, middle and end), are shown in Figure 5. The UV-Vis absorption spectra of AlPcCl with the time for various initial concentrations of F-127 (or different [F-127mic]/[AlPcCl] ratios) are shown in Figure 6-a; the number of micelles was calculated based on the aggregation number of F-127 (Nagg) of 37 at 25.0 ºC.21 For the [F-127mic]/[AlPcCl] ratio of approximately 3 per AlPcCl molecule, the spectral profile had broad bands, indicating that AlPcCl was in self-aggregated state. Now, for the ratio of approximately 7, the spectra showed peaks characteristic of AlPcCl in monomeric state with a high absorptivity in the 675 nm region (Figure 6-a). However, enlargement of the Soret and Q bands still indicated the presence of PS self-aggregated forms in this copolymer.
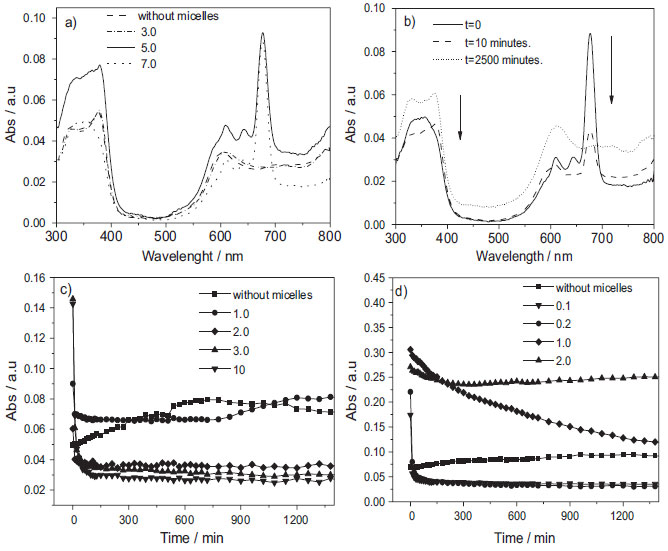 Figure 6. Absorption spectra of a) AlPcCl (2.5x10-6 mol L-1) at various [F-127mic]/ [AlPcCl] ratios (varying from no micelle to 7.0 and [F-127] from 0 to 6.2 x10-4 mol L-1), T= 25.0 ºC b) absorption spectra of AlPcCl (2.5x10-6 mol L-1) in F-127 (6.2x10-4 mol L-1; [F-127mic]/[AlPcCl] ratio = 7.0) at different times, T = 25.0 ºC. Temporal variation of absorbance of AlPcCl (2.5x10-6 mol L-1) in band QIII (λmax = 677 nm) at different [copolymermic] /[AlPcCl] ratios, c) F-127 (0 to 6.2 x10-4 mol L-1) and d) P-123 (0 to 1.5x10-3 mol L-1) at 25.0 ºC, pH = 6.9
Similarly, for the P-123 system at the initial time, we observed the same dependency. For example, at the higher [P-123mic]/[AlPcCl] ratio, more monomers were present. However, when compared to the F-127 system (Figure 6-a), the region above 500 nm resulted in a smaller band widening, more similar to the spectrum of AlPcCl in monomeric state (not shown). This result indicates a greater stabilization of the AlPcCl monomers at the initial time for the AlPcCl/P-123 system, in agreement with the results of previous studies on AlPcCl/copolymer interaction, which showed an apparent greater interaction between AlPcCl and the more hydrophobic copolymer. The electronic absorption spectra of AlPcCl (2.5x10-6 mol L-1) in F-127 (6.0x10-4 mol L-1, [F-127mic]/[AlPcCl] ratio= 7 obtained at different times are shown in Figure 6-b. The characteristic PS monomer peaks can be observed initially at t= 0. Concurrently with the decrease in the characteristic monomer bands, a broad band appears at 610 nm for the self-aggregated forms of AlPcCl. The electronic absorption spectrum profiles of AlPcCl in P-123 monitored versus time (not shown) were similar to those of the AlPcCl/F-127 system; however, the decrease in the absorbance bands related to monomers, as well as the appearance and increase in respect to the self-aggregates, was significantly slower in this copolymer which reinforces that P-123 is a better copolymer to stabilize AlPcCl in a monomeric state. For a better visualization of the temporal aggregation effect of AlPcCl in both copolymers, Figures 6-c and 6-d present the kinetic profiles in the QIII band (λmax= 677 nm) for different [copolymer]/[AlPcCl] ratios. The self-aggregation kinetic profile of AlPcCl (Figure 7-d) at higher [P-123mic]/[AlPcCl] ratios (mostly 1.0 and 2.0) is generally slower when compared to that of F-127. In addition, the aggregation kinetics in systems containing P-123 was dependent on the [P-123mic]/[AlPcCl] ratio. In this case, the aggregation kinetics was slower in systems containing a greater amount of micelles per PS molecule. In turn, in the AlPcCl/F-127 system kinetics, the self-aggregation speed was rapid and independent of the micellar concentration (Figure 6-c). Additionally, small fluctuations in the absorption intensities of AlPcCl for long times (t> 500 min) and various [copolymer]/[AlPcCl] ratios can be observed in Figures 6-c and 8-d, which, although relatively small, can also indicate reorganization processes between different forms of self-aggregated AlPcCl.
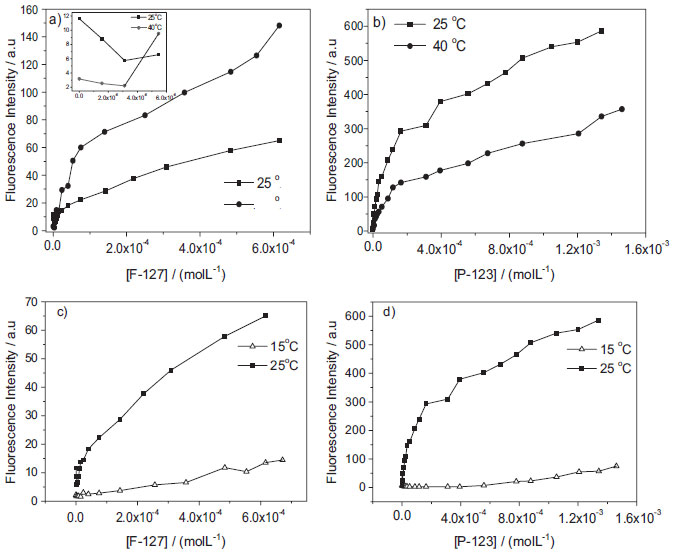 Figure 7. Binding isotherms of AlPcCl (8.0x10-7 mol L-1; λmax=680 nm) for different concentrations of a) [F-127]; inset: isotherm behavior at 25 and 40.0 ºC in the low F-127 concentration region (<6x10-6 mol L-1) and b) [P-123] obtained at 40 (●) and 25.0 ºC (■), c) and d) [F-127], [P-123 ] at 15 ºC (Δ) and 25.0 ºC (●), pH = 6.9
Additionally, we have tried to fit the kinetic profile curves of AlPcCl for different [copolymer]/[AlPcCl] ratios to known kinetic models and observed that the system is inherently complex and does not provide reliable kinetic relations. The kinetic effects observed in the AlPcCl/copolymers interaction (Figures 6-c and 6-d) reinforced the hypothesis of diffusional processes directly associated with the interaction of different forms of self-organized AlPcCl (dependent on the [AlPcCl]/[micelle] ratio and [micelle]) in micro-environments of different types of polymer micelles. The relatively faster aggregation of AlPcCl in F-127, together with the spectroscopic results of the interaction between phthalocyanine and the polymeric micelles (binding isotherm), showed the dependence of the AlPcCl-poloxamer interaction on the polymer micelle structure, which has a relatively more hydrophilic region formed by the organization of PEO groups when compared to P-123. In addition to the absolute size of the polymeric micelles, one should also consider that both copolymers possess different aggregation numbers (number of unimers in the copolymer per micelle) at 25.0 ºC (Nagg/F-127=37 and Nagg/P-123 = 86 at 25.0 ºC.20,21 The higher aggregation number of P-123 makes the micelles a more hydrophobic micro-environment at a particular temperature in this copolymer when compared to F-127, which can solubilize the PS, thereby justifying an increased interaction of AlPcCl in this copolymer. These results that shown a high stability of AlPcCl in P-123, depending of ratio [P-123mic]/AlPcCl, are interesting once provides an alternative to the of use AlPcCl this copolymer in monomeric form in future applications. Influence of temperature on the binding properties of AlPcCl with P-123 and F-127 polymeric micelles Additional studies of the binding behavior of isotherms were conducted with AlPcCl in F-127 and P-123 varying the temperature of the system. The structural organization of copolymers in aqueous medium is known to depend on the temperature. For example, variations in the number of micellar aggregates (Nagg)20,21 promote significant changes in polymeric micelle volume and size, which can modulate the micelle-drug interaction.39 Figures 7-a and 7-b shows the comparison between the binding isotherms of AlPcCl in P-123 and F-127 at 25 and 40.0 ºC. The comparison of the isotherm profiles of P-123 and F-127 at 25 and 40.0 ºC (Figures 7-a and 7-b) shows that the higher temperature binding profiles slightly differ from those at 25.0 ºC, primarily with the variation of the emission intensity of two copolymers with an increasing temperature. For low concentrations of F-127 at 25 and 40.0 ºC in the AlPcCl/F-127 system, (Figure 7-a inset), a small decrease in magnitude of the intensity emission of AlPcCl was initially observed in these conditions, being most evident at 25.0 ºC. As previously discussed, this result suggests a primary AlPcCl's self-aggregation effect for low copolymer concentrations (higher [AlPcCl]/ [F-127mic] ratios). However, with subsequent additions of copolymer and lower [AlPcCl]/[F-127mic] ratios, one can see in the isotherm for 40.0 ºC (Figure 7-a) that the increase in temperature promotes an increase in the fluorescence emission intensity, indicating a possible phthalocyanine disaggregation effect concomitant to the breakdown of its association with the restructured polymer micelle.20 This effect can also be verified through the AlPcCl electronic absorption spectra (not shown), which shows a small increase in absorptivity in the QIII band region of AlPcCl (λ = 677 nm), together with an apparent decrease in the band assigned to the self-aggregates (λ = 610 nm; similar to Figure 4-b). Interestingly, the fluorescence emission intensities of the AlPcCl/P-123 system at 40.0 ºC (Figure 7-b) showed a behavior opposite to that of the AlPcCl/F-127 system. The emission intensity of the AlPcCl/P-123 system at 40.0 ºC was lower than the intensity obtained at 25.0 ºC. Additionally, although the electronic absorption spectra for 40.0 ºC of this system (not shown) was similar to those obtained at 25.0 ºC (Figure 4-b), they presented an additional decrease in absorbance intensity of about 30% in the QIII band region (λ ~ 677 nm), indicative of an increase in the AlPcCl self-aggregation increasing the temperature. The effects observed are discussed primarily in terms of micelle structural reorganization for different copolymer modulation of the photosensitizer interaction with increasing temperature, but also regarding the phthalocyanine association mechanism in these systems at the initial time. During the relatively short time between the addition of the copolymer to the aqueous solution containing AlPcCl and the measurement of the spectra, the phthalocyanine molecules are probably located in the boundary region between the chain of PEO groups and water, or in the interfacial region on the hydrophilic polymeric micelles. At the initial time, the time for the stabilization of AlPcCl in the innermost region containing the micro domains formed by the more hydrophobic PPO chain in these micelles is insufficient. On the other hand, considering the structural reorganization of the two types of copolymers in micelle form, the P-123 hydrophilic PEO chain is approximately five times shorter than that of F-127 (Figure 1-b).21 As a result, the interfacial volume and surface area of the hydrophilic polymer micelle shell of F-127 are larger than those of P-123, despite the increase in the aggregation number caused by the increase in the temperature. The aggregation number of this more hydrophobic copolymer is 2 and 3 times greater for P-123 and F-127, respectively, in this temperature range.20 This result makes the core formed by the hydrophobic PPO chain of the micelle of P-123 relatively higher when compared to F-127. Thus, the greater self-aggregation effect of AlPcCl in the AlPcCl/P-123 system, demonstrated by the behavior of the isotherms at initial time at 40 ºC relative to 25.0 ºC, is probably due to AlPcCl molecule distribution in a smaller volume and area for this hydrophilic copolymer shell in micellar form in relation to F-127. In F-127, in turn, AlPcCl molecules have less access to water molecules, thus favoring disaggregation. In fact, as observed in studies of dynamic light scattering of the microenvironment and copolymer micellization process, the copolymer PEO groups are usually hydrated with large amounts of water due to hydrogen bonding involving water hydrogen and PEO group oxygen and also due to physical entrapment of some molecules in the same micellar microenvironment.20,40 The same study was also performed at 15.0 ºC, which is lower than the CMT of both copolymers in the concentration range used.41 That is, at this temperature the copolymer molecules are in unimer form in solution; thus, this system is not thought to form micelles.39 Figures 7-c and 7-d show a comparison of the isotherms of the AlPcCl/F-127 and AlPcCl/P-123 systems obtained at 25.0 ºC with the association profile of AlPcCl and the copolymers at 15.0 ºC at the initial time. First, for the AlPcCl/copolymers system at 15.0 ºC (Figures 7-c and 7-d, F-127 and P-123, respectively), one can see that a binding isotherm profile (Figure 5) was not detected, showing different stages of association between PS and the copolymers dependent on their concentrations and/or the [AlPcCl]/[copolymer] ratio. In turn, the association profile of the AlPcCl/poloxamer system at 15.0 ºC shows a small variation in the emission intensity of AlPcCl with increasing concentrations of both copolymers. This is probably due to a weaker bond between AlPcCl and the copolymer unimers in the solution at this lower temperature than that of the association profile of AlPcCl and micelles, which had hydrophobic and hydrophilic micro-environments, enabling a more significant interaction of AlPcCl. One can also observe in the behavior of AlPcCl/P-123 at 15.0 ºC (Figure 7-d), primarily with a higher copolymer concentration, that the emission intensity was about five times greater than that obtained with greater concentrations in the AlPcCl/F-127 system. This was due to a greater interaction between AlPcCl and P-123 unimers, which have more hydrophobic domains than F-127.42 Additionally, the absorption spectra of AlPcCl in both copolymers at 15.0 ºC (not shown) remained practically unchanged as the copolymer concentrations increase, giving a typical AlPcCl self-aggregate spectrum. These results suggest that the AlPcCl-copolymer-unimer interactions of F-127 and P-123 are not strong enough to promote changes in the self-aggregation state of AlPcCl at this low temperature. Therefore, the distinct effects observed in the interaction profiles of AlPcCl in copolymers at temperatures above and below the CMT provided evidence that modulation of the AlPcCl-copolymer interaction results from changes in the structure of the copolymer in solution due to temperature variations.43
CONCLUSIONS We studied the spectroscopic properties of photosensitizer compound aluminum chloride phthalocyanine (AlPcCl) in solvents of different polarities and solubilized in micro-heterogeneous media of polymeric micelles of PluronicTM surfactants P-123 and F-127. Was observed that AlPcCl suffers self-aggregation process in media rich of water, presenting J-type aggregates in elevated contents of water (>50% V/V). This aggregation changes the mainly photo-physical properties of AlPcCl, unfeasing its use in photodynamic therapy. So it is indispensable the solubilization of AlPcCl in a drug delivery system adequate which is capable to encapsulate the phthalocyanine in a monomeric form and delivery it in a specific cellular target site. So we studied the solubilization properties of AlPcCl in a nanostructured and biocompatible polymeric micelles of triblock copolymers P-123 and F-127, where the mainly difference between both surfactants is in structure. Both copolymers has the same number of repetitive unities hydrophobic PPO in their structures, but F-127 has approximately five times greater number of hydrophilic groups PEO, being more hydrophilic than P-123. The studies of interaction AlPcCl/copolymers showed that AlPcCl has a complex binding profile with polymeric micelles, independently of temperature, probably involving association of different forms of phthalocyanine (self-aggregate and monomeric form). However our studies showed that AlPcCl has more interaction with the more hydrophobic copolymer P-123 than in F-127, including showing a monomeric profile in this copolymer, depending of ratio AlPcCl/copolymer. In this case P-123 it is a more effective copolymer to solubilize this molecule once has more adequate physical chemistry properties necessary to solubilize AlPcCl as lower polarity and higher aggregation number in comparison to F-127. Several studies in literature showed that it is not easy the solubilization of AlPcCl in a monomeric form in a drug delivery system. Liposomes are more indicated to solubilize this phthalocyanine, but the problem is that is a more expensive carrier to commercial applications. However we verified that P-123 besides to be cheap and biocompatible, also is a viable system to solubilize AlPcCl in a monomeric form (in higher ratio [P-123mic]/[AlPcCl]), adequate to applications of this system in future studies involving the Photodynamic Inactivation of microorganisms and Photodynamic Therapy against several types of injuries.
SUPPLEMENTARY MATERIAL This article contains supplementary information that can be freely accessed at http://quimicanova.sbq.org.br/.
REFERENCES 1. Leznoff, C. C.; Lever, A. B. P.; Phthalocyanines, Properties and Applications, 4th ed., Wiley: New York, 1996. 2. Kadish, K.; Smith, K. M.; Guillard, R.; The Porphyrin Handbook, 15, Academic Press: Boston, 2003. 3. Saka, E. T.; Gol, C.; Durmus, M.; Kantekin, H.; Bıyıklıoğlu, Z.; J. Photochem. Photobiol., A 2012, 241, 67. DOI: http://dx.doi.org/10.1016/j.jphotochem.2012.05.023 4. Castano, A. P.; Demidova, T. N.; Hamblim, M. R.; Photodiagn. Photodyn. Ther. 2005, 1, 279. DOI: http://dx.doi.org/10.1016/S1572-1000(05)00007-4 5. Lee, S. J.; Park, K.; Oh, Y.; Kwon, S.; Her, S.; Kim, I.; Biomaterials 2009, 30, 2929. DOI: http://dx.doi.org/10.1016/j.biomaterials.2009.01.058 PMID: 19254811 6. Ribeiro, M. G.; Azzellini, G. C.; J. Braz. Chem. Soc. 2003, 6, 914. DOI: http://dx.doi.org/10.1590/S0103-50532003000600008 7. Dhami, S.; de Mello, A. J.; Rumbles, G.; Bishop, S. M.; Phillips, D.; Beeby, A.; Photochem Photobiol. 1994, 61, 341. DOI: http://dx.doi.org/10.1111/j.1751-1097.1995.tb08619.x 8. Martin, P. C.; Gouterman, M.; Pepich, B. V.; Renzoni, G. E.; Schindele, D. C.; Inorg. Chem. 1991, 30, 3305. DOI: http://dx.doi.org/10.1021/ic00017a016 9. Moreira, L. M.; Lima, A.; Soares, R. R. S.; Batistela, V. R.; Gerola, A. P.; Hioka, N.; Bonacin, J. A.; Severino, D.; Baptista, M. S.; da Hora, A. E.; Rodrigues, M. R.; Codognoto, L.; Oliveira, H. P. M.; J. Braz. Chem. Soc. 2009, 9, 1653. DOI: http://dx.doi.org/10.1590/S0103-50532009000900013 10. Wiederkehr, N. A.; J. Braz. Chem. Soc. 1996, 1, 7. DOI: http://dx.doi.org/10.5935/0103-5053.19960002 11. Nunes, S. M. T.; Sguilla, F. S.; Tedesco, A. C.; Braz. J. Med. Biol. Res. 2004, 37, 273. DOI: http://dx.doi.org/10.1590/S0100-879X2004000200016 PMID: 14762584 12. Van Nostrum, C. F.; Adv. Drug Delivery Rev. 2004, 56, 9. DOI: http://dx.doi.org/10.1016/j.addr.2003.07.013 13. Batrakova, E. V.; Kabanov, A. V.; J. Controlled Release 2008, 130, 98. DOI: http://dx.doi.org/10.1016/j.jconrel.2008.04.013 14. Casarano, R.; Petri, D. F. S.; Jaffe, M.; Catalani, L. H.; J. Braz. Chem. Soc. 2009, 20, 1414. DOI: http://dx.doi.org/10.1590/S0103-50532009000800005 15. Kataoka, K.; Harada, A.; Nagasaki, Y.; Adv. Drug Delivery Rev. 2001, 47, 113. DOI: http://dx.doi.org/10.1016/S0169-409X(00)00124-1 16. Jiang, X.; Li, L.; Liu, J.; Zhuo, R.; J. Controlled Release 2011, 152, 36. DOI: http://dx.doi.org/10.1016/j.jconrel.2011.08.107 17. Zhao, Y-Z.; Sun, C-Z.; Lu, C-T.; Dai, D. D.; Lv, H. F.; Wu, Y.; Wan, C. W.; Chen, L. J.; Lin, M.; Li, X. K.; Cancer Lett. 2011, 311, 187. DOI: http://dx.doi.org/10.1016/j.canlet.2011.07.016 PMID: 21872982 18. Miyata, K.; Christie, R. J.; Kataoka, K.; React. Funct. Polym. 2011, 71, 227. DOI: http://dx.doi.org/10.1016/j.reactfunctpolym.2010.10.009 19. Harada, Y.; Yamamoto, T.; Sakai, M.; Saiki, T.; Kawano, K.; Maitani, Y.; Yokoyama, M.; Int. J. Pharm. 2011, 404, 271. DOI: http://dx.doi.org/10.1016/j.ijpharm.2010.10.043 PMID: 21093556 20. Alexandridis, P.; Holzwarth, J. F.; Hatton, T. A.; Macromolecules 1994, 27, 2414. DOI: http://dx.doi.org/10.1021/ma00087a009 21. Ough, E. A.; Gasyna, Z.; Stillman, M. J.; Inorg. Chem. 1991, 30, 2301. DOI: http://dx.doi.org/10.1021/ic00010a016 22. Dolphin, D.; The Porphyrins: Physical Chemistry, 5th ed., Academic Press Inc,: Boston, 1978. 23. Khairutdinov, R.; Serpone, N.; J. Phys. Chem. B. 1999, 103, 761. DOI: http://dx.doi.org/10.1021/jp980869s 24. Gracetto, A. C.; Tessaro, A. L.; de Souza, V. R.; Caetano, W.; Pontes, R. M.; Batistela, V. R.; de Oliveira, H. P.; Hioka, N; Appl. Spectrosc. 2011, 65, 604. DOI: http://dx.doi.org/10.1366/10-06161 PMID: 21639981 25. Ambrosek, D.; Köhn, A.; Schulze, J.; Kühn, O.; J. Phys. Chem. A 2012, 116, 11451. DOI: http://dx.doi.org/10.1021/jp3069706 PMID: 22946964 26. Lakowicz, J. R.; Principles of Fluorescence Spectroscopy, 3th ed., Springer: New York, 2006. 27. Simplicio, F. I.; Soares, R. R. S.; Maionchi, F.; Santin, O. F.; Hioka, N.; J. Phys. Chem. A 2004, 108, 9384. DOI: http://dx.doi.org/10.1021/jp048542g 28. Tessaro, A. L.; Batistela, V. R.; Gracetto, A. C.; de Oliveira, H. P. M.; Sernaglia, R. L.; de Souza, V. R.; Caetano, W.; Hioka, N.; J. Phys. Org. Chem. 2011, 24, 155. DOI: http://dx.doi.org/10.1002/poc.1721 29. Eaton, D. F.; J. Photochem. Photobiol., B 1988, 2, 523. DOI: http://dx.doi.org/10.1016/1011-1344(88)85081-4 30. Alexandridis, P.; Hatton, T. A.; Colloids Surf., A 1995, 96, 1. DOI: http://dx.doi.org/10.1016/0927-7757(94)03028-X 31. Wanka, G.; Hoffmann, H.; Ulbricht, W.; Macromolecules 1994, 27, 4145. DOI: http://dx.doi.org/10.1021/ma00093a016 32. Croy, S. R.; Kwon, G. S.; Curr. Pharm. Des. 2006, 12, 4669. DOI: http://dx.doi.org/10.2174/138161206779026245 PMID: 17168771 33. Moore, S. A.; Harris, A. A.; Palepu, R. M.; Fluid Phase Equilib. 2007, 251, 110. DOI: http://dx.doi.org/10.1016/j.fluid.2006.11.009 34. Hosseinzadeh, R.; Maleki, R.; Matin, A. A.; Youseff, N.; Spectrochim. Acta, Part A 2008, 69, 1183. DOI: http://dx.doi.org/10.1016/j.saa.2007.06.022 35. Caetano, W.; Tabak, M.; Spectrochim. Acta, Part A 1999, 55, 2513. DOI: http://dx.doi.org/10.1016/S1386-1425(99)00043-8 36. Du, W.; Teng, T.; Zhou, C-C.; Xi, L.; Wang, J-Z.; J. Lumin. 2012, 132, 1207. DOI: http://dx.doi.org/10.1016/j.jlumin.2011.07.025 37. Bakshi, M. S.; Sachar, S.; Colloids Surf., A 2006, 276, 146. DOI: http://dx.doi.org/10.1016/j.colsurfa.2005.10.032 38. Liu, K.; Wang, Y.; Yao, J.; Luo, Y.; Chem. Phys. Lett. 2007, 438, 36. DOI: http://dx.doi.org/10.1016/j.cplett.2007.02.048 39. Nishiyama, N.; Kataoka, K.; Pharmacol. Ther. 2006, 112, 630. DOI: http://dx.doi.org/10.1016/j.pharmthera.2006.05.006 PMID: 16815554 40. Zhou, Z.; Chu, B.; J. Colloid Interface Sci. 1988, 126, 171. DOI: http://dx.doi.org/10.1016/0021-9797(88)90111-7 41. Riess, G.; Prog. Polym. Sci. 2003, 28, 1107. DOI: http://dx.doi.org/10.1016/S0079-6700(03)00015-7 42. Chen, S.; Li, Y.; Guo, C.; Wang, J.; Ma, J.; Liang, X.; Yang, L. R.; Liu, H. Z.; Langmuir 2007, 23, 12669. DOI: http://dx.doi.org/10.1021/la702049d PMID: 17988160 43. Su, Y.; Wang, J.; Liu, H.; Macromolecules 2002, 35, 6426. DOI: http://dx.doi.org/10.1021/ma0105284 |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access